
A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree–Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake


Abstract
:
1. Introduction
| BDE | |
| IP | |
| EA | |
| PA |
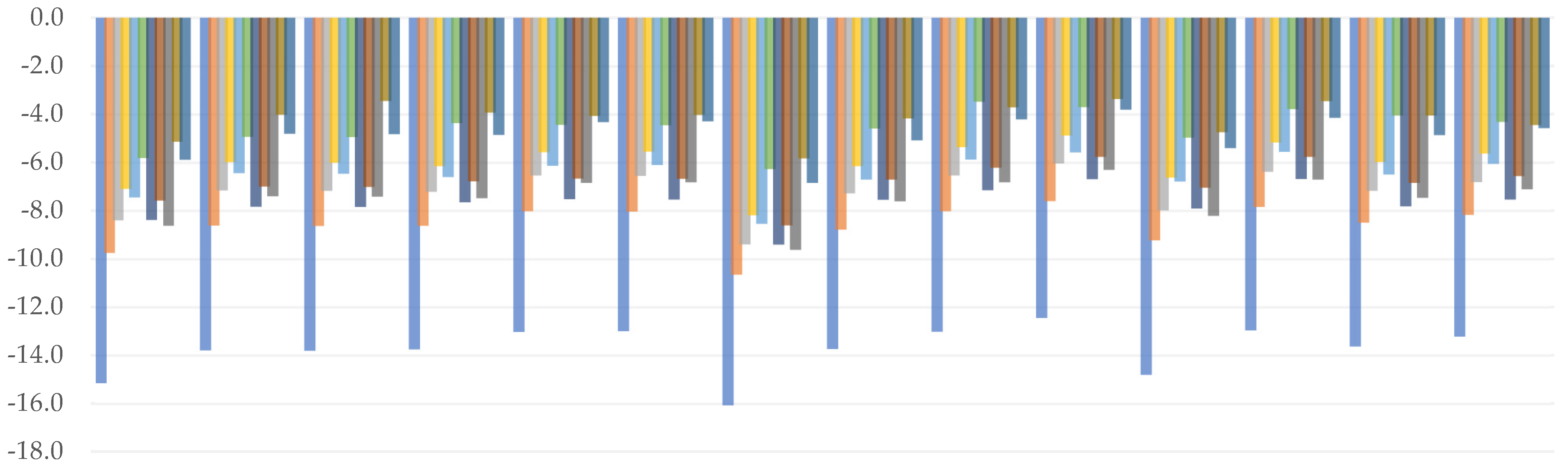
2. Results
3. Discussion
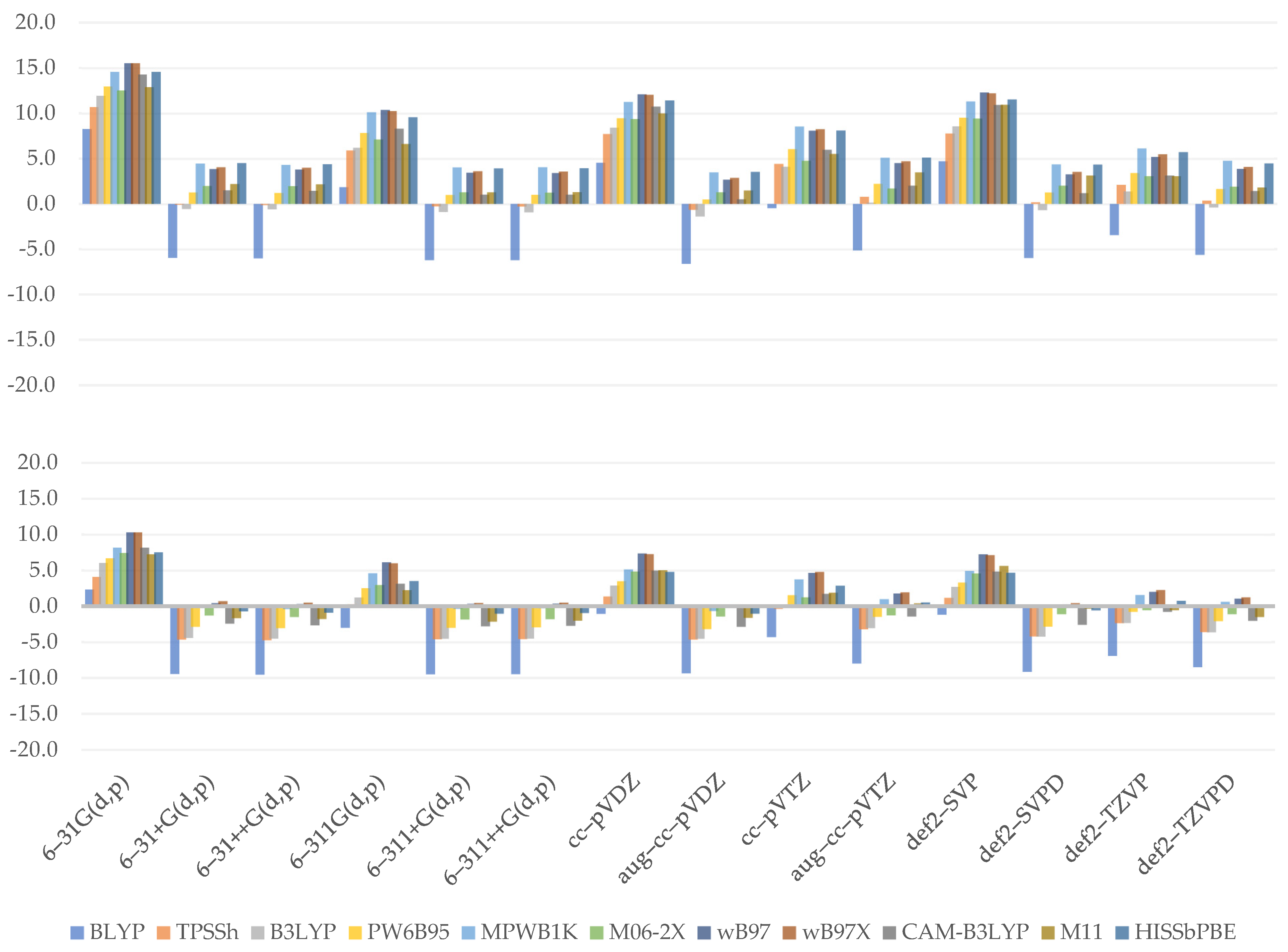
3.1. Bond Dissociation Enthalpy
3.2. Adiabatic Ionization Potential
3.3. Adiabatic Electron Affinity
3.4. Proton Affinity
3.5. Section Conclusions
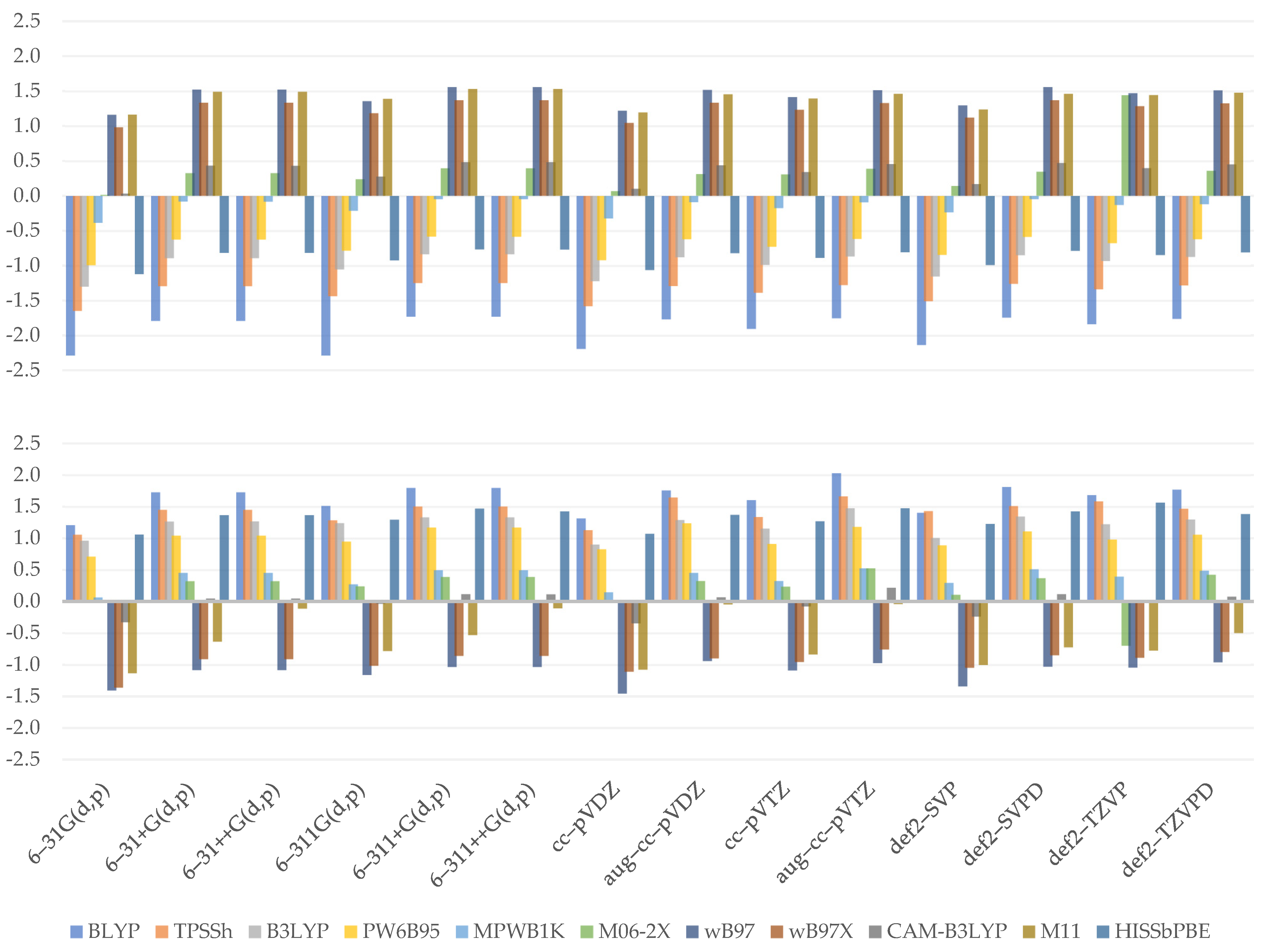
3.6. Performance Evaluation
3.7. Janak’s Theorem Applicability
4. Materials and Methods
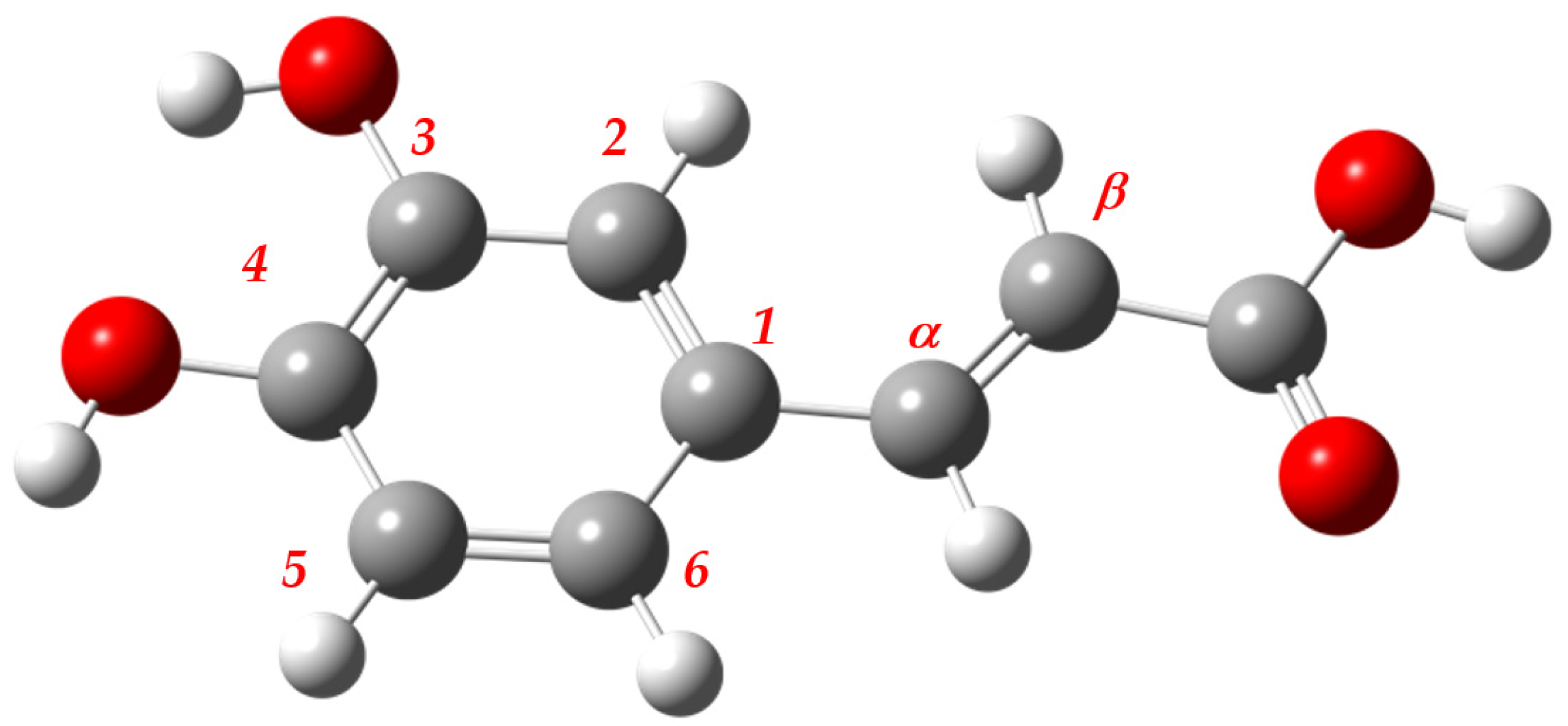
4.1. Caffeic Acid as a Reference Structure
4.2. On Functionals and Basis Set Choice
4.3. DFT Calculations
4.4. Linear Regression Models
- %HF at short range (SR, [0, 100]);
- %HF at middle range (MR, [0, 100]);
- %HF at long range (LR, [0, 100]);
- Number of basis functions (NBF, N > 0);
- Presence of valence double basis set (ζ, 0 ∨ 1);
- presence of diffuse function (D, 0 ∨ 1).
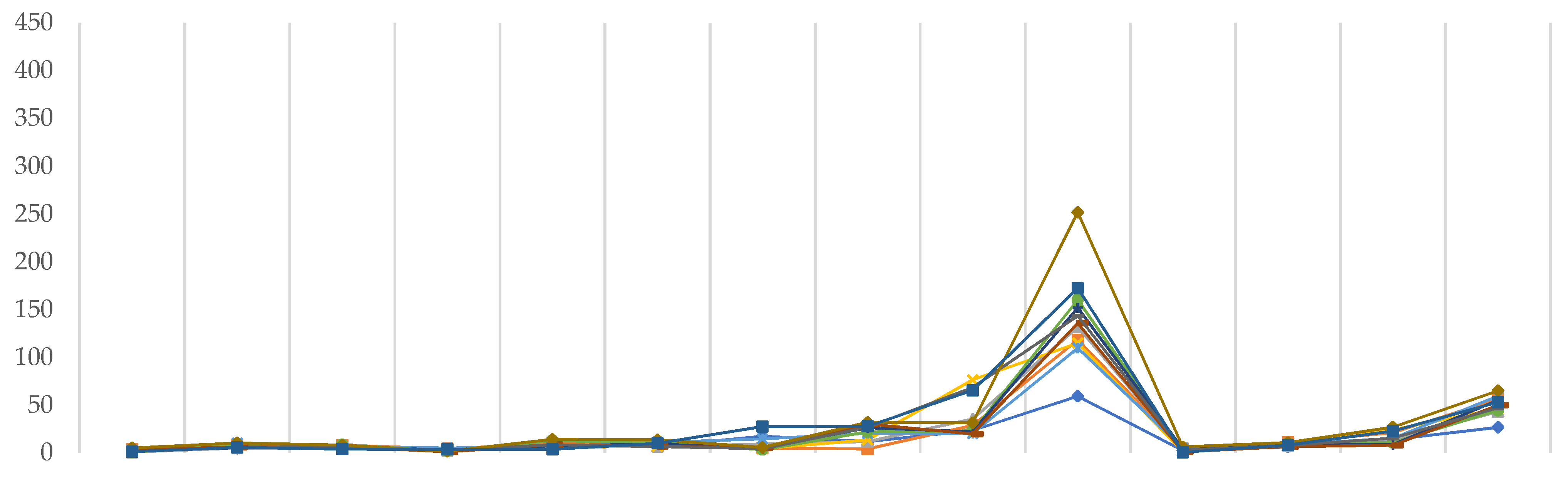
4.5. Computational Performance
4.6. Janak’s Theorem Revisited
4.7. Scoring Function
- Hydroxyl bond length at C3;
- Hydroxyl bond length at C4;
- Hydrogen bond length;
- Bond dissociation enthalpy at C3;
- Bond dissociation enthalpy at C4;
- Adiabatic electron affinity;
- Adiabatic ionization potential;
- Proton affinity at C3;
- Proton affinity at C4.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food Antioxidants: Chemical Insights at the Molecular Level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Nimse, S.B.; Pal, D. Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.R. Reactive oxygen species or reactive sulfur species: Why we should consider the latter. J. Exp. Biol. 2020, 223, jeb196352. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicryl-hydrazyl (DPPH) for estimating anti-oxidant activity. Songklanakarin J. Sci. Technol. 2004, 26, 211–219. [Google Scholar]
