
Quantitative Structure–Activity Relationship Evaluation of MDA-MB-231 Cell Anti-Proliferative Leads

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. GA-MLR QSAR Models
2.1.1. Model-1.1 (Divided Set: Training Set–80% and Prediction Set–20%)
2.1.2. Model-1.2 (Divided Set: Training Set–80% and Prediction Set–20%)
3. Discussion
- 1.
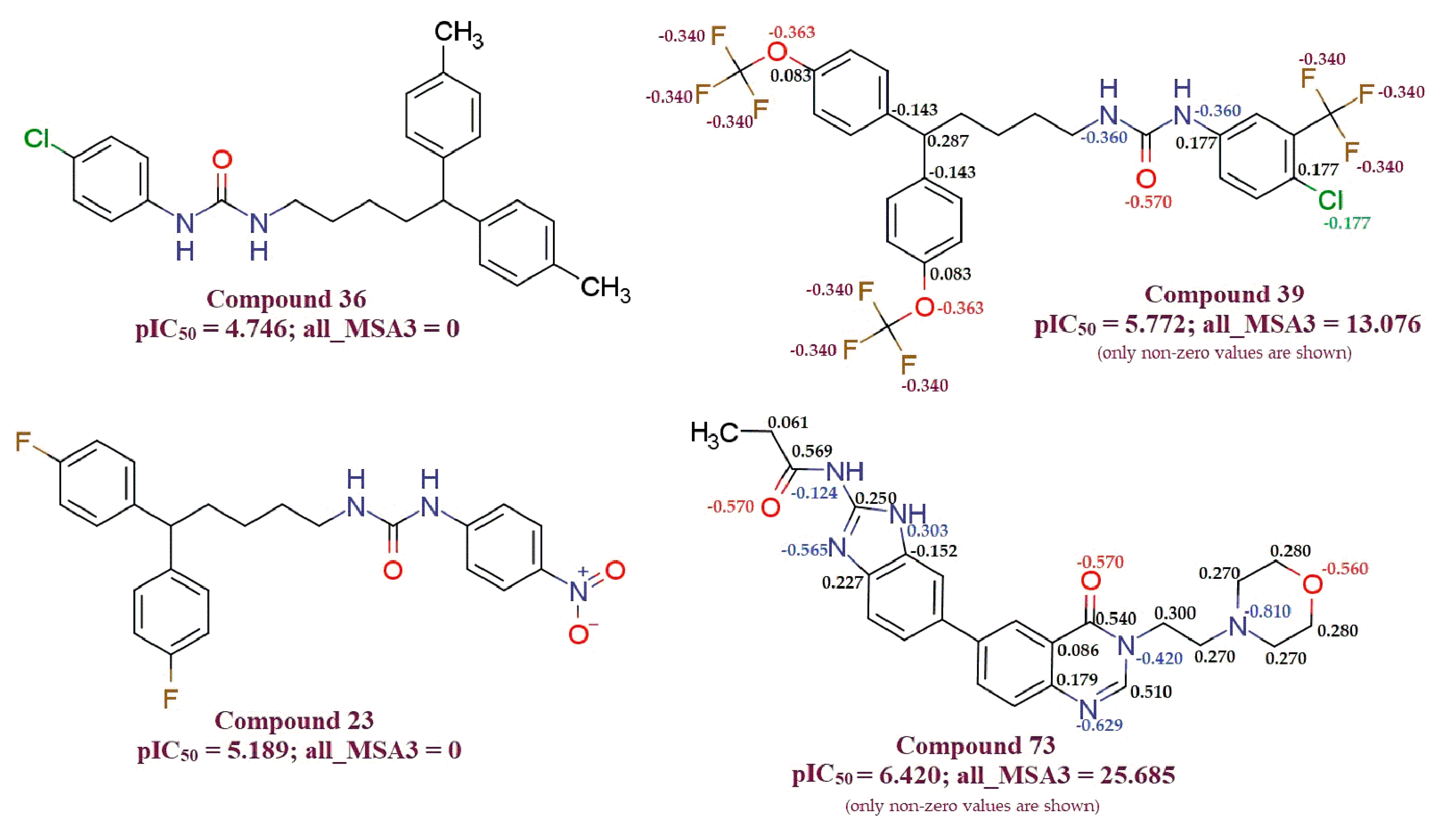
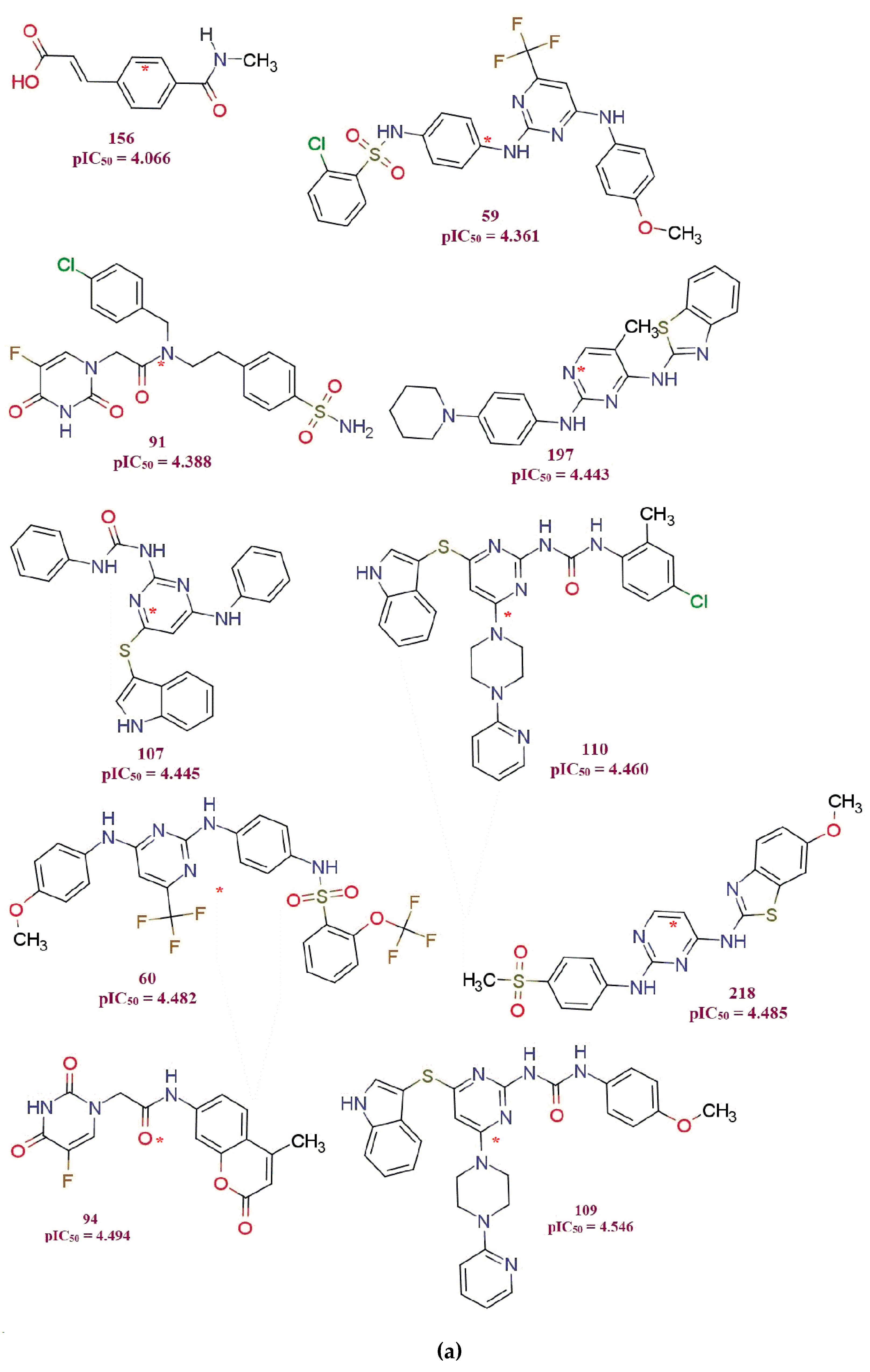
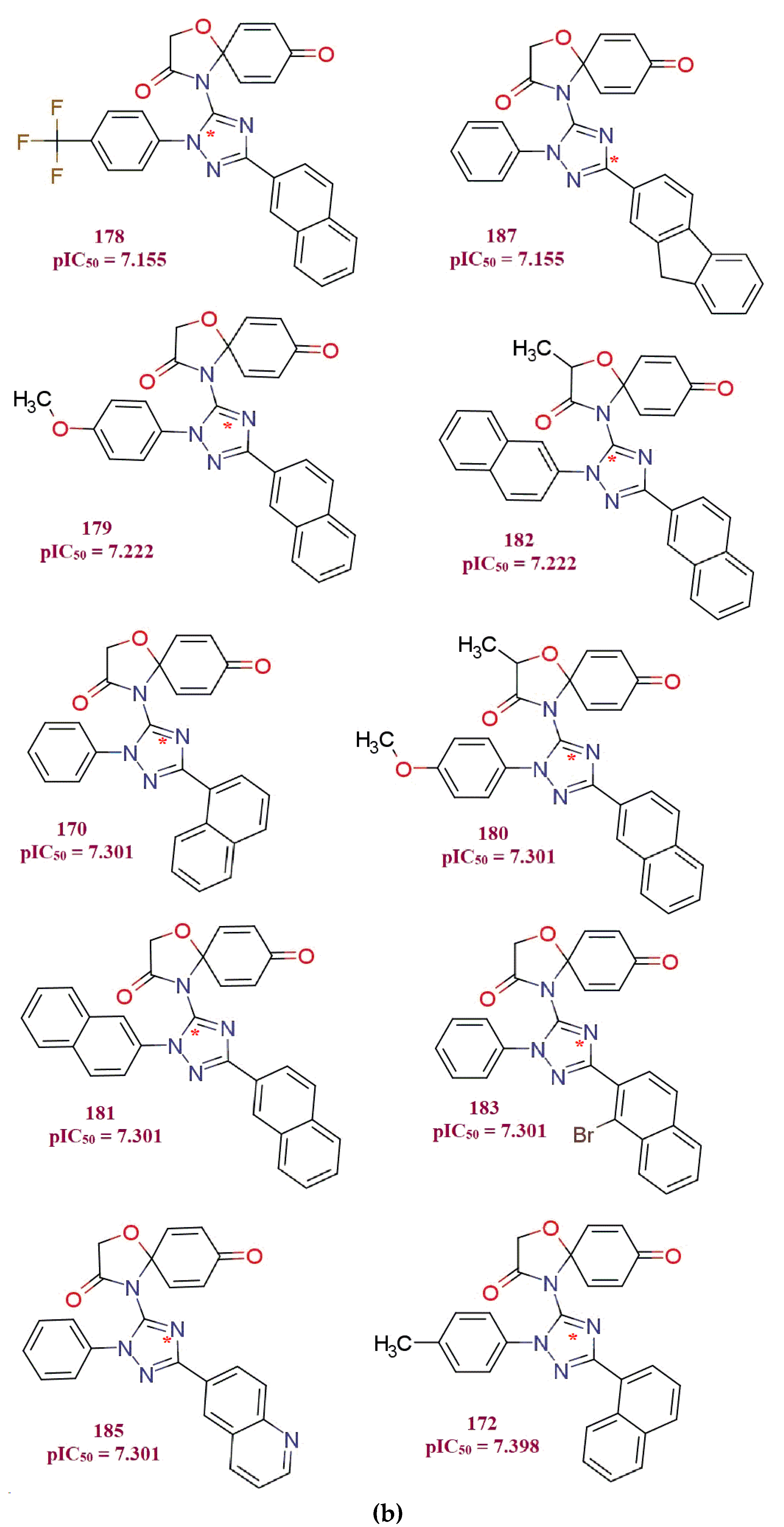
