
Species Differences in Metabolism of Soluble Epoxide Hydrolase Inhibitor, EC1728, Highlight the Importance of Clinically Relevant Screening Mechanisms in Drug Development


 ,
, 
Abstract
:1. Introduction
2. Results
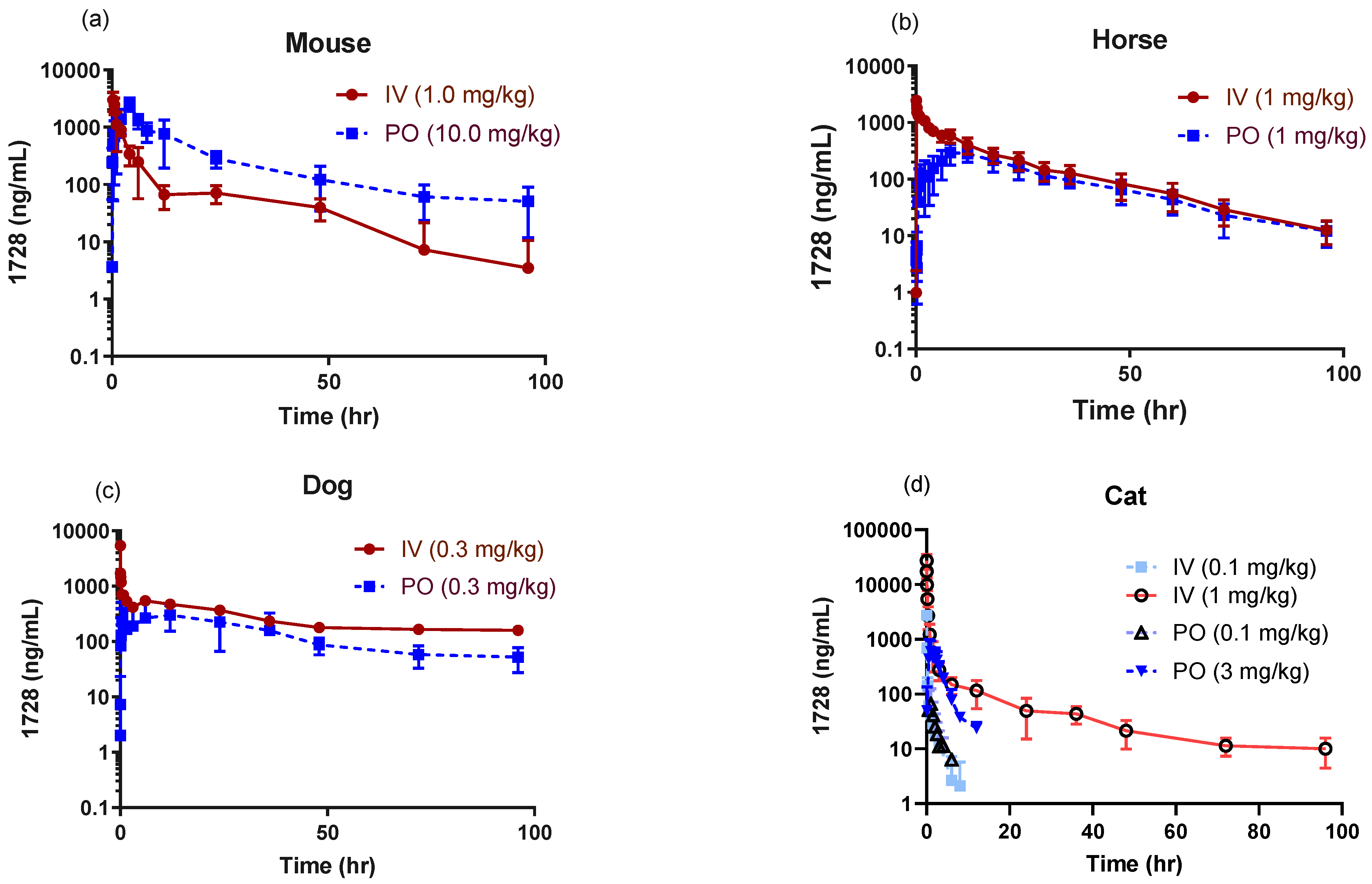
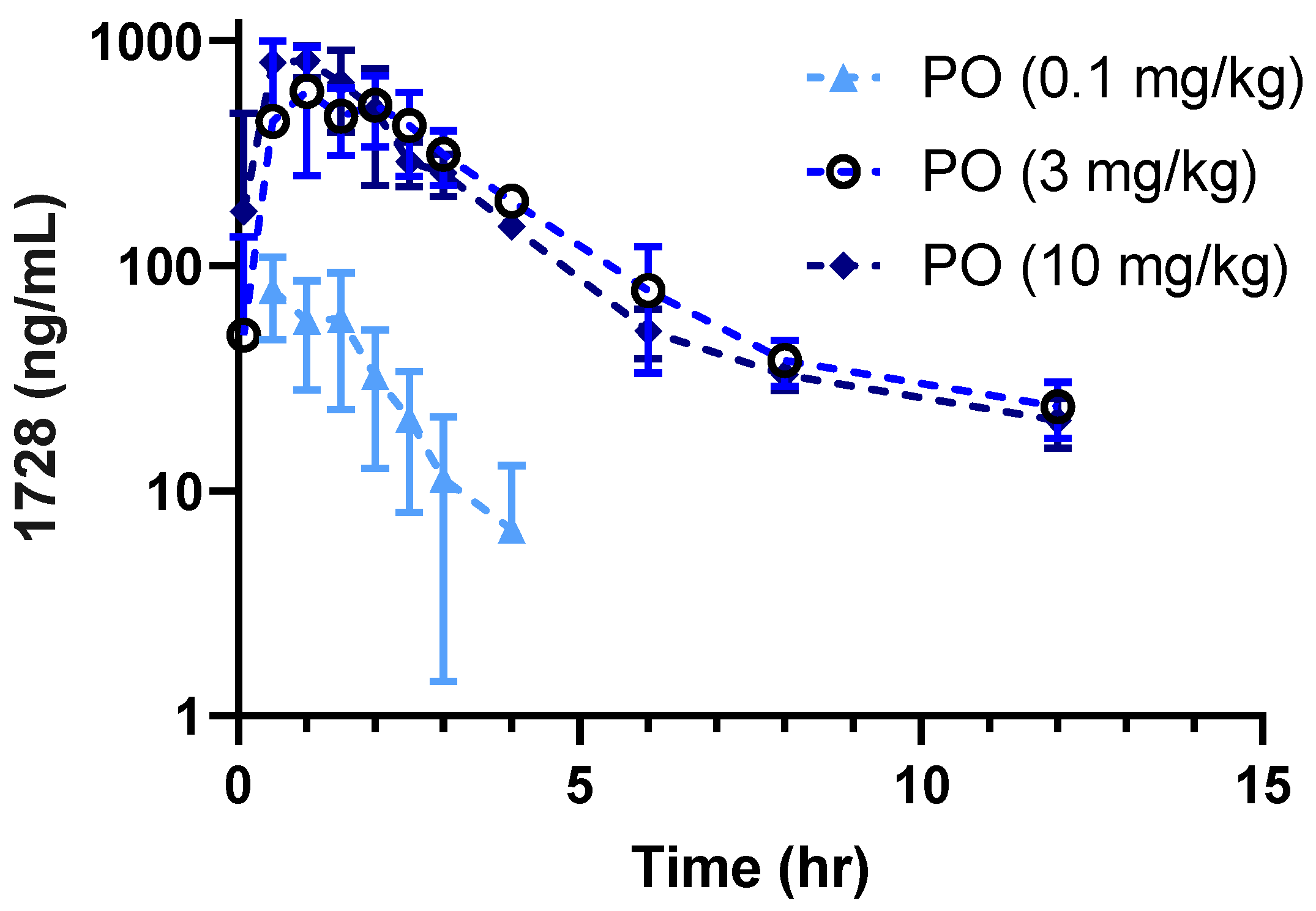
2.1. Pharmacokinetic Profiles in Companion Animals
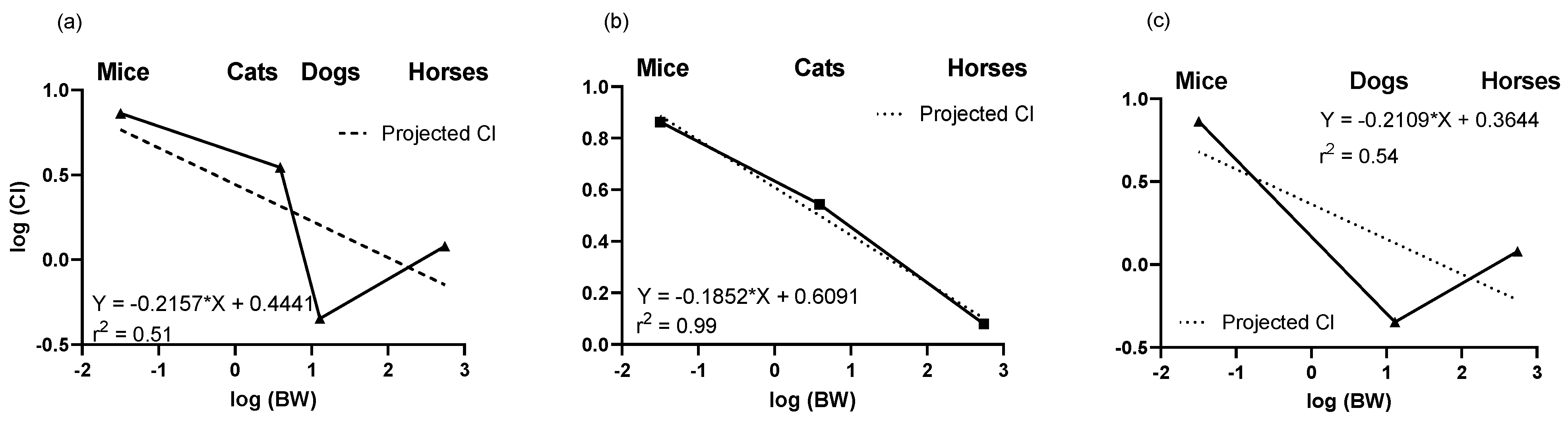
2.2. Predictions of Clearance Based on Allometry
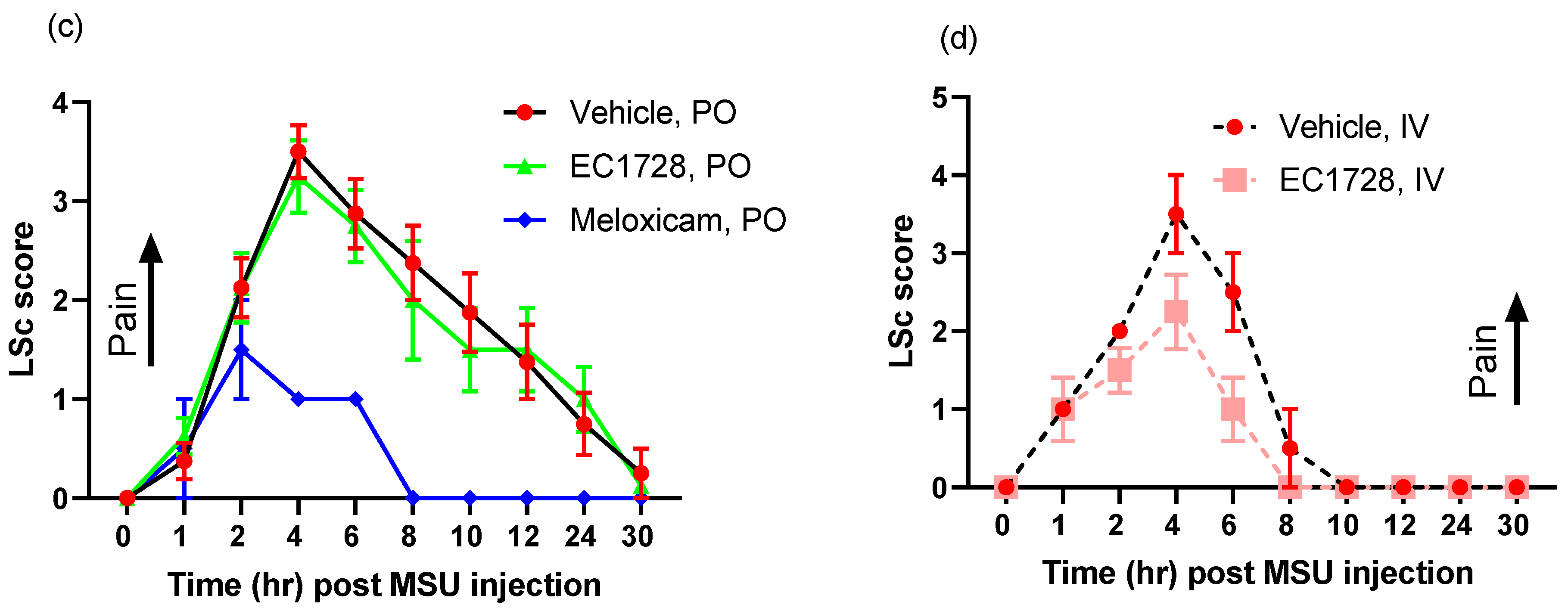
2.3. Efficacy of EC1728 in a Monosodium Urate Model of Inflammatory Pain
3. Discussion
4. Materials and Methods
4.1. Solubility and Potency
4.2. Test Article and Preparation of Dosing Solution
4.3. In Vitro Metabolism and Plasma Protein Binding
4.4. In-Vivo Studies
4.5. Feline Efficacy Studies
Anaesthesia
4.6. Pharmacokinetic Parameter Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Modric, S. Regulatory framework for the availability and use of animal drugs in the United States. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 1005–1012. [Google Scholar] [CrossRef]
- Richardson, S.J.; Bai, A.; Kulkarni, A.A.; Moghaddam, M.F. Efficiency in Drug Discovery: Liver S9 Fraction Assay as a Screen for Metabolic Stability. Drug Metab. Lett. 2016, 10, 83–90. [Google Scholar] [CrossRef] [Green Version]
- Court, M.H. Canine cytochrome P-450 pharmacogenetics. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Visser, M.; Weber, K.L.; Lyons, L.A.; Rincon, G.; Boothe, D.M.; Merritt, D.A. Identification and quantification of domestic feline cytochrome P450 transcriptome across multiple tissues. J. Vet. Pharmacol. Ther. 2019, 42, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Court, M.H. Feline drug metabolism and disposition: Pharmacokinetic evidence for species differences and molecular mechanisms. Vet. Clin. N. Am. Small Anim. Pract. 2013, 43, 1039–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugonnard, M.; Leblond, A.; Keroack, S.; Cadoré, J.L.; Troncy, E. Attitudes and concerns of French veterinarians towards pain and analgesia in dogs and cats. Vet. Anaesth. Analg. 2004, 31, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Capner, C.A.; Lascelles, B.D.; Waterman-Pearson, A.E. Current British veterinary attitudes to perioperative analgesia for dogs. Vet. Rec. 1999, 145, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Sanders, K.M. Opiates, the Pylorus, and Gastroparesis. Gastroenterology 2020, 159, 414–421. [Google Scholar] [CrossRef]
