
Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1)

 , and
, and
Abstract
:1. Introduction
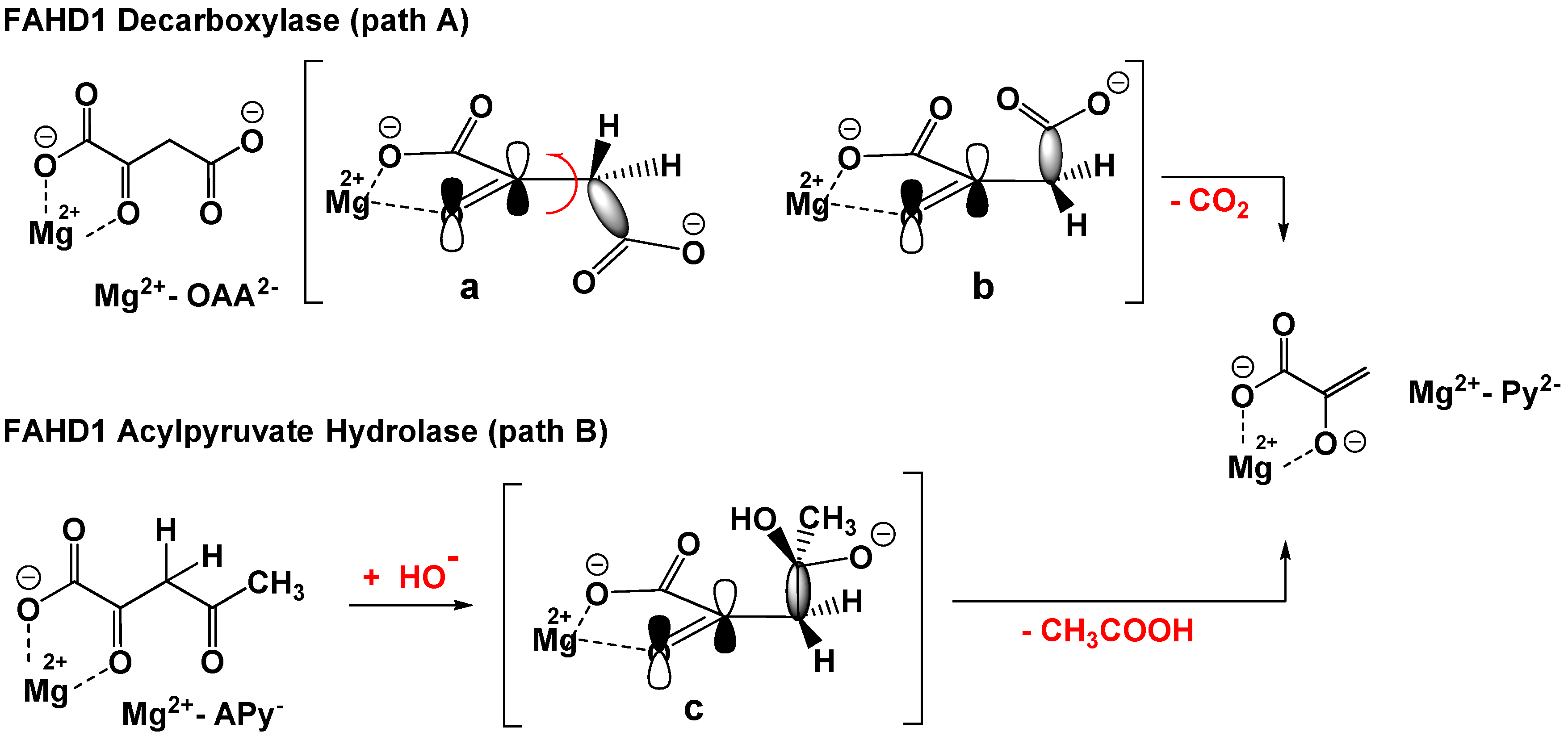
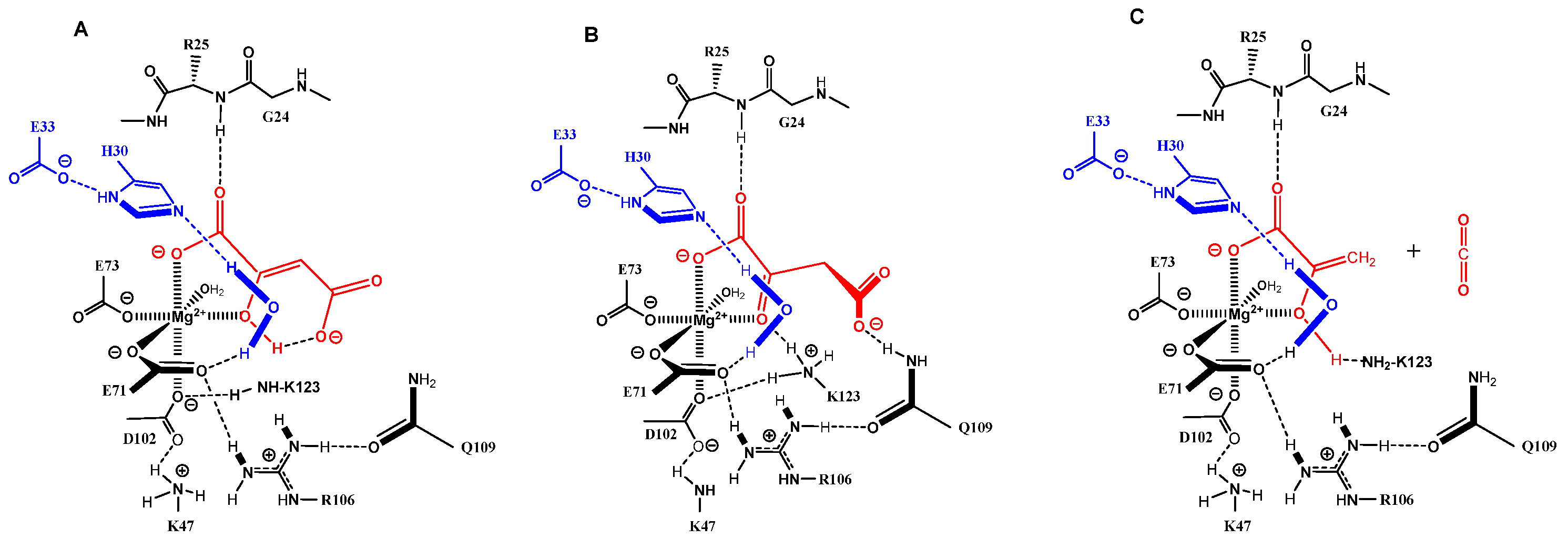
1.1. The C–C Bond Cleavage Mechanism in ODx- and ApH-Activity of FAHD1
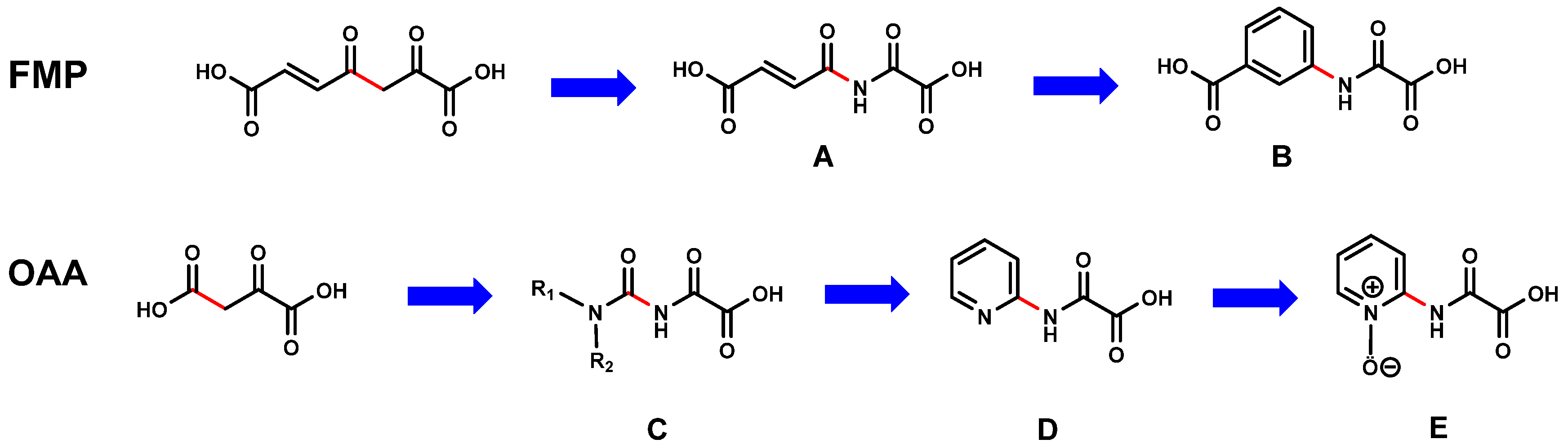
1.2. Design Steps of First Generation of FAHD1 Inhibitors
2. Results and Discussion
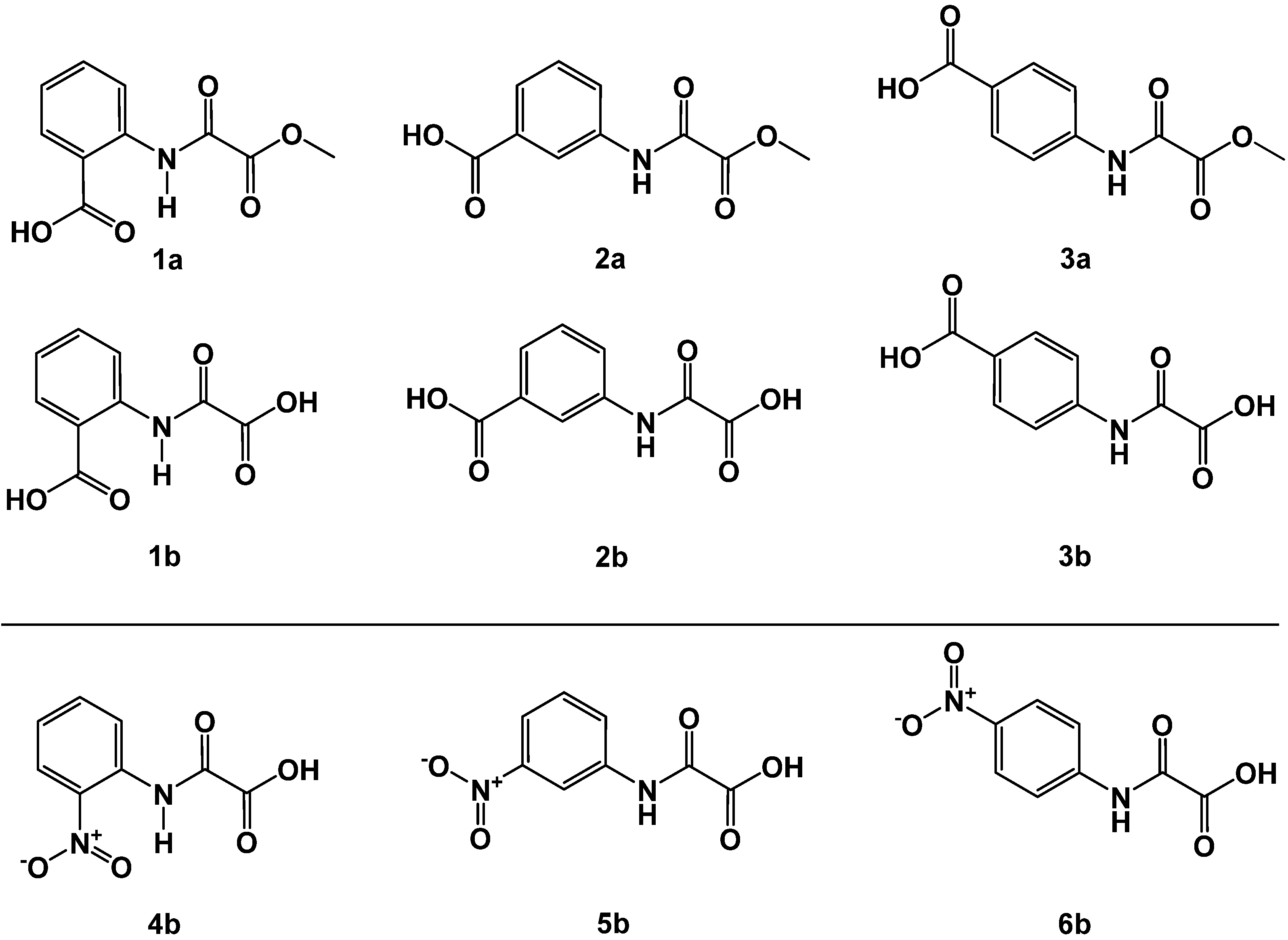
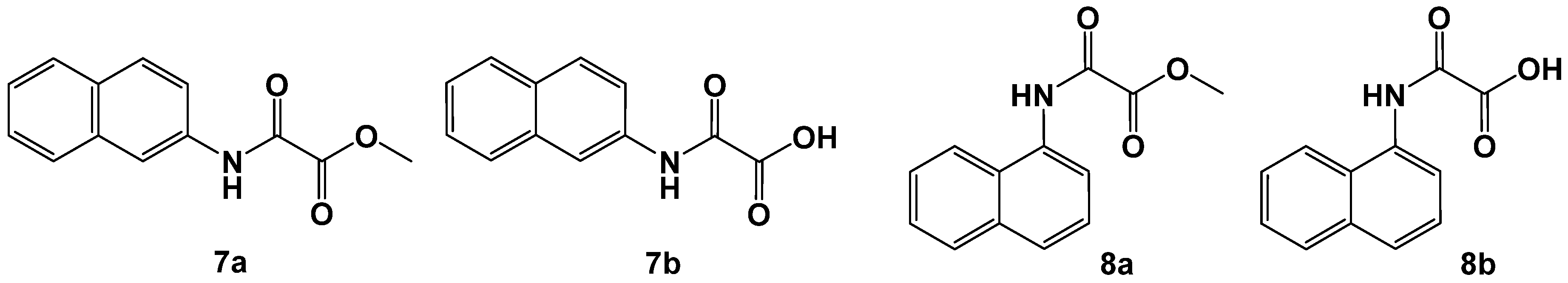
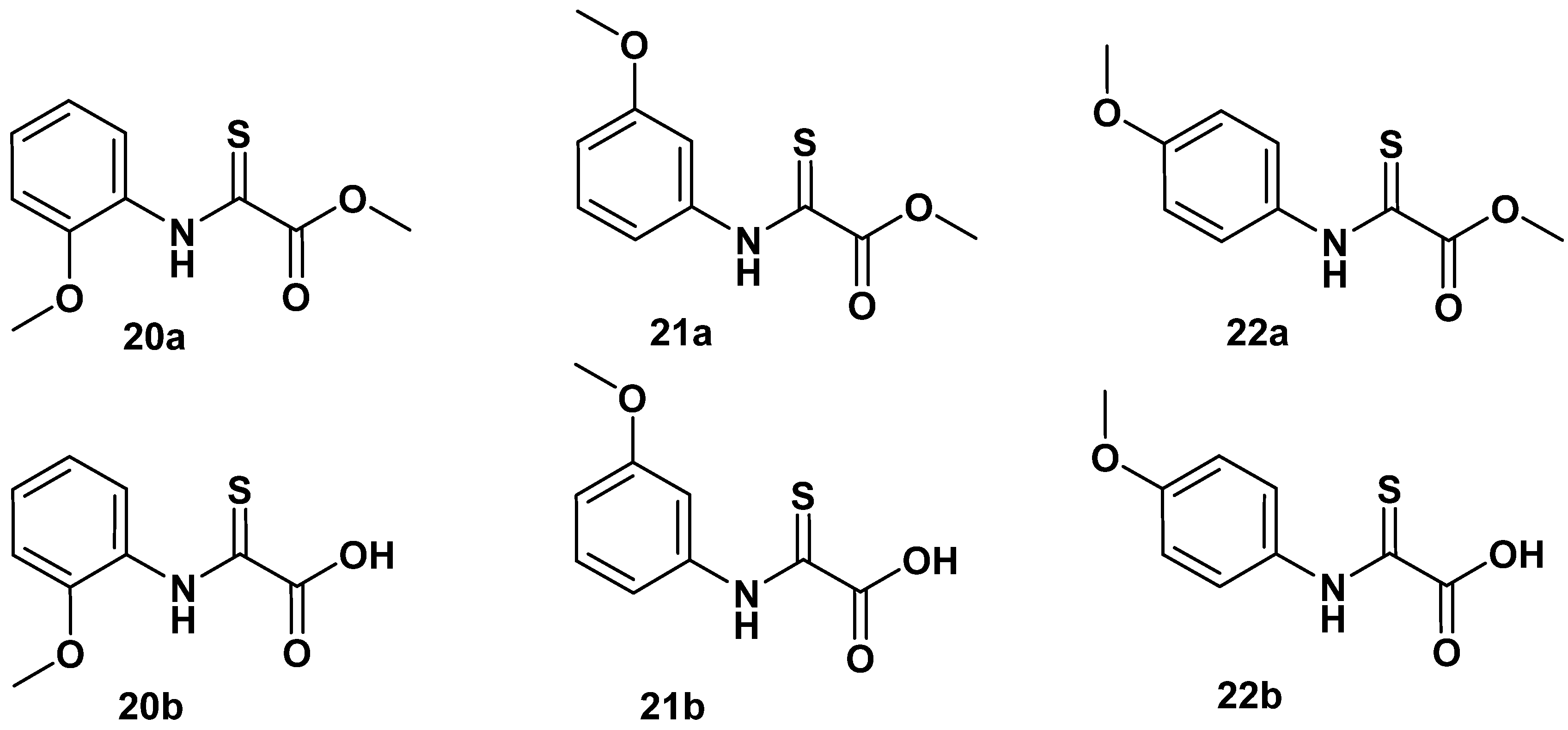
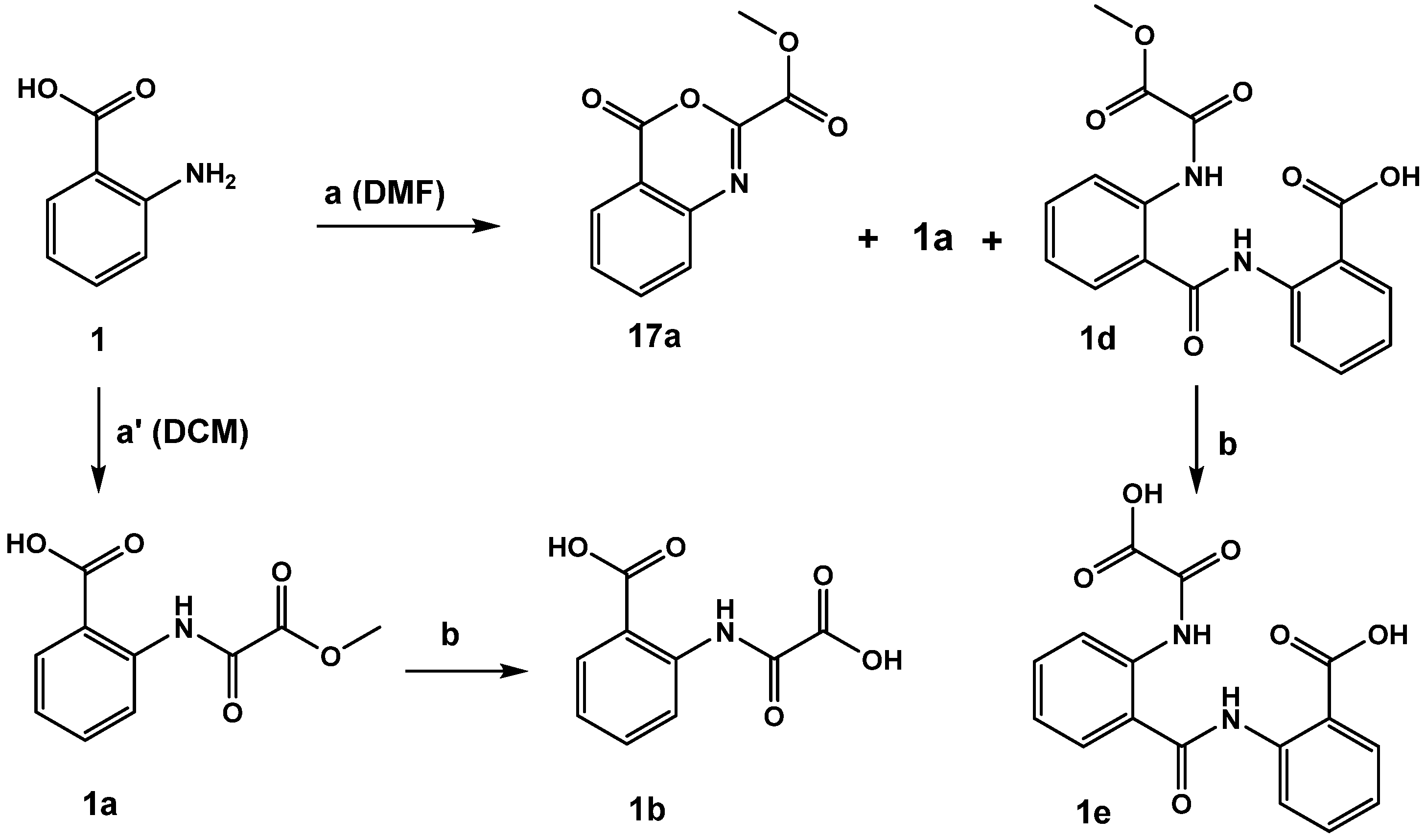
2.1. N-Aryl-Oxalamic Acids as FAHD1 Inhibitors
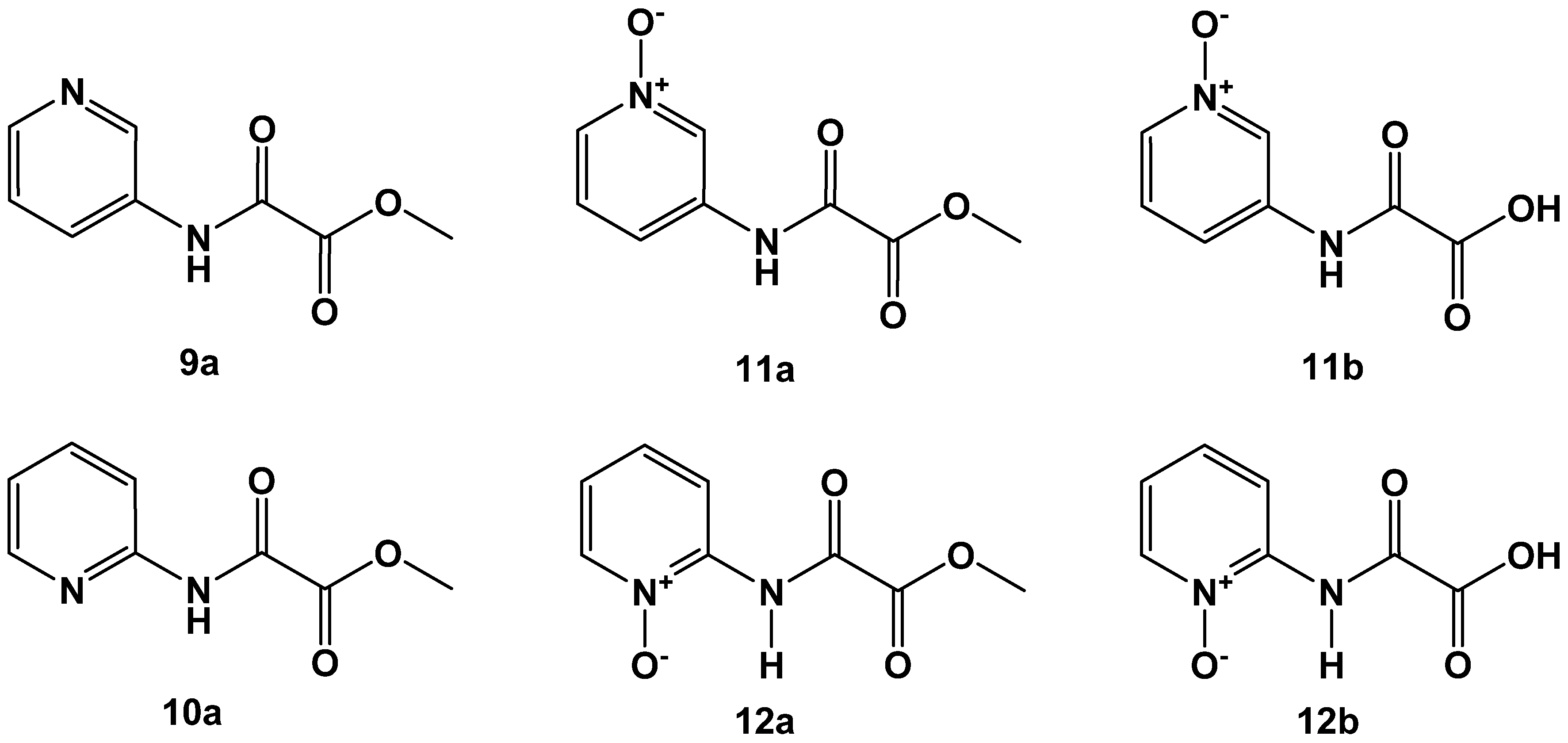
2.2. N-Pyridyl Substituted Oxalamic Acid Derivatives and Their N-Oxides as FAHD1 Inhibitors
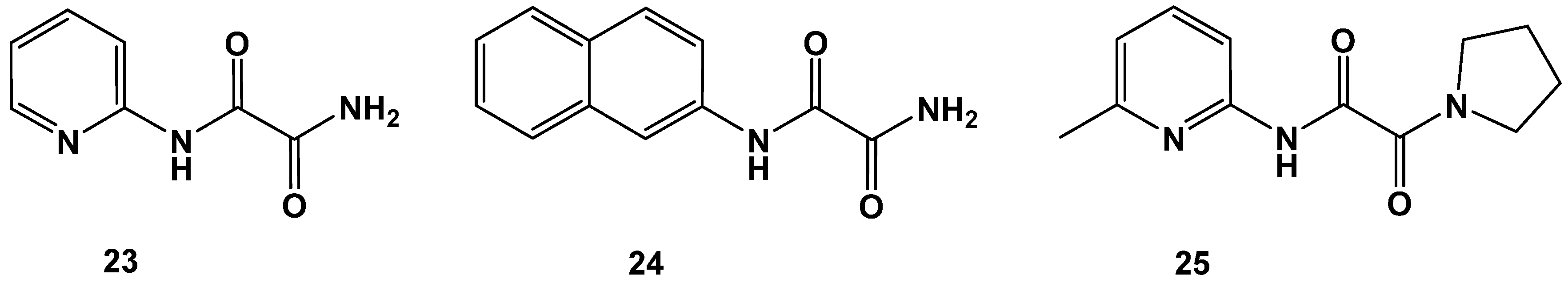
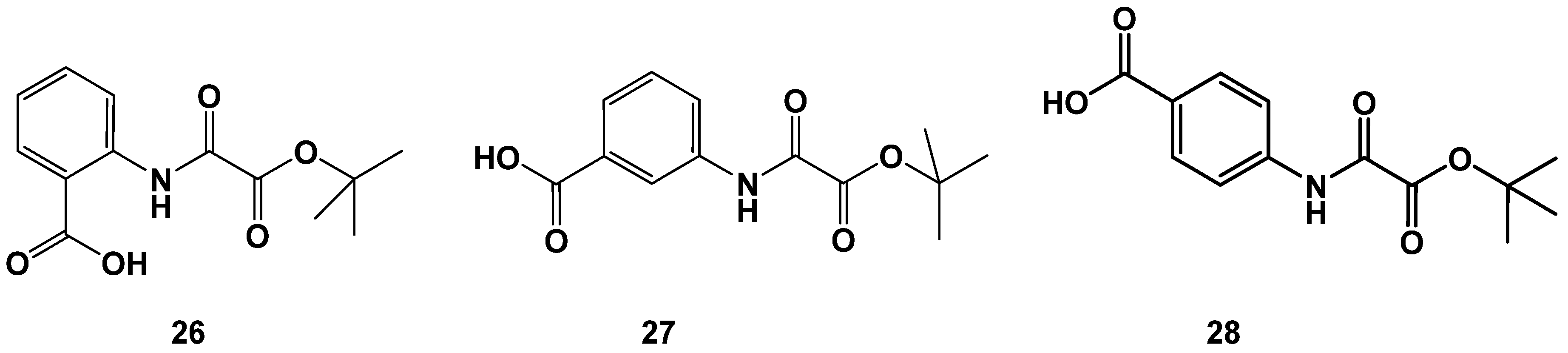
2.3. Variations Affecting the 1,2-Dicarbonyl Motif in Oxalamic Acid Derivatives
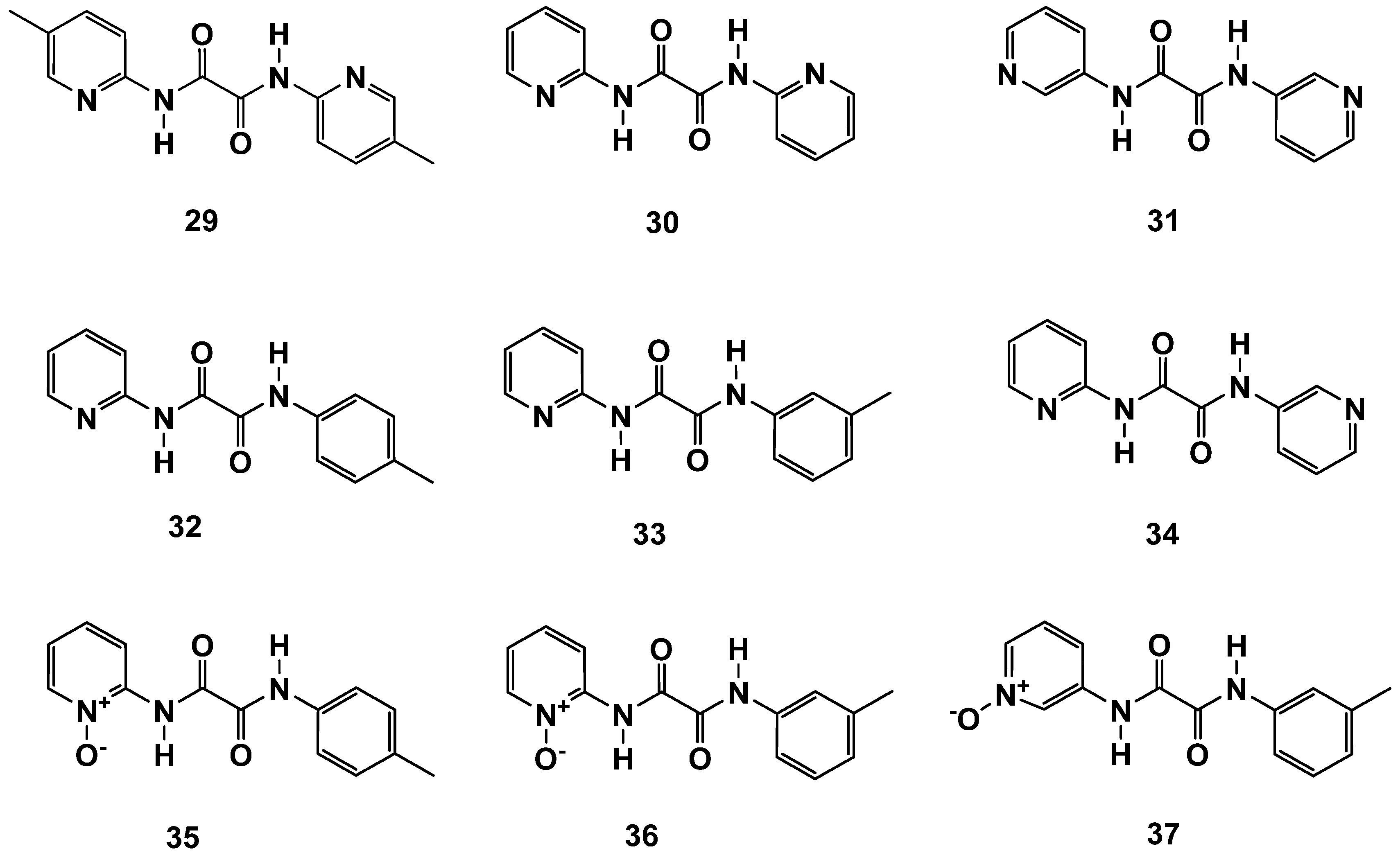
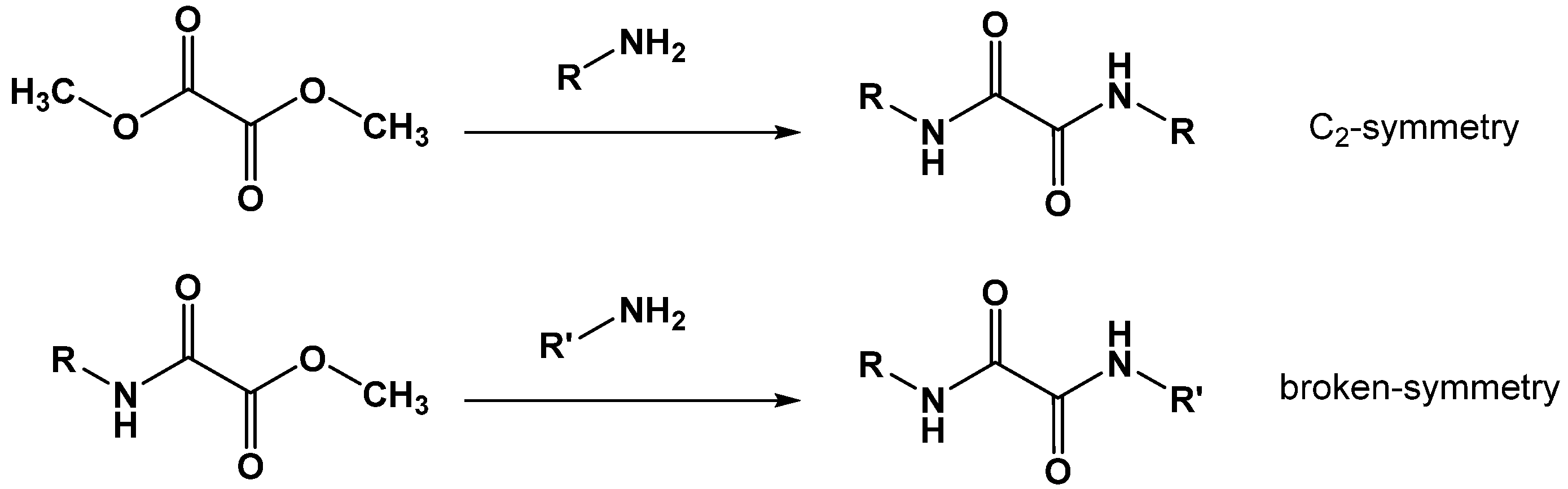
2.4. N,N′-Diaryl-Oxalamic Acid Diamides
3. Materials and Methods
3.1. Reaction Monitoring and Purification of Compounds
3.1.1. Thin-Layer Chromatography (TLC)
3.1.2. Middle Pressure Liquid Chromatography (MPLC)
3.2. Analytical Characterization
3.2.1. High-Resolution Mass Spectroscopy (HRMS)