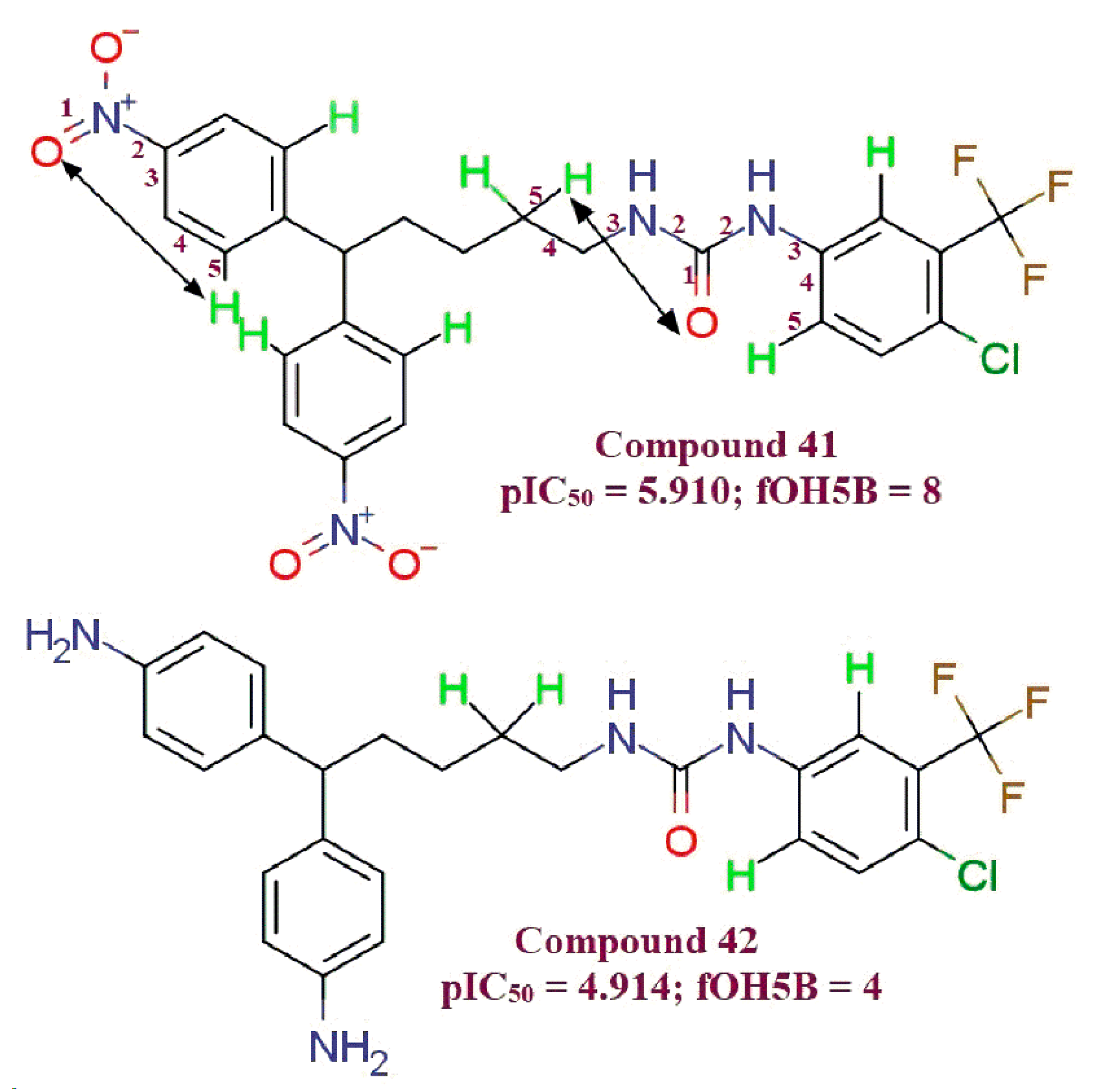
- all_MSA3, fHringC4B, fOH5B and com_sp2N_2A: All of these molecular descriptors have positive values of the coefficient and increase in the values of these molecular descriptors, which increases MDA-MB-231 anti-proliferative activity.
- 2.
- com_Splus_7A, com_don_6A and fringNC8B: These three molecular descriptors have negative coefficients and hence decrease in their values possibly will increase MDA-MB-231 anti-proliferative activity.