- Kamata, M.; Nagahama, S.; Kakishima, K.; Sasaki, N.; Nishimura, R. Comparison of behavioral effects of morphine and fentanyl in dogs and cats. J. Vet. Med. Sci. 2012, 74, 231–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clutton, R.E. Opioid Analgesia in Horses. Vet. Clin. N. Am. Equine Pract. 2010, 26, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Simon, B.T.; Steagall, P.V. The present and future of opioid analgesics in small animal practice. J. Vet. Pharmacol. Ther. 2017, 40, 315–326. [Google Scholar] [CrossRef]
- APCC. Announcing: The Top 10 Pet Toxins! Available online: https://www.aspca.org/news/announcing-top-10-pet-toxins (accessed on 21 April 2021).
- Khan, S.A.; McLean, M.K. Toxicology of frequently encountered nonsteroidal anti-inflammatory drugs in dogs and cats. Vet. Clin. N. Am. Small Anim. Pract. 2012, 42, 289–306. [Google Scholar] [CrossRef]
- Knych, H.K. Nonsteroidal Anti-inflammatory Drug Use in Horses. Vet. Clin. N. Am. Equine Pract. 2017, 33, 1–15. [Google Scholar] [CrossRef]
- Adrian, D.; Papich, M.G.; Baynes, R.; Stafford, E.; Lascelles, B.D.X. The pharmacokinetics of gabapentin in cats. J. Vet. Intern. Med. 2018, 32, 1996–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, J.R.; Grubb, T.L.; Green, S.; Cox, S.; Villarino, N.F. Plasma disposition of gabapentin after the intragastric administration of escalating doses to adult horses. J. Vet. Intern. Med. 2020, 34, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Guedes, A.G.P.; Meadows, J.M.; Pypendop, B.H.; Johnson, E.G.; Zaffarano, B. Assessment of the effects of gabapentin on activity levels and owner-perceived mobility impairment and quality of life in osteoarthritic geriatric cats. J. Am. Vet. Med. Assoc. 2018, 253, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Philbrick, A. Pain Management in Companion Animals. In America’s Pharmacist; National Community Pharmacists Association: Alexandria, VA, USA, 2015; pp. 43–52. [Google Scholar]