3.2.2. Melting Points
3.2.3. Nuclear Magnetic Resonance Spectroscopy (NMR)
3.3. Inhibitor Assay on the 96-Well Plate
3.4. General Synthetic Procedures
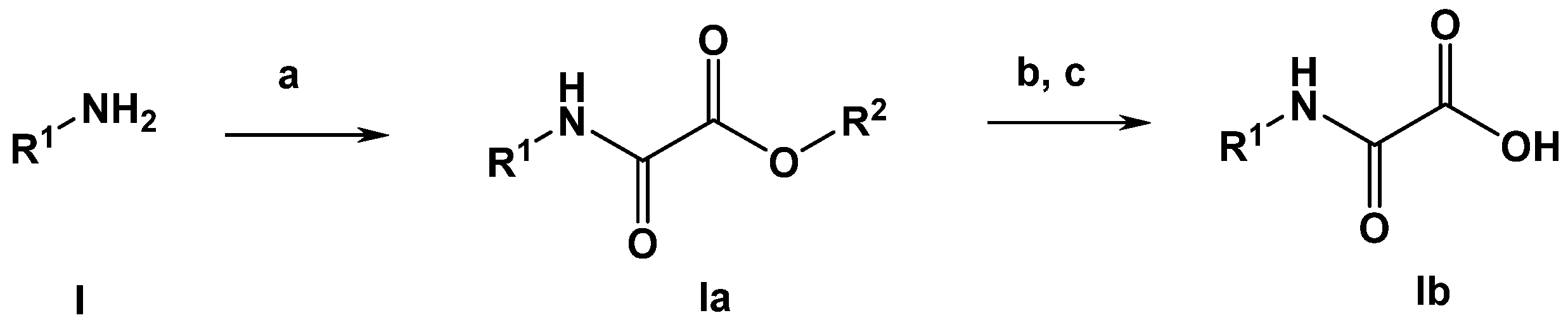
3.4.1. General Synthesis of Oxalamic Acid Derivatives
3.4.2. Saponification of Methyl Ester Derivatives
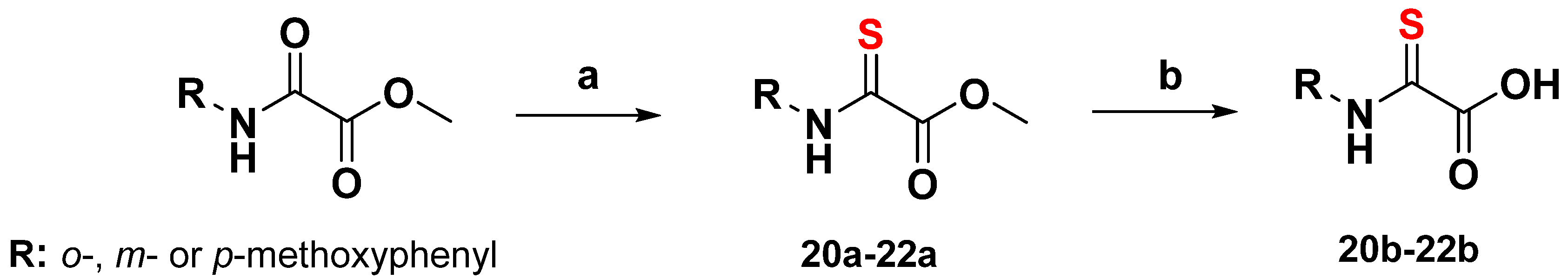
3.4.3. General Synthesis of 2-Thiooxoacetate Derivatives
3.4.4. Synthesis of Pyridine and Quinoline N-Oxides
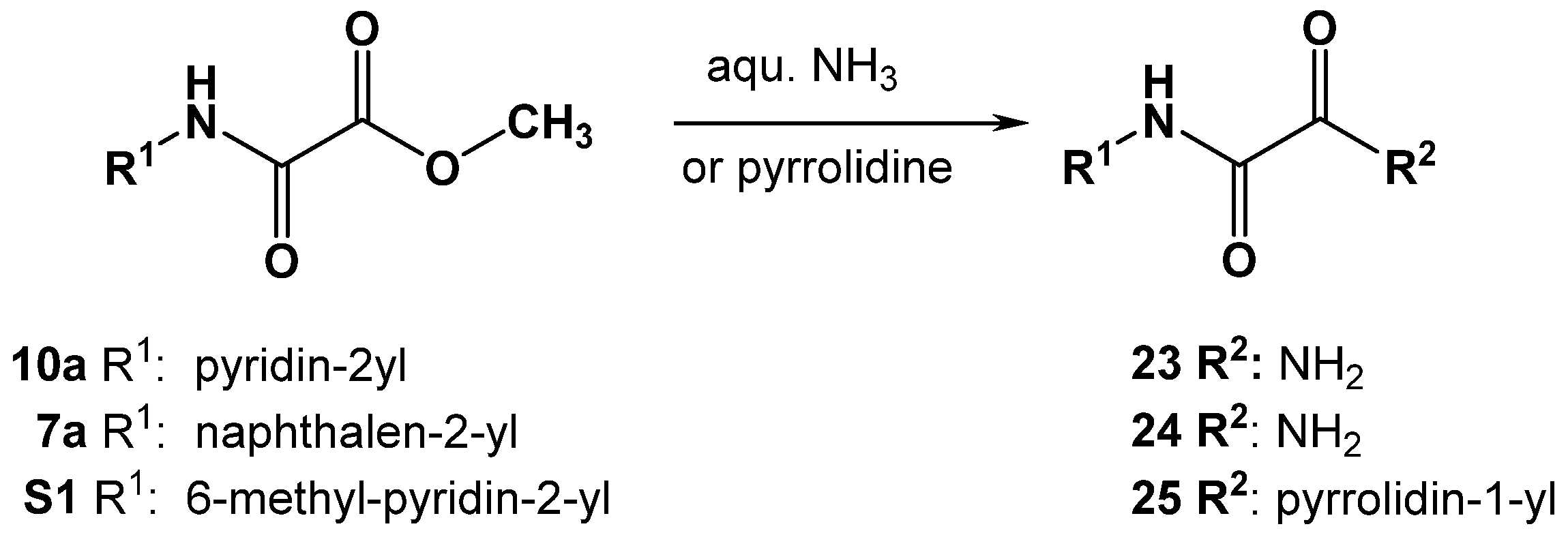
3.4.5. Synthesis of Oxalamic Acid Amides
3.4.6. Synthesis of N,N′-Diaryl-Oxalamic Acid Diamides
Synthesis of Oxalic Acid Diamides by Aminolysis of DIMOX
Synthesis of Oxalic Acid Diamides by Aminolysis of Methyl Esters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pircher, H.; von Grafenstein, S.; Diener, T.; Metzger, C.; Albertini, E.; Taferner, A.; Unterluggauer, H.; Kramer, C.; Liedl, K.R.; Jansen-Dürr, P. Identification of FAH Domain-containing Protein 1 (FAHD1) as Oxaloacetate Decarboxylase. J. Biol. Chem. 2015, 290, 6755–6762. [Google Scholar] [CrossRef] [Green Version]
- Pircher, H.; Straganz, G.D.; Ehehalt, D.; Morrow, G.; Tanguay, R.M.; Jansen-Dürr, P. Identification of Human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a Novel Mitochondrial Acylpyruvase. J. Biol. Chem. 2011, 286, 36500–36508. