
Antitumor Mechanism of Hydroxycamptothecin via the Metabolic Perturbation of Ribonucleotide and Deoxyribonucleotide in Human Colorectal Carcinoma Cells

 and
and 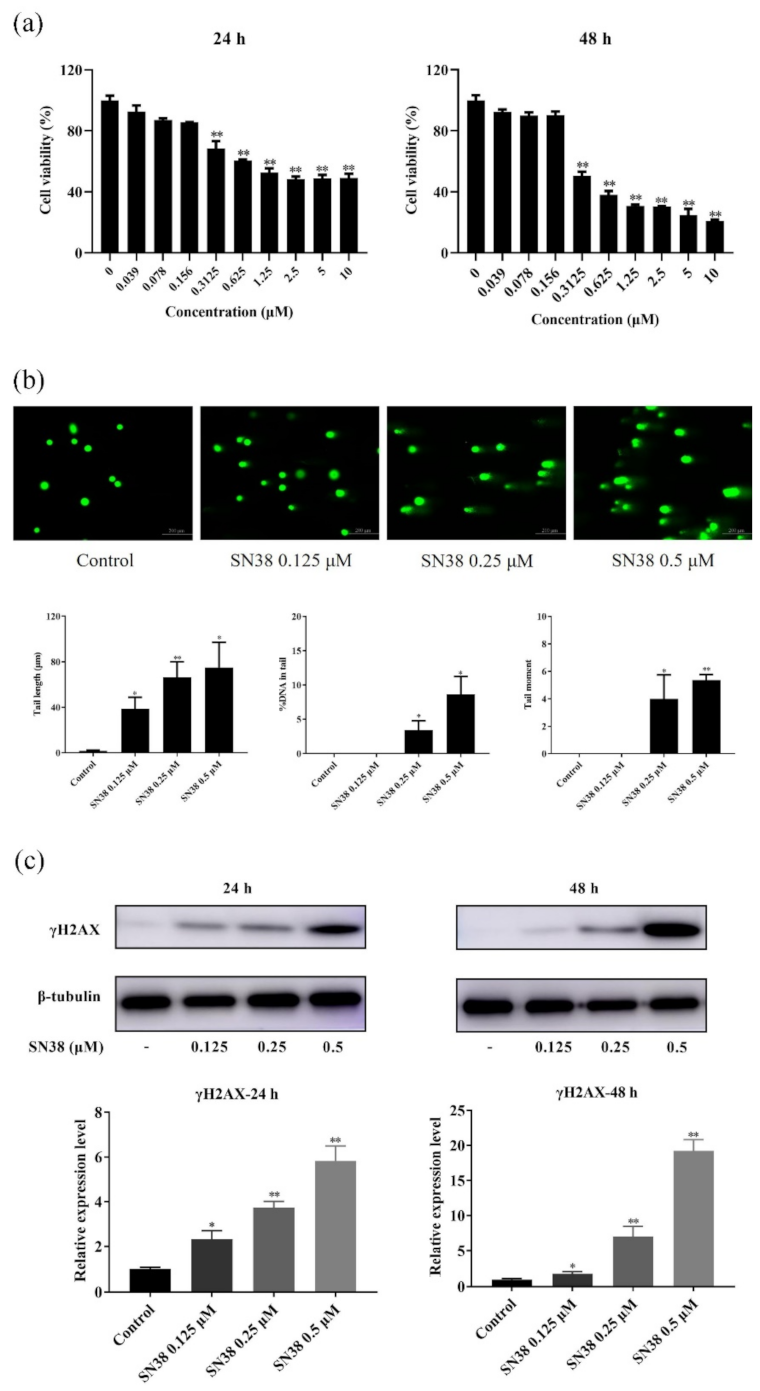{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
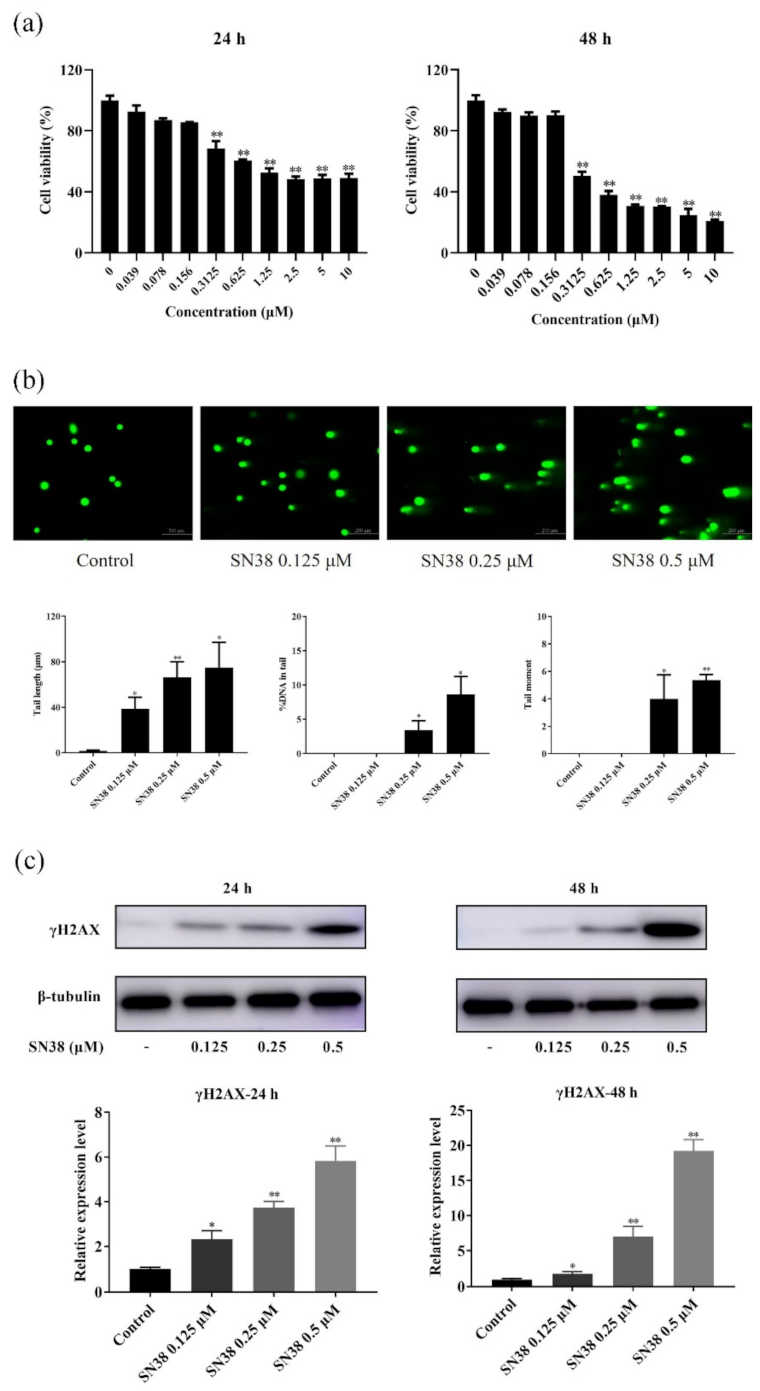
2.1. SN38 Decreased the Viability of the HCT 116 Cell Line
2.2. SN38 Caused DNA Damage of the HCT 116 Cell Line
2.3. SN38 Induced Cell Cycle Arrest in the HCT 116 Cell Line
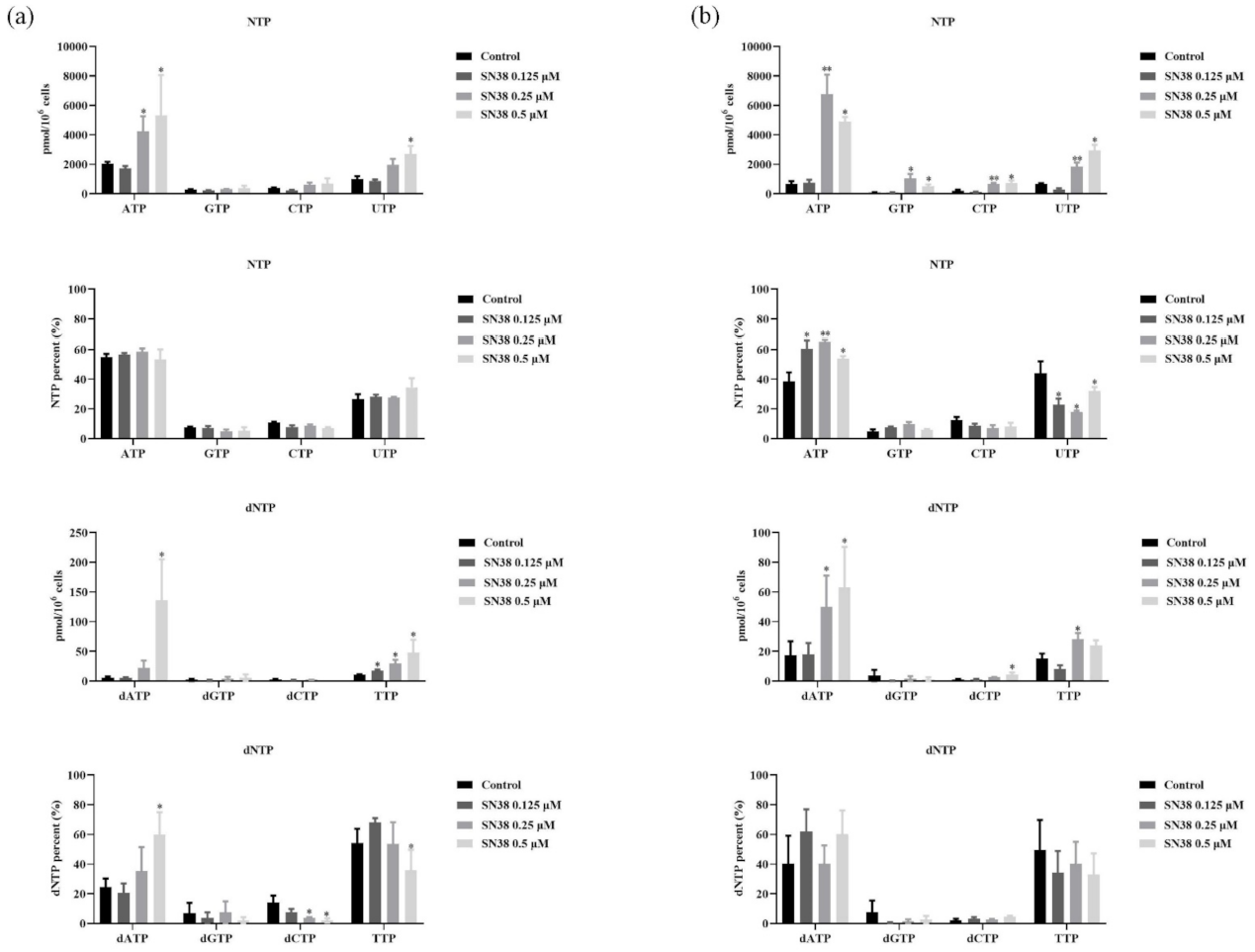
2.4. SN38 Leads to the Perturbation of RNs and dRNs Pools in the HCT 116 Cell Line
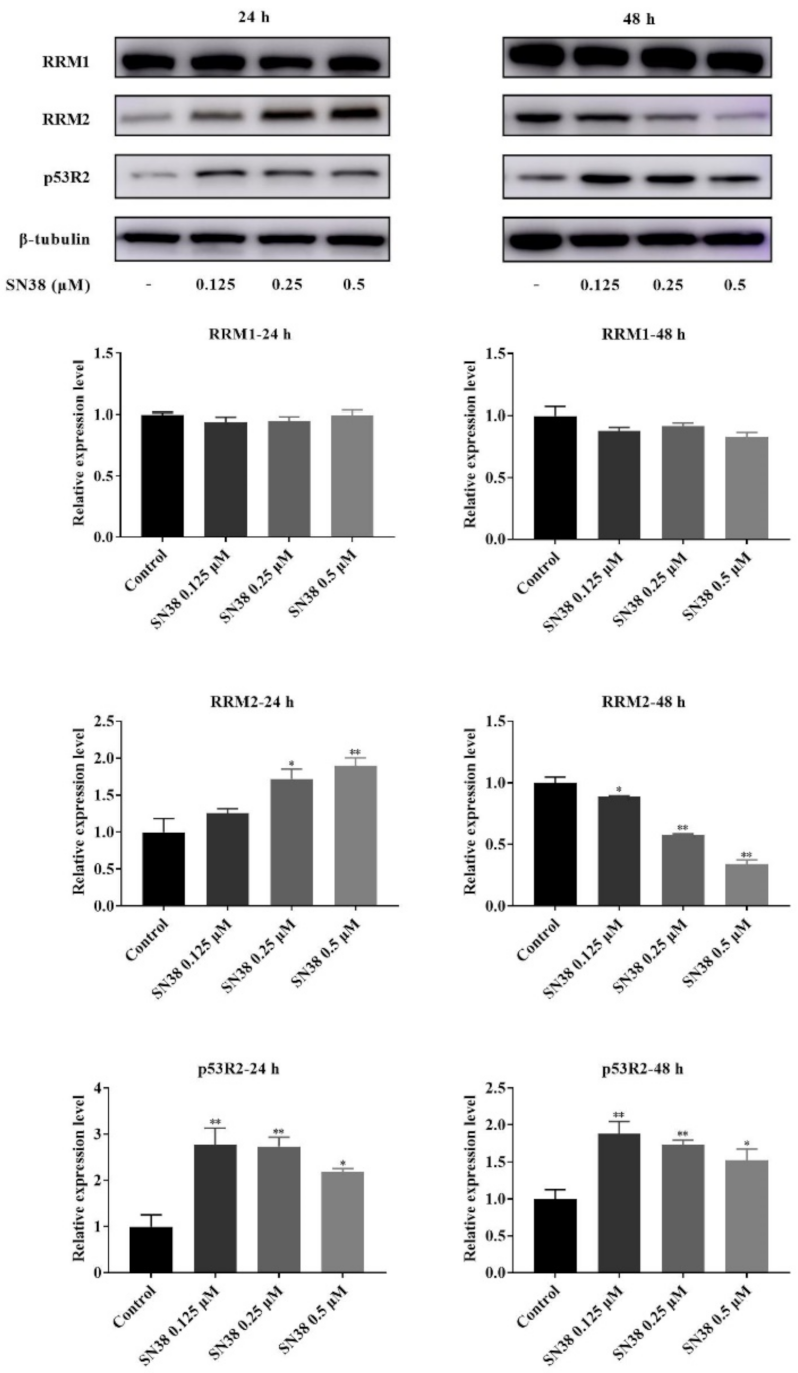
2.5. Western Blot of RNR
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture and Measurement of Cell Viability
4.3. Comet Assay Analysis
4.4. Cell Cycle Distribution Analysis
4.5. LC-MS/MS Analysis
4.6. Western Blot Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Poddar, S. ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. In Identification of Modulators of Deoxyribonucleotide Pools and Replication Stress in Cancer; University of California: Los Angeles, CA, USA, 2019; pp. 2–62. [Google Scholar]
- Meuth, M. The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp. Cell Res. 1989, 181, 305–316. [Google Scholar] [CrossRef]
- Weinberg, G.; Ullman, B.; Martin, D.W. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA 1981, 78, 2447–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabosseau, P.; Buhagiar-Labarchède, G.; Onclercq-Delic, R.; Lambert, S.; Debatisse, M.; Brison, O.; Amor-Guéret, M. Pyrimidine pool imbalance induced by BLM helicase deficiency contributes to genetic instability in Bloom syndrome. Nat. Commun. 