- Peck, C. A retrospective questionnaire study. In The Adverse Effect Profile of Gabapentin in Dogs; Department of Clinical Science, Swedish University of Agricultural Sciences: Uppsala, Sweden, 2017. [Google Scholar]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Inceoglu, B.; Hammock, B.D. Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat. 2011, 96, 76–83. [Google Scholar] [CrossRef] [Green Version]
- McReynolds, C.; Morisseau, C.; Wagner, K.; Hammock, B. Epoxy Fatty Acids Are Promising Targets for Treatment of Pain, Cardiovascular Disease and Other Indications Characterized by Mitochondrial Dysfunction, Endoplasmic Stress and Inflammation. Adv. Exp. Med. Biol. 2020, 1274, 71–99. [Google Scholar] [CrossRef]
- Shihadih, D.S.; Harris, T.R.; Kodani, S.D.; Hwang, S.H.; Lee, K.S.S.; Mavangira, V.; Hamamoto, B.; Guedes, A.; Hammock, B.D.; Morisseau, C. Selection of Potent Inhibitors of Soluble Epoxide Hydrolase for Usage in Veterinary Medicine. Front. Vet. Sci. 2020, 7, 580. [Google Scholar] [CrossRef]
- Guedes, A.; Galuppo, L.; Hood, D.; Hwang, S.H.; Morisseau, C.; Hammock, B.D. Soluble epoxide hydrolase activity and pharmacologic inhibition in horses with chronic severe laminitis. Equine Vet. J. 2017, 49, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McReynolds, C.B.; Hwang, S.H.; Yang, J.; Wan, D.; Wagner, K.; Morisseau, C.; Li, D.; Schmidt, W.K.; Hammock, B.D. Pharmaceutical Effects of Inhibiting the Soluble Epoxide Hydrolase in Canine Osteoarthritis. Front. Pharmacol. 2019, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.; Inceoglu, B.; Dong, H.; Yang, J.; Hwang, S.H.; Jones, P.; Morisseau, C.; Hammock, B.D. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur. J. Pharmacol. 2013, 700, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.; Yang, J.; Inceoglu, B.; Hammock, B.D. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J. Pain Off. J. Am. Pain Soc. 2014, 15, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Adamska, M.; Cronin, A.; Di Giallonardo, F.; Burgener, J.; Marowsky, A.; Falck, J.R.; Morisseau, C.; Hammock, B.D.; Gruzdev, A.; et al. EH3 (ABHD9): The first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J. Lipid Res. 2012, 53, 2038–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulu, A.; Appt, S.; Morisseau, C.; Hwang, S.H.; Jones, P.D.; Rose, T.E.; Dong, H.; Lango, J.; Yang, J.; Tsai, H.J.; et al. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br. J. Pharmacol. 2012, 165, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Liu, J.Y.; Wagner, K.M.; Pakhomova, S.; Dong, H.; Morisseau, C.; Fu, S.H.; Yang, J.; Wang, P.; Ulu, A.; et al. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem. 2014, 57, 7016–7030. [Google Scholar] [CrossRef] [PubMed]
- Guedes, A.G.P.; Aristizabal, F.; Sole, A.; Adedeji, A.; Brosnan, R.; Knych, H.; Yang, J.; Hwang, S.H.; Morisseau, C.; Hammock, B.D. Pharmacokinetics and antinociceptive effects of the soluble epoxide hydrolase inhibitor t-TUCB in horses with experimentally induced radiocarpal synovitis. J. Vet. Pharmacol. Ther. 2018, 41, 230–238. [Google Scholar] [CrossRef]
- Jones, P.D.; Wolf, N.M.; Morisseau, C.; Whetstone, P.; Hock, B.; Hammock, B.D. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal. Biochem. 2005, 343, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Sams, R.A. Principles of drug disposition in the horse. Vet. Clin. N. Am. Equine Pract. 1987, 3, 221–250. [Google Scholar] [CrossRef]
- Visser, M.; Zaya, M.J.; Locuson, C.W.; Boothe, D.M.; Merritt, D.A. Comparison of predicted intrinsic hepatic clearance of 30 pharmaceuticals in canine and feline liver microsomes. Xenobiotica Fate Foreign Compd. Biol. Syst. 2019, 49, 177–186. [Google Scholar] [CrossRef]
- Lautz, L.S.; Jeddi, M.Z.; Girolami, F.; Nebbia, C.; Dorne, J.L.C.M. Metabolism and pharmacokinetics of pharmaceuticals in cats (Felix sylvestris catus) and implications for the risk assessment of feed additives and contaminants. Toxicol. Lett. 2021, 338, 114–127. [Google Scholar] [CrossRef]
- Liu, J.Y.; Lin, Y.P.; Qiu, H.; Morisseau, C.; Rose, T.E.; Hwang, S.H.; Chiamvimonvat, N.; Hammock, B.D. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2013, 48, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Hammock, B.D.; McReynolds, C.B.; Wagner, K.; Buckpitt, A.; Cortes-Puch, I.; Croston, G.; Lee, K.S.S.; Yang, J.; Schmidt, W.K.; Hwang, S.H. Movement to the Clinic of Soluble Epoxide Hydrolase Inhibitor EC5026 as an Analgesic for Neuropathic Pain and for Use as a Nonaddictive Opioid Alternative. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Chen, D.; Whitcomb, R.; MacIntyre, E.; Tran, V.; Do, Z.N.; Sabry, J.; Patel, D.V.; Anandan, S.K.; Gless, R.; Webb, H.K. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J. Clin. Pharmacol. 2012, 52, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Shugarts, S.; Benet, L.Z. The role of transporters in the pharmacokinetics of orally administered drugs. Pharm. Res. 2009, 26, 2039–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Benet, L.Z. Predicting drug disposition via application of BCS: Transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Goh, L.B.; Spears, K.J.; Yao, D.; Ayrton, A.; Morgan, P.; Roland Wolf, C.; Friedberg, T. Endogenous drug transporters in in vitro and in vivo models for the prediction of drug disposition in man. Biochem. Pharmacol. 2002, 64, 1569–1578. [Google Scholar] [CrossRef]
- Cho, S.M.; Park, S.W.; Kim, N.H.; Park, J.A.; Yi, H.; Cho, H.J.; Park, K.H.; Hwang, I.; Shin, H.C. Expression of intestinal transporter genes in beagle dogs. Exp. Ther. Med. 2013, 5, 308–314. [Google Scholar] [CrossRef]
- Lee, K.S.; Morisseau, C.; Yang, J.; Wang, P.; Hwang, S.H.; Hammock, B.D. Forster resonance energy transfer competitive displacement assay for human soluble epoxide hydrolase. Anal. Biochem. 2013, 434, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Lee, K.S.S.; Yang, J.; Hammock, B.D. Target-Mediated Drug Disposition-A Class Effect of Soluble Epoxide Hydrolase Inhibitors. J. Clin. Pharmacol. 2021, 61, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, S.; Sharkey, M.; Mealey, K.; Ostrander, E.A.; Martinez, M. Pharmacogenetic and metabolic differences between dog breeds: Their impact on canine medicine and the use of the dog as a preclinical animal model. AAPS J. 2008, 10, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dance, A. Why the sexes don’t feel pain the same way. Nature 2019, 567, 448–450. [Google Scholar] [CrossRef] [Green Version]