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.K.H.; Loeffler, J.R.; Liedl, K.R.; Gstach, H.; Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: Multifunctional enzymes from microbes to mitochondria. Biochem. Soc. Trans. 2018, 46, 295–309. [Google Scholar] [CrossRef]
- Hong, H.; Seo, H.; Park, W.; Kim, K.K.-J. Sequence, structure and function-based classification of the broadly conserved FAH superfamily reveals two distinct fumarylpyruvate hydrolase subfamilies. Environ. Microbiol. 2020, 22, 270–285. [Google Scholar] [CrossRef]
- Weiss, A.K.H.; Albertini, E.; Holzknecht, M.; Cappuccio, E.; Dorigatti, I.; Krahbichler, A.; Damisch, E.; Gstach, H.; Jansen-Dürr, P. Regulation of cellular senescence by eukaryotic members of the FAH superfamily—A role in calcium homeostasis? Mech. Ageing Dev. 2020, 190, 111284. [Google Scholar] [CrossRef] [PubMed]
- Etemad, S.; Petit, M.; Weiss, A.K.H.; Schrattenholz, A.; Baraldo, G.; Jansen-Dürr, P. Oxaloacetate decarboxylase FAHD1—A new regulator of mitochondrial function and senescence. Mech. Ageing Dev. 2019, 177, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.K.H.; Naschberger, A.; Loeffler, J.R.; Gstach, H.; Bowler, M.W.; Holzknecht, M.; Cappuccio, E.; Pittl, A.; Etemad, S.; Dunzendorfer-Matt, T.; et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochem. J. 2018, 475, 3561–3576. [Google Scholar] [CrossRef]