- Galano, A.; Raúl Alvarez-Idaboy, J. Computational strategies for predicting free radical scavengers’ protection against oxidative stress: Where are we and what might follow? Int. J. Quantum Chem. 2019, 119, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Alvarez-Idaboy, J.R. Kinetics of radical-molecule reactions in aqueous solution: A benchmark study of the performance of density functional methods. J. Comput. Chem. 2014, 35, 2019–2026. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar]
- Perdew, J.P. Accurate Density Functional for the Energy: Real-Space Cutoff of the Gradient Expansion for the Exchange Hole. Phys. Rev. Lett. 1985, 55, 1665–1668. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Roussel, M.R. Exchange holes in inhomogeneous systems: A coordinate-space model. Phys. Rev. A 1989, 39, 3761–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Dilabio, G.A.; Otero-de-la-Roza, A. Noncovalent Interactions in Density Functional Theory. Rev. Comput. Chem. 2016, 29, 1–97. [Google Scholar]
- Mata, R.A.; Suhm, M.A. Benchmarking Quantum Chemical Methods: Are We Heading in the Right Direction? Angew. Chem.-Int. Ed. 2017, 56, 11011–11018. [Google Scholar] [CrossRef]
- La Rocca, M.V.; Rutkowski, M.; Ringeissen, S.; Gomar, J.; Frantz, M.C.; Ngom, S.; Adamo, C. Benchmarking the DFT methodology for assessing antioxidant-related properties: Quercetin and edaravone as case studies. J. Mol. Model. 2016, 22. [Google Scholar] [CrossRef]
- De Souza, G.L.C.; Peterson, K.A. Benchmarking Antioxidant-Related Properties for Gallic Acid through the Use of DFT, MP2, CCSD, and CCSD(T) Approaches. J. Phys. Chem. A 2021, 125, 198–208. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Denisova, T.G.; Denisov, E.T. Dissociation energies of O-H bonds in natural antioxidants. Russ. Chem. Bull. 2008, 57, 1858–1866. [Google Scholar] [CrossRef]
- Ye, L.; Zhou, S.; Liu, L.; Liu, L.; Waters, D.L.E.; Zhong, K.; Zhou, X.; Ma, X.; Liu, X. Phenolic compounds and antioxidant capacity of brown rice in China. Int. J. Food Eng. 2016, 12, 537–546. [Google Scholar] [CrossRef]