2011, 2, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Guo, R.; Huang, Q.; Yen, Y. Chromosomal instability triggered by Rrm2b loss leads to IL-6 secretion and plasmacytic neoplasms. Cell Rep. 2013, 3, 1389–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammann, A.J. Purine nucleotide imbalance in immunodeficiency disorders. In Genetic Consequences of Nucleotide Pool Imbalance; Springer: Boston, MA, USA, 1985; pp. 487–502. [Google Scholar]
- Boss, G.R.; Seegmiller, J.E. Genetic defects in human purine and pyrimidine metabolism. Annu. Rev. Genet. 1982, 16, 297–328. [Google Scholar] [CrossRef] [PubMed]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef]
- Mathews, C.K. DNA precursor metabolism and genomic stability. FASEB J. 2006, 20, 1300–1314. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Takeda, S.; Sagiya, Y.; Gotoh, M.; Nakamura, Y.; Arakawa, H. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 2003, 34, 440–445. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Pitceathly, R.D.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002, 3, 415–424. [Google Scholar] [CrossRef]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.P.; Zhong, J.; Tan, J.H.; Jiang, X.H.; Qiao, M.M.; Wu, Y.X.; Jiang, S.H. Induction of apoptosis by arsenic trioxide and hydroxy camptothecin in gastriccancer cells in vitro. World J. Gastrol. 2000, 6, 532–539. [Google Scholar]
- Fu, Y.R.; Yi, Z.J.; Yan, Y.R.; Qiu, Z.Y. Hydroxycamptothecin-induced apoptosis in hepatoma SMMC-7721 cells and the role of mitochondrial pathway. Mitochondrion 2006, 6, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonero, R.; Supko, J.G. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin. Cancer Res. 2002, 8, 641–661. [Google Scholar]