- Weiss, A.K.H.; Naschberger, A.; Cappuccio, E.; Metzger, C.; Mottes, L.; Holzknecht, M.; von Velsen, J.; Bowler, M.W.; Rupp, B.; Jansen-Dürr, P. Structural and functional comparison of fumarylacetoacetate domain containing protein 1 in human and mouse. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Tate, S.S.; Grzybowski, A.K.; Datta, S.P. 266. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25°. J. Chem. Soc. 1964, 1381–1389. [Google Scholar] [CrossRef]
- Creighton, D.J.; Rose, I.A. Studies on the mechanism and stereochemical properties of the oxalacetate decarboxylase activity of pyruvate kinase. J. Biol. Chem. 1976, 251, 61–68. [Google Scholar] [CrossRef]
- Tate, S.S.; Grzybowski, A.K.; Datta, S.P. 265. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25°. J. Chem. Soc. 1964, 1372–1380. [Google Scholar] [CrossRef]
- Mulholland, A.J.; Richards, W.G. Calculations on the substrates of citrate synthase-I. Oxaloacetate. J. Mol. Struct. 1998, 429, 13–21. [Google Scholar] [CrossRef]
- Ran, T.; Gao, Y.; Marsh, M.; Zhu, W.; Wang, M.; Mao, X.; Xu, L.; Xu, D.; Wang, W. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. Biochem. J. 2012, 449, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kresge, A.J. Generation and study of enols and other reactive species. Pure Appl. Chem. 1991, 63, 213–221. [Google Scholar] [CrossRef]
- Andersen, H.S.; Iversen, L.F.; Jeppesen, C.B.; Branner, S.; Norris, K.; Rasmussen, H.B.; Møller, K.B.; Møller, N.P. 2-(oxalylamino)-benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J. Biol. Chem. 2000, 275, 7101–7108. [Google Scholar] [CrossRef] [Green Version]
- Mfuh, A.M.; Larionov, O.V. Heterocyclic N-Oxides—An Emerging Class of Therapeutic Agents. Curr. Med. Chem. 2015, 22, 2819–2857. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.M. Synthesis and reactions of 2-hetero-4H-3,1-benzoxazin-4-ones. J. Heterocycl. Chem. 2000, 37, 1369–1388. [Google Scholar] [CrossRef]
- Liao, B.-L.; Pan, Y.-J.; Zhang, W.; Pan, L.-W. Four Natural Compounds Separated from Folium Isatidis: Crystal Structures and Antibacterial Activity. Chem. Biodivers. 2018, 15, e1800152. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Wang, F.; Zhang, G.-L.; Xiao, X. A triclinic polymorph with Z = 3 of N, N ′-bis(2-pyridyl)oxamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o972–o973. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.K.H.; Holzknecht, M.; Cappuccio, E.; Dorigatti, I.; Kreidl, K.; Naschberger, A.; Rupp, B.; Gstach, H.; Jansen-Dürr, P. Expression, Purification, Crystallization, and Enzyme Assays of Fumarylacetoacetate Hydrolase Domain-Containing Proteins. J. Vis. Exp. 2019, e59729. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 1a | 47 ± 1 |
| 2a | 13 ± 1 |
| 3a | 85 ± 2 |
| 1b | 11 ± 1 |
| 2b | 14 ± 1 |
| 3b | 32 ± 2 |
| 4b | 29 ± 2 |
| 5b | 31 ± 2 |
| 6b | 789 ± 6 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 7a | 11 ± 1 |
| 7b | 26 ± 2 |
| 8a | 280 ± 2 |
| 8b | 158 ± 1 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 9a | 27 ± 2 |
| 10a | 249 ± 1 |
| 11a | 29 ± 2 |
| 11b | 4 ± 2 |
| 12a | 168 ± 1 |
| 12b | 65 ± 1 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
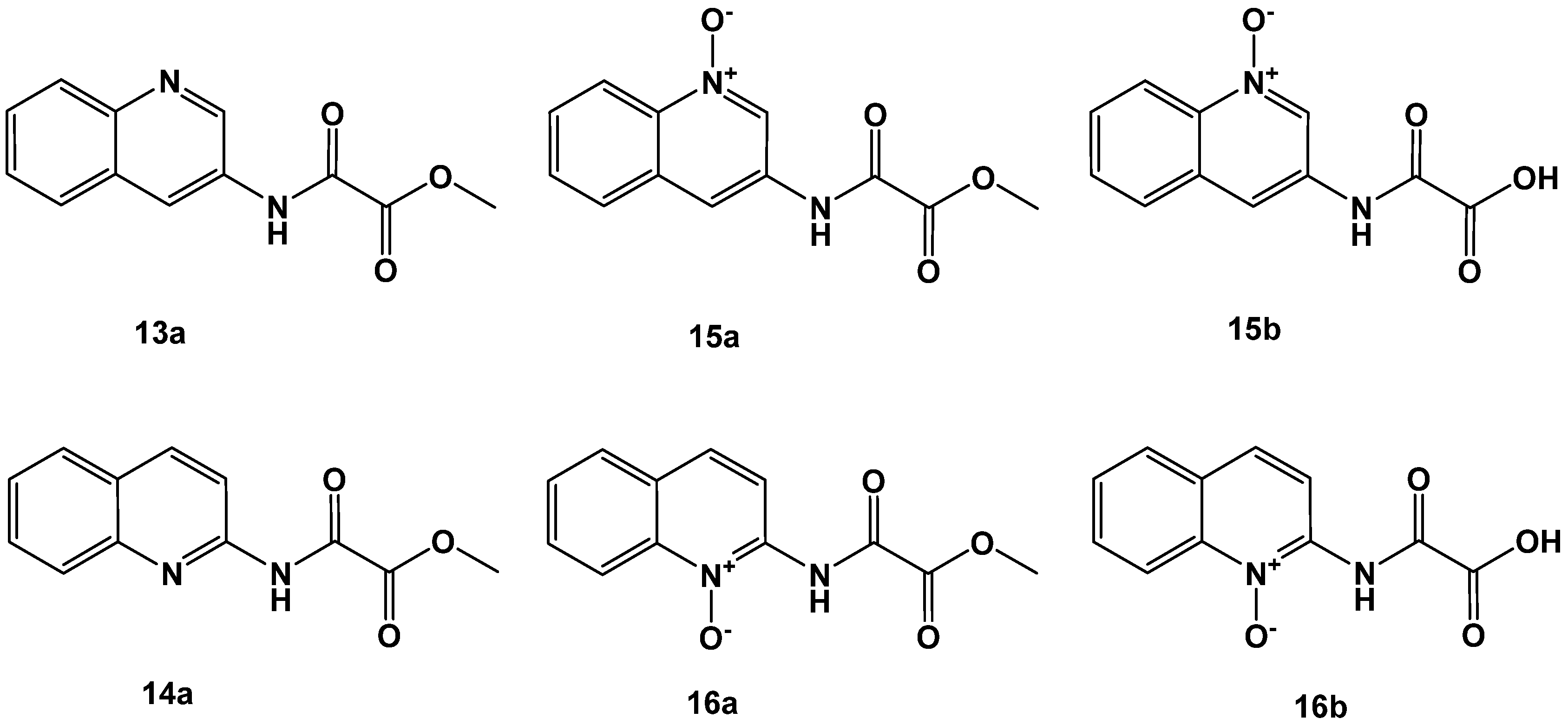
| 13a | 33 ± 2 |
| 14a | 33 ± 2 |
| 15a | 31 ± 2 |
| 15b | 8 ± 1 |
| 16a | 51 ± 1 |
| 16b | 31 ± 2 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
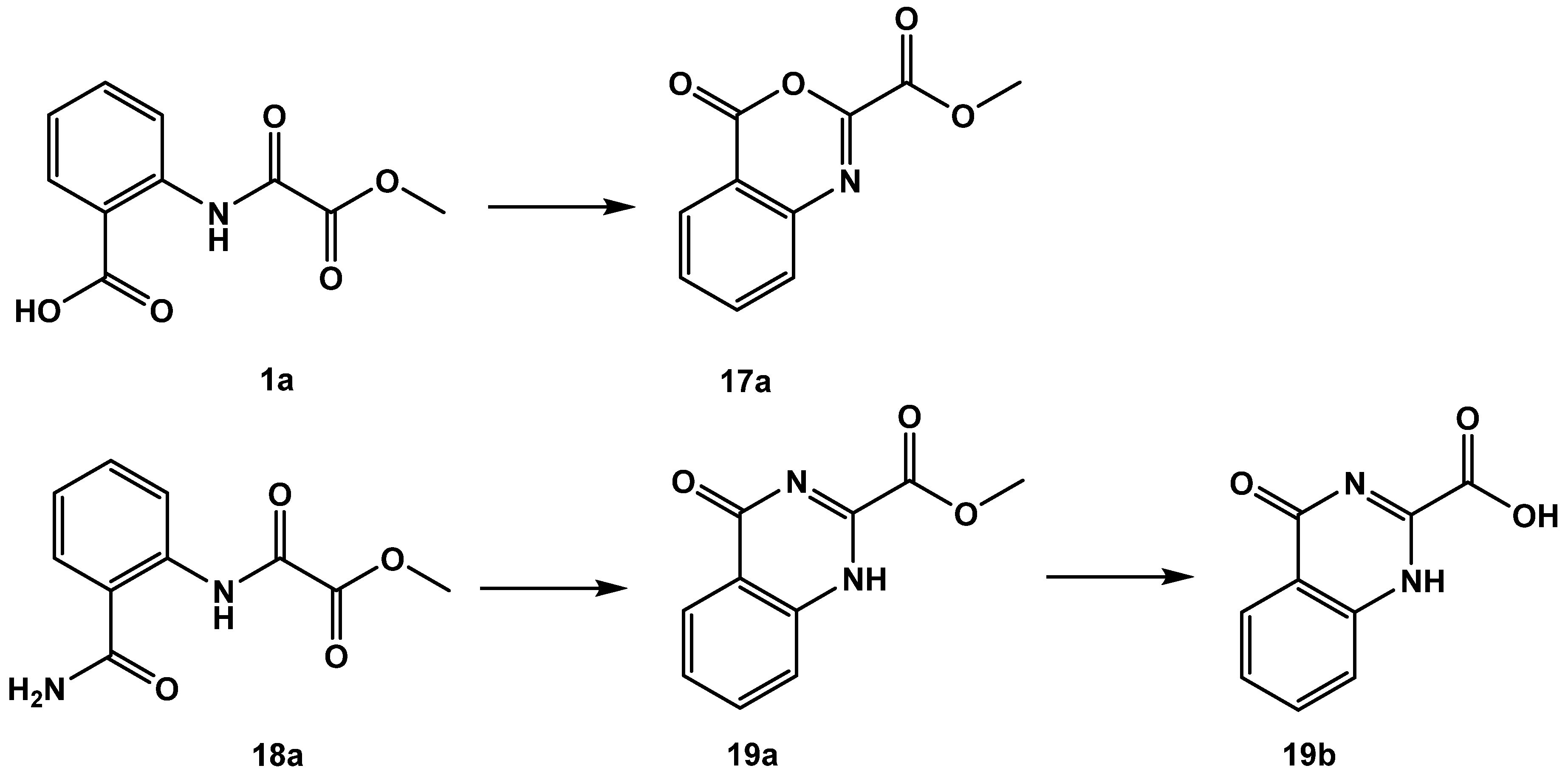
| 1a | 47 ± 1 |
| 17a | 11 ± 2 |
| 18a | 59 ± 2 |
| 19a | 43 ± 2 |
| 19b | 30 ± 1 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 20a | - |
| 21a | 85 ± 3 |
| 21b | 63 ± 2 |
| 22a | 130 ± 1 |