- Hwang, S.H.; Tsai, H.J.; Liu, J.Y.; Morisseau, C.; Hammock, B.D. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J. Med. Chem. 2007, 50, 3825–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecic, S.; Zeki, A.A.; Xu, X.; Jin, G.Y.; Zhang, S.; Kodani, S.; Halim, M.; Morisseau, C.; Hammock, B.D.; Deng, S.X. Novel piperidine-derived amide sEH inhibitors as mediators of lipid metabolism with improved stability. Prostaglandins Other Lipid Mediat. 2018, 136, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, I.; Martinez, M.; Hunter, R.P. Interspecies allometric scaling. Part I: Prediction of clearance in large animals. J. Vet. Pharmacol. Ther. 2006, 29, 415–423. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solubility (µg/mL) a | Melting Point (°C) | cLogP b | Potency (Ki) (ng/mL) c | |||
|---|---|---|---|---|---|---|---|
| Mouse | Cat | Dog | Horse | ||||
EC1728 (t-TUCB)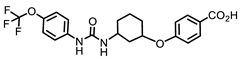 | 5 | 240–244 (242) | 4.7 | 11 | 0.18 ± 0.004 | 0.39 ± 0.02 | 25.87 |
| Piperidine Series | |||||||
UC1770 (TPPU) | 60 | 198.2–200.8 (199.5) | 1.5 | 2.9 ± 0.72 | 155 ± 80 | 1138 ± 696 | 25 ± 9 |
AR9281 (UC1153, APAU)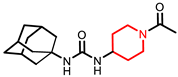 | 277 | 205–206 (205.5) | 0.8 | 2.8 | 144 | 160 | 39 |
| Parameter 1 | Mouse | Cat | Dog | Horse | |
|---|---|---|---|---|---|
| Dose (mg/kg) | |||||
| IV | 1 | 0.1 | 1 | 0.3 | 1 |
| PO | 10 | 0.1 | 3 | 0.3 | 1 |
| Cmax (ng/mL) | |||||
| IV | 3087 ± 1089 | 2076 ± 1194 | 27,241 ± 8114 | 1566 ± 1128 | 2436 ± 304 |
| PO | 2570 ± 670 | 91.5 ± 14.6 | 802 ± 241 | 403 ± 232 | 336 ± 83 |
| IV-Dose adjusted (Cmax/dose) | 3087 ± 1089 | 20,760 ± 11,942 | 27,241 ± 8114 | 5219 ± 3761 | 2436 ± 304 |
| PO-Dose adjusted (Cmax/dose) | 257 ± 67 | 915 ± 146 | 267 ± 80 | 1343 ± 772 | 336 ± 83 |
| AUC (h·ng/mL) | |||||
| IV | 8488 ± 1319 | 271 ± 88 | 8182 ± 3083 | 12,662 ± 8309 | 16,604 ± 4535 |
| PO | 28,624 ± 6008 | 140 ± 62 | 2081 ± 137 | 14,443 ± 4852 | 9190 ± 2921 |
| IV-Dose adjusted (AUC/dose) | 8488 ± 1319 | 2716 ± 878 | 8182 ± 3083 | 96,388 ± 63,239 | 16,604 ± 4535 |
| PO-Dose adjusted (AUC/dose) | 2862 ± 601 | 1402 ± 619 | 693 ± 46 | 48,142 ± 16,173 | 9190 ± 2921 |
| Tmax (h) | |||||
| IV | 0.38 ± 0.14 | 0.04 ± 0.04 | 0.025 ± 0 | 0.04 ± 0.03 | 0.08 ± 0 |
| PO | 4.0 ± 0.0 | 0.8 ± 0.6 | 1.2 ± 0.8 | 10.3 ± 9.3 | 10.6 ± 3.8 |