- Håkansson, P.; Hofer, A.; Thelander, L. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem. 2006, 281, 7834–7841. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.K.; Spinelli, J.B.; Asara, J.M.; Toker, A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 2017, 7, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Cao, M.; Fan, M.; Wu, H.; Huang, W.; Zhang, Y.; Hu, Z.; Jin, X. AMPK-mTOR-ULK1 axis activation-dependent autophagy promotes hydroxycamptothecin-induced apoptosis in human bladder cancer cells. J. Cell. Physiol. 2020, 235, 4302–4315. [Google Scholar] [CrossRef]
- Mah, L.; El-Osta, A.; Karagiannis, T. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Dandrea, T.; Hellmold, H.; Jonsson, C.; Zhivotovsky, B.; Hofer, T.; Wärngård, L.; Cotgreave, I. The transcriptosomal response of human A549 lung cells to a hydrogen peroxide-generating system: Relationship to DNA damage, cell cycle arrest, and caspase activation. Free Radic. Biol. Med. 2004, 36, 881–896. [Google Scholar] [CrossRef]
- Du, L.; Yang, F.; Fang, H.; Sun, H.; Chen, Y.; Xu, Y.; Li, H.; Zheng, L.; Zhou, B.B.S. AICAr suppresses cell proliferation by inducing NTP and dNTP pool imbalances in acute lymphoblastic leukemia cells. FASEB J. 2019, 33, 4525–4537. [Google Scholar] [CrossRef]
- Liu, B.; Großhans, J. The role of dNTP metabolites in control of the embryonic cell cycle. Cell Cycle 2019, 18, 2817–2827. [Google Scholar] [CrossRef]
- Yousefi, B.; Samadi, N.; Ahmadi, Y. Akt and p53R2, partners that dictate the progression and invasiveness of cancer. DNA Repair. 2014, 22, 24–29. [Google Scholar] [CrossRef]