| 22b | 167 ± 1 |
| 20b | 295 ± 2 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 23 | 137 ± 3 |
| 24 | 107 ± 1 |
| 25 | 324 ± 2 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 26 | 12 ± 3 |
| 27 | 261 ± 2 |
| 28 | 15 ± 2 |
| OAA | 6 ± 2 |
| Compound ID | IC50 ± Std. Error (µM) |
|---|---|
| 29 | 3 ± 2 |
| 30 | 8 ± 2 |
| 31 | 131 ± 1 |
| 32 | 3 ± 2 |
| 33 | 8 ± 2 |
| 34 | inactive |
| 35 | 17 ± 1 |
| 36 | 14 ± 2 |
| 37 | inactive |
| OAA | 6 ± 2 |
| Well Nr. | Buffer | FAHD1 (40 µM) (2 µM Final; ~5 µg) | DMSO (1% Final) | Inhibitor (in DMSO) (1% Final DMSO) | OAA (20 mM) (1 mM Final) |
|---|---|---|---|---|---|
| 1 | 94 µL | --- | 1 µL | --- | 5 µL |
| 2 | 94 µL | --- | --- | 1 µL of dilution “0” | 5 µL |
| 3 | 94 µL | 5 µL | --- | 1 µL of dilution “0” | --- |
| 4 | 99 µL | --- | --- | 1 µL of dilution “0” | --- |
| 5 | 89 µL | 5 µL | 1 µL | --- | 5 µL |
| 6 | 89 µL | 5 µL | --- | 1 µL of dilution “6” | 5 µL |
| 7 | 89 µL | 5 µL | --- | 1 µL of dilution “5” | 5 µL |
| 8 | 89 µL | 5 µL | --- | 1 µL of dilution “4” | 5 µL |
| 9 | 89 µL | 5 µL | --- | 1 µL of dilution “3” | 5 µL |
| 10 | 89 µL | 5 µL | --- | 1 µL of dilution “2” | 5 µL |
| 11 | 89 µL | 5 µL | --- | 1 µL of dilution “1” | 5 µL |