- Vulić, J.J.; Ćebović, T.N.; Ćanadanović-Brunet, J.M.; ĆEtković, G.S.; Čanadanović, V.M.; Djilas, S.M.; Tumbas Šaponjac, V.T. In vivo and in vitro antioxidant effects of beetroot pomace extracts. J. Funct. Foods 2014, 6, 168–175. [Google Scholar] [CrossRef]
- Farah, A.; Donangelo, C.M. Phenolic compounds in coffee. Braz. J. Plant Physiol. 2006, 18, 23–36. [Google Scholar] [CrossRef]
- Jeremić, S.; Radenković, S.; Filipović, M.; Antić, M.; Amić, A.; Marković, Z. Importance of hydrogen bonding and aromaticity indices in QSAR modeling of the antioxidative capacity of selected (poly)phenolic antioxidants. J. Mol. Graph. Model. 2017, 72, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Leopoldini, M.; Marino, T.; Russo, N.; Toscano, M. Antioxidant properties of phenolic compounds: H-atom versus electron transfer mechanism. J. Phys. Chem. A 2004, 108, 4916–4922. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Softwares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- VanBesiena, E.; Marques, M.P.M. Ab initio conformational study of caffeic acid. J. Mol. Struct. Theochem. 2003, 625, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Design of density functionals that are broadly accurate for thermochemistry, thermochemical kinetics, and nonbonded interactions. J. Phys. Chem. A 2005, 109, 5656–5667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Da Chai, J.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Peverati, R.; Truhlar, D.G. Improving the accuracy of hybrid meta-GGA density functionals by range separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scuseria, G.E.; Savin, A. Assessment of a middle-range hybrid functional. J. Chem. Theory Comput. 2008, 4, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, K.