| Clint (mL/min·kg) | 2.0 ± 0.3 | 6.6 ± 2.1 | 2.3 ± 0.8 | 0.7 ± 0.6 | 1.0 ± 0.3 |
| Vss (L/kg) | 1.5 ± 0.9 | 0.4 ± 0.2 | 2.0 ± 0.8 | 2.6 ± 2.0 | 1.2 ± 0.2 |
| %F | 34 ± 8 | 52 ± 51 | 29 ± 6 * | 42 ± 14 | 50 ± 8 |
| T1/2 (h) | 1.4 ± 0.4 | 16.5 ± 2.3 | |||
| IV α | α = 1.20 ± 0.34 | α = 0.23 ± 0.13 | α = 0.10 ± 0.08 | ||
| β | β = 44 ± 23 | β = 20.5 ± 7.4 | β = 47 ± 10 | ||
| PO | 16.3 ± 2.8 | 0.9 ± 0.2 | 4.6 ± 4.1 | 42 ± 20 | 18.0 ± 4.3 |
| PPB (%) | 96.04 ± 0.99 | 98.32 ± 0.61 | 98.32 ± 0.61 | 98.0 ± 1.4 | 98.75 ± 0.24 |
| Cl(hep) (ml/min·kg) | 7.30 ± 0.97 | 8.60 ± 2.12 | 3.5 ± 1.1 | 0.45 ± 0.38 | 1.2 ± 0.33 |
| Body weight (kg) | 0.032 ± 0.002 | 3.9 ± 0.3 | 12.9 ± 1.6 | 554 ± 22 | |
| Route | PO | ||
|---|---|---|---|
| Parameter 1 | |||
| Dose (mg/kg) | 0.1 | 3.0 | 10.0 |
| Cmax (ng/mL) | 92 ± 15 | 803 ± 241 | 1115 ± 541 |
| AUC (h·ng/mL) | 140 ± 62 | 2369 ± 517 | 2278 ± 245 |
| %F Based on 0.1 mg/kg | 52 ± 23.0 | 29 ± 6.0 | 8 ± 1.0 |
| T1/2 (h) | 0.88 ± 0.2 | 4.62 ± 4.1 | 4.50 ± 2.2 |
| Tmax (h) | 0.83 ± 0.6 | 1.17 ± 0.8 | 0.83 ± 0.3 |
| Clint (mL/min·kg) | 14.4 ± 8.4 | 24.1 ± 1.6 | 73.7 ± 7.8 |
| Kel (1/h) | 0.64 ± 0.35 | 0.28 ± 0.05 | 0.32 ± 0.06 |
| Vehicle | Dose (mg/kg): Concentration (mg/mL) | |
|---|---|---|
| Mouse | IV: DMSO:PEG300:Saline (2:25:73) PO: PEG400 (100%) | 1.0 mg/kg: 0.06 mg/mL, IV 10 mg/kg: 1 mg/mL, PO |
| Cat | IV (0.1 mg/kg) and PO: DMSO: PEG400 (20:80) PEG400 for 1 mg/kg IV | 0.1 mg/kg: 2 mg/mL, IV and PO 1 mg/kg: 20 mg/mL, IV 3.0 mg/kg: 60 mg/mL, PO 10 mg/kg: 100 mg/mL, PO |
| Dog | IV: PEG400 (100%) PO: PEG400 (100%) | 0.3 mg/kg: 1 mg/mL, IV 0.3 mg/kg: 1 mg/mL, PO |
| Horse | IV: DMSO (100%) PO: PEG400 (100%) | 1 mg/kg: 20 mg/mL, IV 1 mg/kg: 20 mg/mL, PO |
| Species | Time (h) |
|---|---|
| Mouse | IV: 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96 (n = 4) PO: 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96 (n = 4) |
| Cat | IV (0.1 mg/kg): 0.017, 0.08, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 6, 8 (n = 3) IV (1 mg/kg): 0.025, 0.05, 0.1, 0.2, 0.38, 0.75, 1.5, 3, 6, 12, 24, 36, 48, 72, 96 (n = 6) PO: 0.08, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12 (n = 3 per group) |