4. Materials and Methods
4.1. Dataset Selection
4.2. Molecular Structure Drawing and Optimization
4.3. Molecular Descriptor Calculation and Molecular Descriptor Pruning
4.4. QSAR Model–Development and Validation
- i.
- As per OECD guidelines, thorough internal as well as external validation of the developed QSAR model(s), for example, is necessarily mandatory. Hence, some molecules from the dataset were randomly kept aside as a prediction set, and remaining molecules (training set) were subjected to SFS treatment to develop the QSAR model. The QSAR model(s) generated is validated using molecules in the prediction set.Random splitting of the dataset using random splitting option in QSARINS v.2.2.4 into an 80% training set (175 molecules in training set) and a 20% prediction set (44 molecules in prediction set) was achieved. The training set was used for QSAR model development, and the prediction set was utilized for external validation.
- ii.
- QSARINS v2.2.4 with default settings and Q2LOO as a fitness function for feature selection was deployed in genesis of the GA-MLR-based QSAR models with double cross validation. Up to six variables, there was a generous increase in the Q2LOO value, but minor augmentation was observed thereafter. Consequently, the selection of the molecular descriptor was confined to a set of six descriptors to foil the danger of over-fitting, and this additionally helped to derive easy and informative QSAR models (see supplementary information Table S3 values for all the selected molecular descriptors present in QSAR models).
- iii.
- Abide by OECD guidelines; for ensured proper validation, all the models were subjected to internal and external validation, Y-randomization and model applicability domain (AD) analysis using QSARINS 2.2.4. Robustness of the GA-MLR-based QSAR model was adjudicated on the basis of (a) internal validation based on Leave-One-Out (LOO) and Leave-Many-Out (LMO) procedure; (b) external validation; (c) Y-randomization and (d) fulfilling of the respective threshold value for the statistical parameters: R2 ≥ 0.6, Q2LOO ≥ 0.5, Q2LMO ≥ 0.6, R2 > Q2, R2ex ≥ 0.6, RMSEtr < RMSEcv, ΔK ≥ 0.05, CCC ≥ 0.80, Q2-Fn ≥ 0.60, r2m ≥ 0.6, 0.9 ≤ k ≤ 1.1, 0.9 ≤ k’ ≤ 1.1 with RMSE and MAE close to zero. All QSAR models which failed to meet any of these criteria were omitted. Two QSAR models (1.1 and 1.2) with best values of these parameters and with best predicative ability (Q2-Fn > 0.71) were selected.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CADD | Computer-Aided Drug Designing |
| SMILES | Simplified Molecular-Input Line-Entry System |
| GA | Genetic Algorithm |
| MLR | Multiple Linear Regression |
| QSAR | Quantitative Structure–Activity Relationship |
| OLS | Ordinary Least Square |
| QSARINS | QSAR Insubria |
| OECD | Organization for Economic Co-operation and Development |
| OFS | Objective Feature Selection |
| SFS | Subjective Feature Selection |
| HER2 | Human Epidermal growth factor Receptor 2 |
| TNBC | Triple Negative Breast Cancer |
| ER | Estrogen Receptor |
| PR | Progesterone Receptor |
| BCCL | Breast Cancer Cell Line |
| LOF | Lack of Fit (Friedmann Parameter) |
| RMSE | Root Mean Square Error |
| MAE | Mean Absolute Error |
| RSS | Residual Sum of Squares |
| CCC | Concordance Correlation Coefficient |
| PRESS | Predictive Residual Sum of Squares |
| LOO | Leave One Out |
| LMO | Leave Many Out |
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F.; Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. 2020. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/20-Breast-fact-sheet.pdf (accessed on 29 July 2021).
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F.; Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 29 July 2021).
- European College of Authenticated Cell cultures Cell line profile MDA-MB-231. Eur. Collect. Authenticated Cell Cult. 2017, 231, 1–3, MDA-MB-231 (ECACC 92020424).