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Accuracy of AH n equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z =11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, H.; Zheng, J.; Liang, G. Structure-thermodynamics-antioxidant activity relationships of selected natural phenolic acids and derivatives: An experimental and theoretical evaluation. PLoS ONE 2015, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, J.; Ma, L.; Li, J.; Shahzad, N.; Kim, C.K. Structure-antioxidant activity relationship of methoxy, phenolic hydroxyl, and carboxylic acid groups of phenolic acids. Sci. Rep. 2020, 10, 2611. [Google Scholar] [CrossRef] [PubMed]
- Tošović, J.; Bren, U. Antioxidative action of ellagic acid—A kinetic DFT study. Antioxidants 2020, 9, 0587. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Chen, D.F.; Deng, G.; Guo, R.; Fu, Z.M. The antioxidative activity of piceatannol and its different derivatives: Antioxidative mechanism analysis. Phytochemistry 2018, 156, 184–192. [Google Scholar] [CrossRef]
- Milenković, D.; Đorović, J.; Petrović, V.; Avdović, E.; Marković, Z. Hydrogen atom transfer versus proton coupled electron transfer mechanism of gallic acid with different peroxy radicals. React. Kinet. Mech. Catal. 2018, 123, 215–230. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Deng, G.; Guo, R.; Chen, D.F.; Fu, Z.M. Substituent effects on the radical scavenging activity of isoflavonoid. Int. J. Mol. Sci. 2019, 20, 397. [Google Scholar] [CrossRef] [Green Version]
- Pérez-González, A.; Alvarez-Idaboy, J.R.; Galano, A. Dual antioxidant/pro-oxidant behavior of the tryptophan metabolite 3-hydroxyanthranilic acid: A theoretical investigation of reaction mechanisms and kinetics. New J. Chem. 2017, 41, 3829–3845. [Google Scholar] [CrossRef]
- Yang, L.; Liu, H.; Xia, D.; Wang, S. Antioxidant properties of camphene-based thiosemicarbazones: Experimental and theoretical evaluation. Molecules 2020, 25, 1192. [Google Scholar] [CrossRef] [Green Version]
- De Wang, Q.; Sun, Y.; Sun, M.M.; Liang, J.H. Chemical kinetics of hydrogen atom abstraction from propargyl sites by hydrogen and hydroxy radicals. Int. J. Mol. Sci. 2019, 20, 3227. [Google Scholar] [CrossRef] [Green Version]