- Engström, Y.; Eriksson, S.; Jildevik, I.; Skog, S.; Thelander, L.; Tribukait, B. Cell cycle-dependent expression of mammalian ribonucleotide reductase. Differential regulation of the two subunits. J. Biol. Chem. 1985, 260, 9114–9116. [Google Scholar] [CrossRef]
- Saiko, P.; Ozsvar-Kozma, M.; Bernhaus, A.; Jaschke, M.; Graser, G.; Lackner, A.; Grusch, M.; Horvath, Z.; Madlener, S.; Krupitza, G. N-hydroxy-N’-(3, 4, 5-trimethoxyphenyl)-3, 4, 5-trimethoxy-benzamidine, a novel resveratrol analog, inhibits ribonucleotide reductase in HL-60 human promyelocytic leukemia cells: Synergistic antitumor activity with arabinofuranosylcytosine. Int. J. Oncol. 2007, 31, 1261–1266. [Google Scholar] [PubMed] [Green Version]
- Fei, B.; Chi, A.L.; Weng, Y. Hydroxycamptothecin induces apoptosis and inhibits tumor growth in colon cancer by the downregulation of survivin and XIAP expression. World J. Surg. Oncol. 2013, 11, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Bio.. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichard, P. Ribonucleotide reductase and deoxyribonucleotide pools. In Genetic Consequences of Nucleotide Pool Imbalance; Springer: Boston, MA, USA, 1985; pp. 33–45. [Google Scholar]
- Van Moorsel, C.; Smid, K.; Voorn, D.; Bergman, A.; Pinedo, H.; Peters, G. Effect of gemcitabine and cisplatinum combinations on ribonucleotide and deoxyribonucleotide pools in ovarian cancer cell lines. Int. J. Oncol. 2003, 22, 201–207. [Google Scholar]