- Mendelsohn, J.; Howley, P.M.; Israel, M.A.; Gray, J.W.; Thompson, C.B. The Molecular Basis of Cancer; Elsevier Saunders: Philadelphia, PA, USA, 2015; ISBN 978-1-4557-4066-6. [Google Scholar]
- Holliday, D.L.; Speirs, V. Choosing correct breast cancer cell line for breast cancer research. Breast Cancer Res. 2011, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Freischel, A.R.; Damaghi, M.; Cunningham, J.J.; Ibrahim-Hashim, A.; Gillies, R.J.; Gatenby, R.A.; Brown, J.S. Frequency-dependent interactions determine outcome of competition between two breast cancer cell lines. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Zhou, W.X.; Chen, C.; Liu, X.Q.; Li, Y.; Lin, Y.L.; Wu, X.T.; Kong, L.Y.; Luo, J.G. Discovery and optimization of withangulatin A derivatives as novel glutaminase 1 inhibitors for the treatment of triple-negative breast cancer. Eur. J. Med. Chem. 2021, 210, 112980. [Google Scholar] [CrossRef] [PubMed]
- Ashraf-Uz-Zaman, M.; Shahi, S.; Akwii, R.; Sajib, M.S.; Farshbaf, M.J.; Kallem, R.R.; Putnam, W.; Wang, W.; Zhang, R.; Alvina, K.; et al. Design, synthesis and structure-activity relationship study of novel urea compounds as FGFR1 inhibitors to treat metastatic triple-negative breast cancer. Eur. J. Med. Chem. 2021, 209, 112866. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Fu, L.; Yuan, Z.; Liu, Y.; Zhang, K.; Chen, Y.; Wang, L.; Sun, D.; Chen, L.; Liu, B.; et al. Discovery of a novel small-molecule inhibitor of Fam20C that induces apoptosis and inhibits migration in triple negative breast cancer. Eur. J. Med. Chem. 2021, 210, 113088. [Google Scholar] [CrossRef]
- Rassias, G.; Leonardi, S.; Rigopoulou, D.; Vachlioti, E.; Afratis, K.; Piperigkou, Z.; Koutsakis, C.; Karamanos, N.K.; Gavras, H.; Papaioannou, D. Potent antiproliferative activity of bradykinin B2 receptor selective agonist FR-190997 and analogue structures thereof: A paradox resolved? Eur. J. Med. Chem. 2021, 210, 112948. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wu, X.; Yan, S.; Liu, T.; Yin, X. Synthesis and in vitro evaluation of novel spiroketopyrazoles as acetyl-CoA carboxylase inhibitors and potential antitumor agents. Eur. J. Med. Chem. 2021, 212, 113036. [Google Scholar] [CrossRef]
- Luo, L.; Jia, J.J.; Zhong, Q.; Zhong, X.; Zheng, S.; Wang, G.; He, L. Synthesis and anticancer activity evaluation of naphthalene-substituted triazole spirodienones. Eur. J. Med. Chem. 2021, 213, 113039. [Google Scholar] [CrossRef]
- An, L.; Wang, C.; Zheng, Y.G.; Liu, J.D.; Huang, T.H. Design, synthesis and evaluation of calix[4]arene-based carbonyl amide derivatives with antitumor activities. Eur. J. Med. Chem. 2021, 210, 112984. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)quinazolin-4(3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Nepali, K.; Chang, T.Y.; Lai, M.J.; Hsu, K.C.; Yen, Y.; Lin, T.E.; Lee, S.B.; Liou, J.P. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur. J. Med. Chem. 2020, 196, 112291. [Google Scholar] [CrossRef] [PubMed]
- Petreni, A.; Bonardi, A.; Lomelino, C.; Osman, S.M.; ALOthman, Z.A.; Eldehna, W.M.; El-Haggar, R.; McKenna, R.; Nocentini, A.; Supuran, C.T. Inclusion of a 5-fluorouracil moiety in nitrogenous bases derivatives as human carbonic anhydrase IX and XII inhibitors produced a targeted action against MDA-MB-231 and T47D breast cancer cells. Eur. J. Med. Chem. 2020, 190, 112112. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide derived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, S.; Zhao, X.; Chen, Y.; Yang, B.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 2020, 188, 112024. [Google Scholar] [CrossRef] [PubMed]