- Manrique-de-la-Cuba, M.F.; Gamero-Begazo, P.; Valencia, D.E.; Barazorda-Ccahuana, H.L.; Gómez, B. Theoretical study of the antioxidant capacity of the flavonoids present in the Annona muricata (Soursop) leaves. J. Mol. Model. 2019, 25, 200. [Google Scholar] [CrossRef] [PubMed]
- Babiaka, S.B.; Nia, R.; Abuga, K.O.; Mbah, J.A.; de Nziko, V.P.N.; Paper, D.H.; Ntie-Kang, F. Antioxidant potential of flavonoid glycosides from Manniophyton fulvum Müll. (Euphorbiaceae): Identification and molecular modeling. Sci. Afr. 2020, 8, e00423. [Google Scholar] [CrossRef]
- Leopoldini, M.; Chiodo, S.G.; Russo, N.; Toscano, M. Detailed Investigation of the OH Radical Quenching by Natural Antioxidant Caffeic Acid Studied by Quantum Mechanical Models. J. Chem. Theory Comput. 2011, 7, 4218–4233. [Google Scholar] [CrossRef]
- Iuga, C.; Alvarez-Idaboy, J.R.; Russo, N. Antioxidant Activity of trans -Resveratrol toward Hydroxyl and Hydroperoxyl Radicals: A Quantum Chemical and Computational Kinetics Study. J. Org. Chem. 2012, 77, 3868–3877. [Google Scholar] [CrossRef] [PubMed]
- Marković, Z.; Amić, D.; Milenković, D.; Dimitrić-Marković, J.M.; Marković, S. Examination of the chemical behavior of the quercetin radical cation towards some bases. Phys. Chem. Chem. Phys. 2013, 15, 7370. [Google Scholar] [CrossRef]
- Kirschner, K.N.; Reith, D.; Heiden, W. The performance of Dunning, Jensen, and Karlsruhe basis sets on computing relative energies and geometries. Soft Mater. 2020, 18, 200–214. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, B.C.; Bennett, C.J. A Comparison of Medium-Sized Basis Sets for the Prediction of Geometries, Vibrational Frequencies, Infrared Intensities and Raman Activities for Water. J. Phys. Conf. Ser. 2019, 1290, 012013. [Google Scholar] [CrossRef]