- Delmas, D.; Lançon, A.; Colin, D.; Jannin, B.; Latruffe, N. Resveratrol as a chemopreventive agent: A promising molecule for fighting cancer. Curr. Drug Targets 2006, 7, 423–442. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.C.; Kearsey, S.E. A critical balance: dNTPs and the maintenance of genome stability. Genes 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Zegerman, P.; Diffley, J.F. DNA replication as a target of the DNA damage checkpoint. DNA Repair. 2009, 8, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, Q.Q.; Lam, C.W.K.; Guo, J.; Zhang, W.J.; Wang, C.Y.; Wong, V.K.W.; Yao, M.C.; Zhang, W. Investigation into perturbed nucleoside metabolism and cell cycle for elucidating the cytotoxicity effect of resveratrol on human lung adenocarcinoma epithelial cells. Chin. J. Nat. Med. 2019, 17, 608–615. [Google Scholar] [CrossRef]
- Gon, S.; Beckwith, J. Ribonucleotide reductases: Influence of environment on synthesis and activity. Antioxid. Redox Sign. 2006, 8, 773–780. [Google Scholar] [CrossRef]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blakley, R.; Vitols, E. The control of nucleotide biosynthesis. Annu. Rev. Biochem. 1968, 37, 201–224. [Google Scholar] [CrossRef]
- Mathews, C.K. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 2015, 15, 528–539. [Google Scholar] [CrossRef]
- Klanova, M.; Lorkova, L.; Vit, O.; Maswabi, B.; Molinsky, J.; Pospisilova, J.; Vockova, P.; Mavis, C.; Lateckova, L.; Kulvait, V. Downregulation of deoxycytidine kinase in cytarabine-resistant mantle cell lymphoma cells confers cross-resistance to nucleoside analogs gemcitabine, fludarabine and cladribine, but not to other classes of anti-lymphoma agents. Mol. Cancer 2014, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Van Meerloo, J.; Kaspers, G.J.