| Dog | IV: 0.025, 0.05, 0.1, 0.2, 0.38, 0.75, 1.5, 3, 6, 12, 24, 36, 48, 72, 96 (n = 5) PO: 0.025, 0.05, 0.1, 0.2, 0.38, 0.75, 1.5, 3, 6, 12, 24, 36, 48, 72, 96 (n = 5) |
| Horse | IV: 0.083, 0.167, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 18, 24, 30, 36, 48, 60, 72, 96 (n = 8) PO: 0.083, 0.167, 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 18, 24, 30, 36, 48, 60, 72, 96 (n = 8) |
| Sequence Group | Period 1 | Wash-Out | Period 2 |
|---|---|---|---|
| S1 (n = 4) | Vehicle € | 7 days | Test Article £ |
| S2 (n = 4) | Test Article * | Reference Article α or Vehicle β |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McReynolds, C.B.; Yang, J.; Guedes, A.; Morisseau, C.; Garcia, R.; Knych, H.; Tearney, C.; Hamamoto, B.; Hwang, S.H.; Wagner, K.; et al. Species Differences in Metabolism of Soluble Epoxide Hydrolase Inhibitor, EC1728, Highlight the Importance of Clinically Relevant Screening Mechanisms in Drug Development. Molecules 2021, 26, 5034. https://doi.org/10.3390/molecules26165034
McReynolds CB, Yang J, Guedes A, Morisseau C, Garcia R, Knych H, Tearney C, Hamamoto B, Hwang SH, Wagner K, et al. Species Differences in Metabolism of Soluble Epoxide Hydrolase Inhibitor, EC1728, Highlight the Importance of Clinically Relevant Screening Mechanisms in Drug Development. Molecules. 2021; 26(16):5034. https://doi.org/10.3390/molecules26165034
Chicago/Turabian StyleMcReynolds, Cindy B., Jun Yang, Alonso Guedes, Christophe Morisseau, Roberto Garcia, Heather Knych, Caitlin Tearney, Briana Hamamoto, Sung Hee Hwang, Karen Wagner, and et al. 2021. "Species Differences in Metabolism of Soluble Epoxide Hydrolase Inhibitor, EC1728, Highlight the Importance of Clinically Relevant Screening Mechanisms in Drug Development" Molecules 26, no. 16: 5034. https://doi.org/10.3390/molecules26165034
APA StyleMcReynolds, C. B., Yang, J., Guedes, A., Morisseau, C., Garcia, R., Knych, H., Tearney, C., Hamamoto, B., Hwang, S. H., Wagner, K., & Hammock, B. D. (2021). Species Differences in Metabolism of Soluble Epoxide Hydrolase Inhibitor, EC1728, Highlight the Importance of Clinically Relevant Screening Mechanisms in Drug Development. Molecules, 26(16), 5034. https://doi.org/10.3390/molecules26165034