- Vikramaditya, T.; Lin, S.-T. Assessing the role of Hartree-Fock exchange, correlation energy and long range corrections in evaluating ionization potential, and electron affinity in density functional theory. J. Comput. Chem. 2017, 38, 1844–1852. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Amorati, R.; Pedulli, G.F.; Cabrini, L.; Zambonin, L.; Landi, L. Solvent and pH effects on the antioxidant activity of caffeic and other phenolic acids. J. Agric. Food Chem. 2006, 54, 2932–2937. [Google Scholar] [CrossRef]
- Guerrero, A.; Baer, T.; Chana, A.; González, J.; Dávalos, J.Z. Gas phase acidity measurement of local acidic groups in multifunctional species: Controlling the binding sites in hydroxycinnamic acids. J. Am. Chem. Soc. 2013, 135, 9681–9690. [Google Scholar] [CrossRef]
- Weigend, F.; Köhn, A.; Hättig, C. Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations. J. Chem. Phys. 2002, 116, 3175–3183. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J. Comput. Chem. 2003, 24, 1740–1747. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Martinez, A.; Martínez, A.; Vargas, R.; Galano, A. Donator—Acceptor Map and Work Function for Linear Polyene-Conjugated Molecules. A. J. Phys. Chem. B 2009, 113, 12113–12120. [Google Scholar] [CrossRef]
- Martínez, A. Donator acceptor map of psittacofulvins and anthocyanins: Are they good antioxidant substances? J. Phys. Chem. B 2009, 113, 4915–4921. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Rodríguez-Girones, M.A.; Barbosa, A.; Costas, M. Donator acceptor map for carotenoids, melatonin and vitamins. J. Phys. Chem. A 2008, 112, 9037–9042. [Google Scholar] [CrossRef]
- Janak, J.F. Proof that ∂E∂ni=ε in density-functional theory. Phys. Rev. B 1978, 18, 7165–7168. [Google Scholar] [CrossRef]
- Tsuneda, T.; Hirao, K. Self-interaction corrections in density functional theory. J. Chem. Phys. 2014, 140, 18A513. [Google Scholar] [CrossRef]
- Amić, A.; Marković, Z.; Klein, E.; Dimitrić Marković, J.M.; Milenković, D. Theoretical study of the thermodynamics of the mechanisms underlying antiradical activity of cinnamic acid derivatives. Food Chem. 2018, 246, 481–489. [Google Scholar] [CrossRef]