| 12 | 89 µL | 5 µL | --- | 1 µL of dilution “0” | 5 µL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, A.K.H.; Wurzer, R.; Klapec, P.; Eder, M.P.; Loeffler, J.R.; von Grafenstein, S.; Monteleone, S.; Liedl, K.R.; Jansen-Dürr, P.; Gstach, H. Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1). Molecules 2021, 26, 5009. https://doi.org/10.3390/molecules26165009
Weiss AKH, Wurzer R, Klapec P, Eder MP, Loeffler JR, von Grafenstein S, Monteleone S, Liedl KR, Jansen-Dürr P, Gstach H. Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1). Molecules. 2021; 26(16):5009. https://doi.org/10.3390/molecules26165009
Chicago/Turabian StyleWeiss, Alexander K. H., Richard Wurzer, Patrycia Klapec, Manuel Philip Eder, Johannes R. Loeffler, Susanne von Grafenstein, Stefania Monteleone, Klaus R. Liedl, Pidder Jansen-Dürr, and Hubert Gstach. 2021. "Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1)" Molecules 26, no. 16: 5009. https://doi.org/10.3390/molecules26165009
APA StyleWeiss, A. K. H., Wurzer, R., Klapec, P., Eder, M. P., Loeffler, J. R., von Grafenstein, S., Monteleone, S., Liedl, K. R., Jansen-Dürr, P., & Gstach, H. (2021). Inhibitors of Fumarylacetoacetate Hydrolase Domain Containing Protein 1 (FAHD1). Molecules, 26(16), 5009. https://doi.org/10.3390/molecules26165009