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.X.; Li, Y.; Lam, C.W.K.; Wang, C.Y.; Zhang, W.J.; Wong, V.K.W.; Pang, S.S.; Yao, M.C.; Zhang, W. Method for quantification of ribonucleotides and deoxyribonucleotides in human cells using (trimethylsilyl) diazomethane derivatization followed by liquid chromatography–tandem mass spectrometry. Anal. Chem. 2018, 91, 1019–1026. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Luo, W.; Zhang, H.; Wang, C.; Yu, C.; Jiang, Z.; Zhang, W. Antitumor Mechanism of Hydroxycamptothecin via the Metabolic Perturbation of Ribonucleotide and Deoxyribonucleotide in Human Colorectal Carcinoma Cells. Molecules 2021, 26, 4902. https://doi.org/10.3390/molecules26164902
Li Y, Luo W, Zhang H, Wang C, Yu C, Jiang Z, Zhang W. Antitumor Mechanism of Hydroxycamptothecin via the Metabolic Perturbation of Ribonucleotide and Deoxyribonucleotide in Human Colorectal Carcinoma Cells. Molecules. 2021; 26(16):4902. https://doi.org/10.3390/molecules26164902
Chicago/Turabian StyleLi, Yan, Wendi Luo, Huixia Zhang, Caiyun Wang, Caiyuan Yu, Zhihong Jiang, and Wei Zhang. 2021. "Antitumor Mechanism of Hydroxycamptothecin via the Metabolic Perturbation of Ribonucleotide and Deoxyribonucleotide in Human Colorectal Carcinoma Cells" Molecules 26, no. 16: 4902. https://doi.org/10.3390/molecules26164902
APA StyleLi, Y., Luo, W., Zhang, H., Wang, C., Yu, C., Jiang, Z., & Zhang, W. (2021). Antitumor Mechanism of Hydroxycamptothecin via the Metabolic Perturbation of Ribonucleotide and Deoxyribonucleotide in Human Colorectal Carcinoma Cells. Molecules, 26(16), 4902. https://doi.org/10.3390/molecules26164902