
Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Optimization of Extraction Protocol
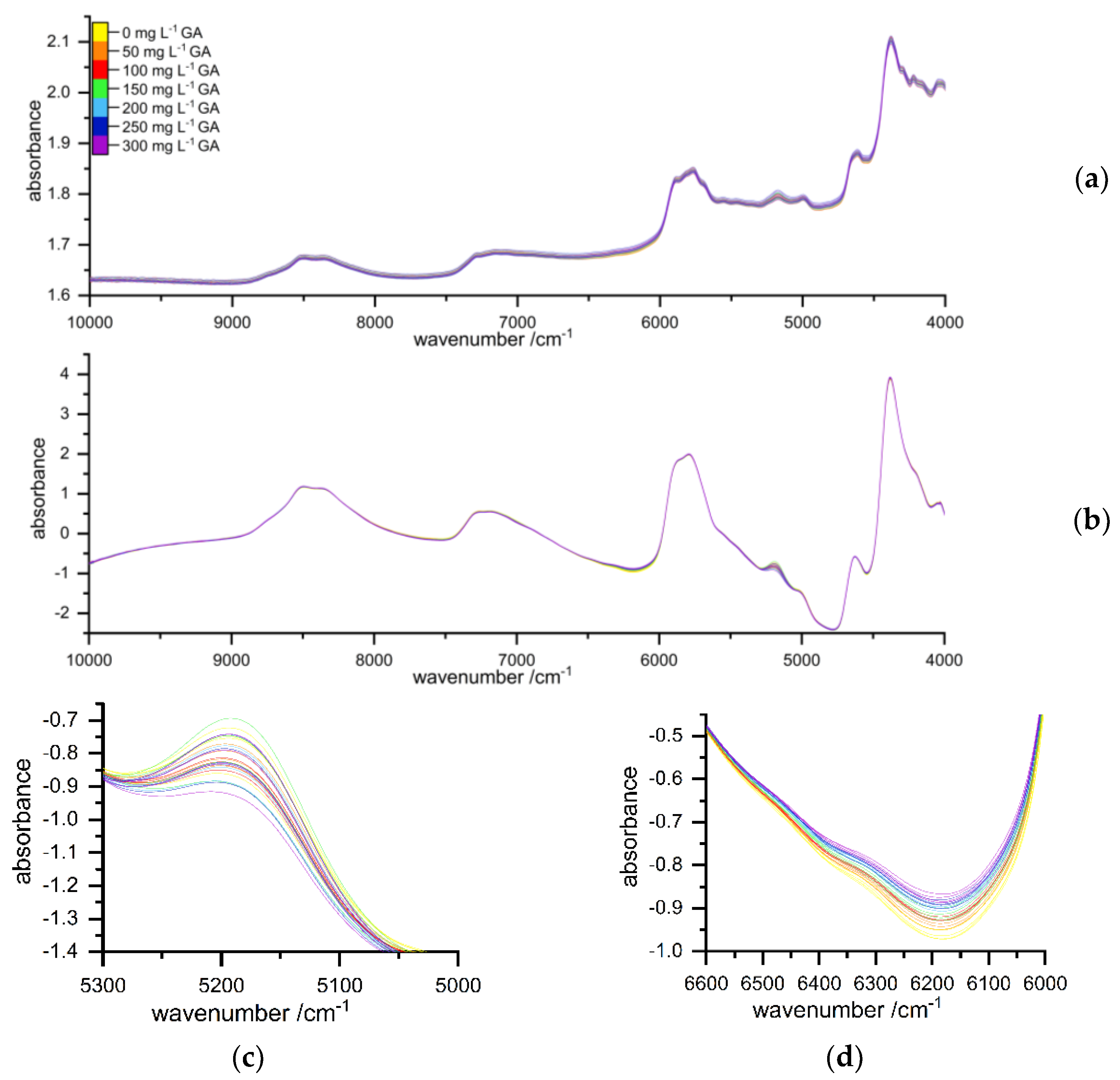
2.2. Spectral Interpretation
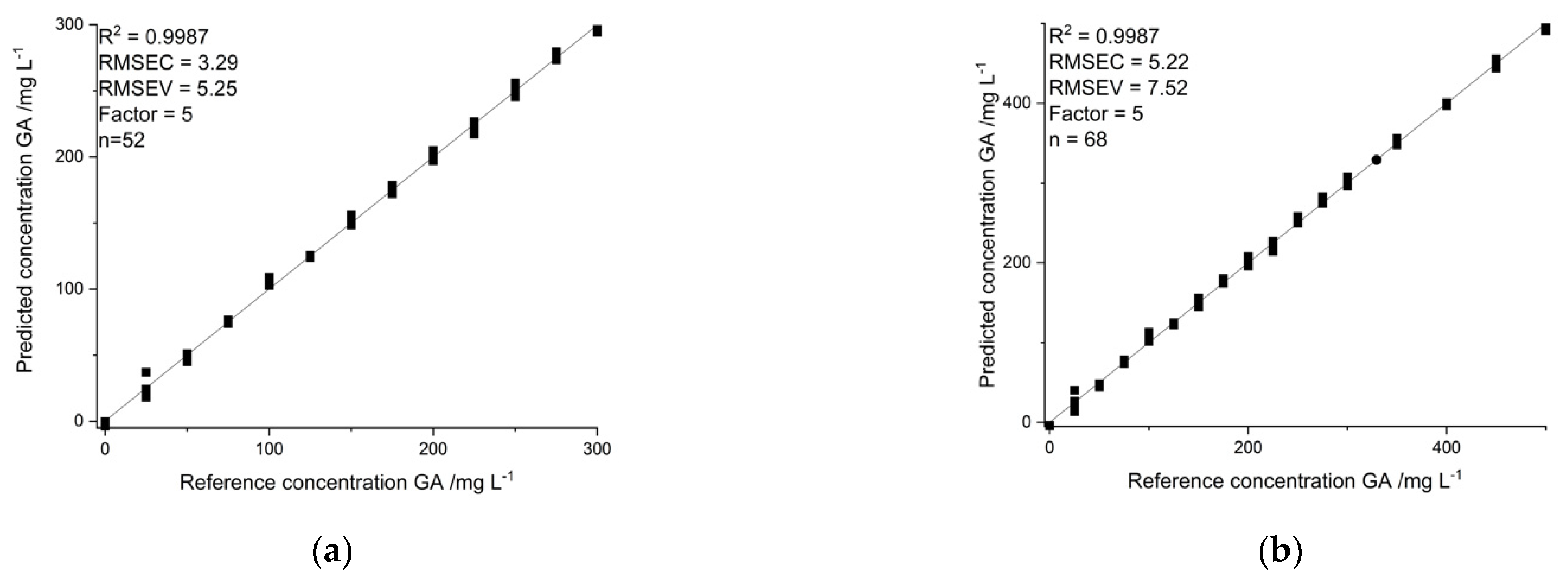
2.3. Calibration
2.4. Method Validation
2.4.1. Selectivity
2.4.2. Repeatability and Accuracy
2.4.3. LOD and LOQ for PLSR
2.4.4. Working Range
2.4.5. Comparison of Novel Established Method to Reference Methods
3. Discussion
4. Materials and Methods
4.1. Chemicals and Samples
4.2. NIR-Spectral Acquisition Using FT-NIR Spectrometer
4.3. Spectral Data Treatment
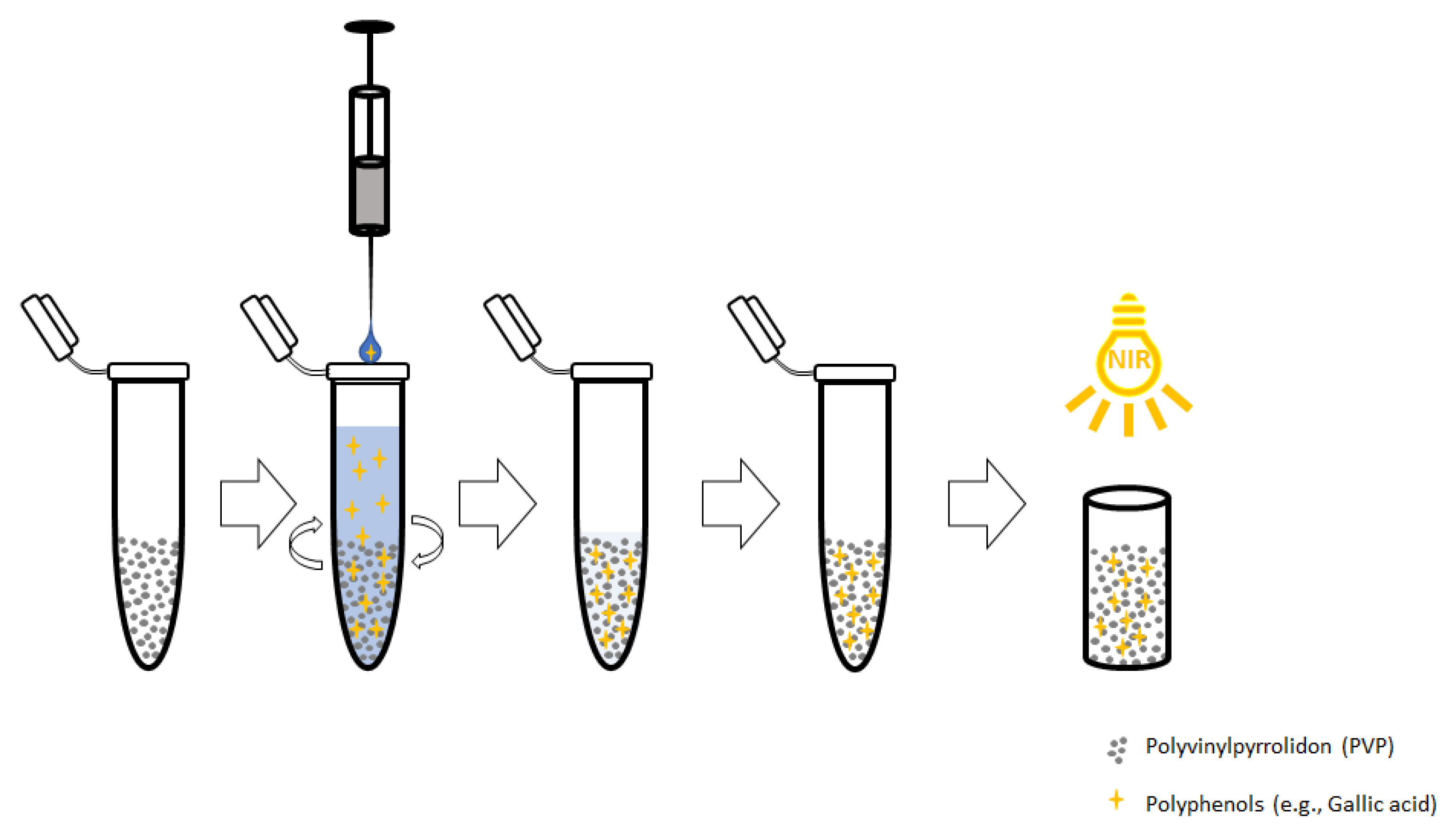
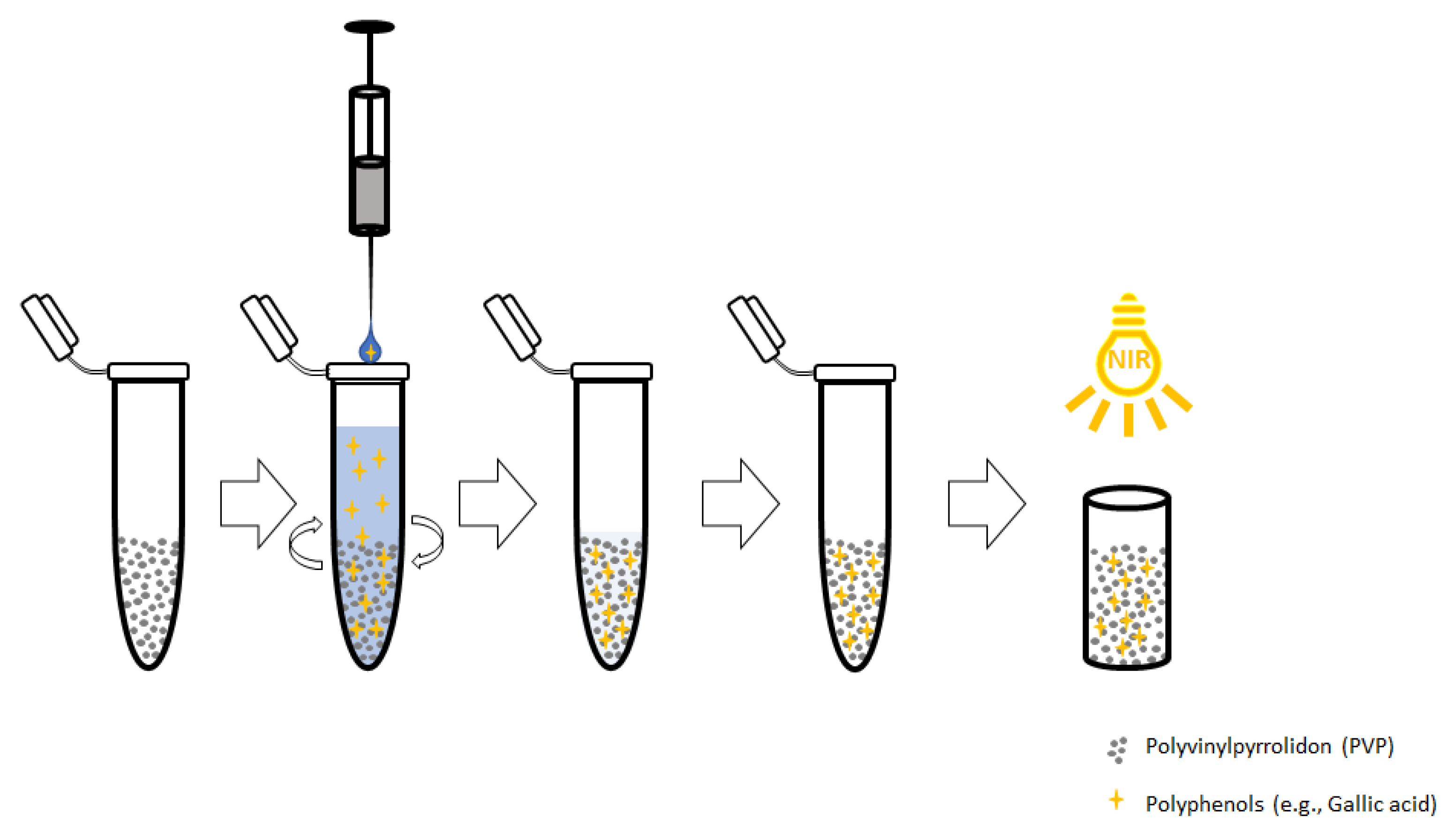