- Urbaniak, A.; Kujawski, J.; Czaja, K.; Szelag, M. Antioxidant properties of several caffeic acid derivatives: A theoretical study. Comptes Rendus Chim. 2017, 20, 1072–1082. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BLYP | TPSSh | B3LYP | PW6B95 | MPWB1K | M06-2X | wB97 | wB97X | CAM–B3LYP | M11 | HISSbPBE | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 6–31G(d,p) | 41% | 41% | 46% | 46% | 41% | 54% | 53% | 51% | 55% | 51% | 46% |
| 6–31+G(d,p) | 32% | 56% | 51% | 61% | 57% | 66% | 61% | 61% | 65% | 70% | 60% |
| 6–31++G(d,p) | 32% | 56% | 51% | 61% | 57% | 66% | 61% | 61% | 63% | 70% | 60% |
| 6–311G(d,p) | 56% | 61% | 60% | 56% | 37% | 78% | 51% | 51% | 61% | 65% | 39% |
| 6–311+G(d,p) | 46% | 53% | 51% | 63% | 52% | 65% | 58% | 58% | 63% | 65% | 58% |
| 6–311++G(d,p) | 46% | 53% | 48% | 63% | 52% | 70% | 63% | 58% | 63% | 65% | 58% |
| cc–pVDZ | 39% | 46% | 51% | 46% | 44% | 53% | 46% | 46% | 47% | 46% | 44% |
| aug–cc–pVDZ | 29% | 51% | 48% | 56% | 42% | 71% | 53% | 56% | 61% | 63% | 51% |
| cc–pVTZ | 44% | 48% | 46% | 48% | 32% | 66% | 53% | 48% | 49% | 63% | 48% |
| aug–cc–pVTZ | 34% | 48% | 48% | 53% | 40% | 63% | 53% | 53% | 58% | 58% | 48% |
| def2–SVP | 41% | 46% | 56% | 46% | 47% | 61% | 46% | 53% | 47% | 56% | 51% |
| def2–SVPD | 39% | 53% | 48% | 63% | 52% | 69% | 58% | 53% | 63% | 68% | 68% |
| def2–TZVP | 32% | 56% | 51% | 56% | 47% | 65% | 51% | 51% | 56% | 63% | 48% |
| def2–TZVPD | 29% | 48% | 44% | 53% | 40% | 64% | 53% | 53% | 58% | 63% | 58% |
| Substance | BDEcalc | BDEexp [22] | Δ(BDEcalc − BDEexp) |
|---|---|---|---|
| Catechin | 76.9 | 83.2 (C4′) | −6.3 |
| Chrysin | 92.6 | 85.4 (C7) | 7.2 |
| (–)-Epicatechin | 82.4 | 82.0 (C4′) | 0.4 |
| (–)-Epigallocatechin | 79.4 | 82.4 (C4′) | −3.0 |
| Fisetin | 86.5 | 83.2 (C4′) | 3.3 |
| Galangin | 92.5 | 86.8 (C7) | 5.7 |
| Gallic acid | 81.0 | 83.0 (C4) | −2.0 |
| Luteolin | 78.1 | 81.9 (C4′) | −3.8 |
| Myricetin | 79.6 | 81.5 (C4′) | −1.9 |
| Quercetin | 78.6 | 82.0 (C4′) | −3.4 |
| Taxifolin | 86.6 | 82.1 (C4′) | 4.5 |
| MAE: 3.8 kcal/mol | RMSE: 4.2 kcal/mol | ||