4.4. Dispersive Solid Phase Extraction
4.5. Method Optimization
4.6. Reference Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Scalbert, A.; Johnson, I.T.; Saltmarsh, M. Polyphenols: Antioxidants and beyond. Am. J. Clin. Nutr. 2005, 81, 215S–217S. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Manach, C.; Morand, C.; Rémésy, C.; Jiménez, L. Dietary polyphenols and the prevention of diseases. Crit. Rev. Food Sci. Nutr. 2005, 45, 287–306. [Google Scholar] [CrossRef] [PubMed]
- Stalikas, C.D. Extraction, separation, and detection methods for phenolic acids and flavonoids. J. Sep. Sci. 2007, 30, 3268–3295. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, T.W.; Morton, R.A. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem. J. 1946, 40, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löwenthal, J. Ueber die Bestimmung des Gerbstoffs. Fresenius Z. Anal. Chem. 1877, 16, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Kim, C.; Neilson, A.P.; Griffin, L.E.; Peck, G.M.; O’Keefe, S.F.; Stewart, A.C. Comparison of Common Analytical Methods for the Quantification of Total Polyphenols and Flavanols in Fruit Juices and Ciders. J. Food Sci. 2019, 84, 2147–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrueta, L.A.; Gallo, B.; Vicente, F. A review of solid phase extraction: Basic principles and new developments. Chromatographia 1995, 40, 474–483. [Google Scholar] [CrossRef]
- Andersen, R.A.; Sowers, J.A. Optimum conditions for bonding of plant phenols to insoluble polyvinylpyrrolidone. Phytochemistry 1968, 7, 293–301. [Google Scholar] [CrossRef]
- Allegrini, F.; Olivieri, A.C. IUPAC-consistent approach to the limit of detection in partial least-squares calibration. Anal. Chem. 2014, 86, 7858–7866. [Google Scholar] [CrossRef] [PubMed]
- The International Union of Pure and Applied Chemistry. IUPAC—Limit of Detection (L03540). Available online: https://goldbook.iupac.org/terms/view/L03540 (accessed on 17 February 2021).
- Geladi, P.; Kowalski, B.R. Partial least-squares regression: A tutorial. Anal. Chim. Acta 1986, 185, 1–17. [Google Scholar] [CrossRef]
- Glenn, J.L.; Kuo, C.C.; Durley, R.C.; Pharis, R.P. Use of insoluble polyvinylpyrrolidone for purification of plant extracts and chromatography of plant hormones. Phytochemistry 1972, 11, 345–351. [Google Scholar] [CrossRef]
- Borneman, Z. Selective removal of polyphenols and brown colour in apple juices using PES/PVP membranes in a single ultrafiltration process. Sep. Purif. Technol. 2001, 22, 53–61. [Google Scholar] [CrossRef]
- Gehrig, H.H.; Winter, K.; Cushman, J.; Borland, A.; Taybi, T. An improved RNA isolation method for succulent plant species rich in polyphenols and polysaccharides. Plant Mol. Biol. Rep. 2000, 18, 369–376. [Google Scholar] [CrossRef]
- Everette, J.D.; Bryant, Q.M.; Green, A.M.; Abbey, Y.A.; Wangila, G.W.; Walker, R.B. Thorough study of reactivity of various compound classes toward the Folin-Ciocalteu reagent. J. Agric. Food Chem. 2010, 58, 8139–8144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barua, D.N.; Roberts, E.A. Methods for the volumetric estimation of tea tannin in green-leaf and black tea. A new alkaline permanganate method. Biochem. J. 1940, 34, 1524–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, R.S. The Standardization of Potassium Permanganate Solution by Sodium Oxalate. 1. J. Am. Chem. Soc. 1912, 34, 393–416. [Google Scholar] [CrossRef] [Green Version]
- Ethenylpyrrolidin-2-One—Substance Information—ECHA. Available online: https://echa.europa.eu/de/substance-information/-/substanceinfo/100.111.937 (accessed on 18 May 2021).

{kind=link}
{kind=link}
{kind=link}
| Method | Analytical Curve | Working Range a | R2 | LOD a | LOQ a |
|---|---|---|---|---|---|
| PVP-extr. | y = 0.99987x + 0.1917 b | 36.8 to >500 | 0.999 | 12.3 to 38.4 c | 36.8 to 115 c |
| F–C d | y = 0.0019x + 0.0096 | 42.9 to 500 | 0.999 | 14.2 | 42.9 |
| Löwenthal titration d | y = 1256.1x + 48.61 | 1.47 to 12,000 | 0.995 | 0.485 | 1.47 |
| Method | Repeatability/% RSD | Recovery/% | CI95% |
|---|---|---|---|
| PVP-extr. | 10.7 | 98.7 ± 10.5 | 138.3 to 157.9 GAE a |
| F–C d | 0.66 | 102.9 ± 0.21 | 307.3 to 309.9 GAE b |
| Löwenthal titration d | 0.70 | 143.5 ± 0.32 | 7145 to 7207 TAE c |
| Method | Control | Ascorbic Acid c | Tyrosine d | Glucose e |
|---|---|---|---|---|
| PVP extr. a | 148.1 ± 15.1 | 149.2 ± 0.07 | 146.0 ± 0.9 | 160.4 ± 22.1 |
| PVP extr. b | 98.7 ± 10.5 | 99.4 ± 0.04 | 97.3 ± 0.6 | 106.9 ± 14.7 |
| F–C a,f | 308.6 ± 0.64 | 1053 ± 2.4 | 398 ± 3.3 | 316 ± 1.2 |
| F–C b,f | 102.9 ± 0.21 | 351.0 ± 0.80 | 132.7 ± 1.1 | 105.3 ± 0.40 |
| L-P a,f | 7176 ± 16 | 7803 ± 43 | 7685 ± 104 | 6745 ± 25 |
| L-P b,f | 102.5 ± 0.23 | 111.5 ± 0.61 | 109.8 ± 1.5 | 96.4 ± 0.36 |
| Method | Red Wine | White Wine | Black Tea |
|---|---|---|---|
| PVP-extr. | 914 ± 33 | 39 ± 6 | 404 ± 30 |
| F–C d | 1954 ± 67 | 261 ± 3 | 726 ± 24 |
| L-P d | 1170 ± 130 | 246 ± 2 | 463 ± 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kappacher, C.; Neurauter, M.; Rainer, M.; Bonn, G.K.; Huck, C.W. Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content. Molecules 2021, 26, 4807. https://doi.org/10.3390/molecules26164807
Kappacher C, Neurauter M, Rainer M, Bonn GK, Huck CW. Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content. Molecules. 2021; 26(16):4807. https://doi.org/10.3390/molecules26164807
Chicago/Turabian StyleKappacher, Christoph, Markus Neurauter, Matthias Rainer, Günther K. Bonn, and Christian W. Huck. 2021. "Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content" Molecules 26, no. 16: 4807. https://doi.org/10.3390/molecules26164807
APA StyleKappacher, C., Neurauter, M., Rainer, M., Bonn, G. K., & Huck, C. W. (2021). Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content. Molecules, 26(16), 4807. https://doi.org/10.3390/molecules26164807