| Method | Type | %HF (SR/MR/LR) |
|---|---|---|
| BLYP [30,31] | GGA | (0%)/0%/(0%) |
| TPSSh [32,33] | GH meta–GGA | (10%)/10%/(10%) |
| B3LYP [31,34] | GH GGA | (20%)/20%/(20%) |
| PW6B95 [35] | GH meta–GGA | (28%)/28%/(28%) |
| MPWB1K [36] | GH meta–GGA | (44%)/44%/(44%) |
| M06–2X [21] | GH meta–GGA | (54%)/54%/(54%) |
| WB97 [37] | RSH GGA | 0%/0%/100% |
| WB97X [37] | RSH GGA | 15.77%/0%/100% |
| CAM–B3LYP [38] | RSH GGA | 19%/0%/65% |
| M11 [39] | RSH meta–GGA | 42.8%/0%/100% |
| HISSbPBE [40] | RSH GGA | 0%/60%/0% |
| Family | Basis Set | Number of Basis Functions |
|---|---|---|
| Pople’s [42,43] | 6–31G(d,p) | 235 |
| 6–31+G(d,p) [41,44,45,46] | 287 | |
| 6–31++G(d,p) | 295 | |
| 6–311G(d,p) | 282 | |
| 6–311+G(d,p) [47,48] | 334 | |
| 6–311++G(d,p) | 342 | |
| Dunning’s [49,50] | cc–pVDZ | 222 |
| aug–cc–pVDZ | 371 | |
| cc–pVTZ | 502 | |
| aug–cc–pVTZ | 782 | |
| Ahlrich’s [51,52] | def2–SVP | 222 |
| def2–SVPD | 336 | |
| def2–TZVP | 451 | |
| def2–TZVPD | 565 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiegel, M.; Gamian, A.; Sroka, Z. A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree–Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake. Molecules 2021, 26, 5058. https://doi.org/10.3390/molecules26165058
Spiegel M, Gamian A, Sroka Z. A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree–Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake. Molecules. 2021; 26(16):5058. https://doi.org/10.3390/molecules26165058
Chicago/Turabian StyleSpiegel, Maciej, Andrzej Gamian, and Zbigniew Sroka. 2021. "A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree–Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake" Molecules 26, no. 16: 5058. https://doi.org/10.3390/molecules26165058
APA StyleSpiegel, M., Gamian, A., & Sroka, Z. (2021). A Statistically Supported Antioxidant Activity DFT Benchmark—The Effects of Hartree–Fock Exchange and Basis Set Selection on Accuracy and Resources Uptake. Molecules, 26(16), 5058. https://doi.org/10.3390/molecules26165058