- Elkhalifa, D.; Siddique, A.B.; Qusa, M.; Cyprian, F.S.; El Sayed, K.; Alali, F.; Al Moustafa, A.E.; Khalil, A. Design, synthesis, and validation of novel nitrogen-based chalcone analogs against triple negative breast cancer. Eur. J. Med. Chem. 2020, 187, 111954. [Google Scholar] [CrossRef] [PubMed]
- Diao, P.C.; Lin, W.Y.; Jian, X.E.; Li, Y.H.; You, W.W.; Zhao, P.L. Discovery of novel pyrimidine-based benzothiazole derivatives as potent cyclin-dependent kinase 2 inhibitors with anticancer activity. Eur. J. Med. Chem. 2019, 179, 196–207. [Google Scholar] [CrossRef]
- Liu, T.; Wan, Y.; Liu, R.; Ma, L.; Li, M.; Fang, H. Improved antiproliferative activities of a new series of 1,3,4-thiadiazole derivatives against human leukemia and breast cancer cell lines. Chem. Res. Chin. Univ. 2016, 32, 768–774. [Google Scholar] [CrossRef]
- Alkhaldi, A.A.M.; Al-Sanea, M.M.; Nocentini, A.; Eldehna, W.M.; Elsayed, Z.M.; Bonardi, A.; Abo-Ashour, M.F.; El-Damasy, A.K.; Abdel-Maksoud, M.S.; Al-Warhi, T.; et al. 3-Methylthiazolo[3,2-a]benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: Design, synthesis, biological and molecular modeling studies. Eur. J. Med. Chem. 2020, 207, 112745. [Google Scholar] [CrossRef]
- Baldi, A. Computational approaches for drug design and discovery: An overview. Syst. Rev. Pharm. 2010, 1, 99–105. [Google Scholar] [CrossRef]
- Joy, S.; Vijayakumar, Y.M.; Sunhye, G. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [Green Version]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.M.; Harten, P.; Young, D.M.; Muratov, E.N.; Golbraikh, A.; Zhu, H.; Tropsha, A. Does rational selection of training and test sets improve the outcome of QSAR modeling? J. Chem. Inf. Model. 2012, 52, 2570–2578. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Winkler, D.A. Understanding the Roles of the “two QSARs”. J. Chem. Inf. Model. 2016, 56, 269–274. [Google Scholar] [CrossRef]
- Huang, J.; Fan, X. Why QSAR fails: An empirical evaluation using conventional computational approach. Mol. Pharm. 2011, 8, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Masand, V.H.; Mahajan, D.T.; Nazeruddin, G.M.; Hadda, T.B.; Rastija, V.; Alfeefy, A.M. Effect of information leakage and method of splitting (rational and random) on external predictive ability and behavior of different statistical parameters of QSAR model. Med. Chem. Res. 2015, 24, 1241–1264. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Roy, P.P.; Kovarich, S.; Yap, C.W.; Papa, E. QSAR modeling is not “Push a button and find a correlation”: A case study of toxicity of (Benzo-)triazoles on Algae. Mol. Inform. 2012, 31, 817–835. [Google Scholar] [CrossRef]
- Gramatica, P. On the development and validation of QSAR models. Methods Mol. Biol. 2013, 930, 499–526. [Google Scholar] [CrossRef]
- Consonni, V.; Ballabio, D.; Todeschini, R. Comments on the Definition of the Q2 Parameter for QSAR Validation. J. Chem. Inf. Model. 2009, 49, 1669–1678. [Google Scholar] [CrossRef]
- Consonni, V.; Todeschini, R.; Ballabio, D.; Grisoni, F. On the Misleading Use of QF32 for QSAR Model Comparison. Mol. Inform. 2019, 38, 1800029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models: How to evaluate It? Comparison of different validation criteria and proposal of using the concordance correlation coefficient. J. Chem. Inf. Model. 2011, 51, 2320–2335. [Google Scholar] [CrossRef] [PubMed]
- Lawal, H.A.; Uzairu, A.; Uba, S. QSAR, molecular docking studies, ligand-based design and pharmacokinetic analysis on Maternal Embryonic Leucine Zipper Kinase (MELK) inhibitors as potential anti-triple-negative breast cancer (MDA-MB-231 cell line) drug compounds. Bull. Natl. Res. Cent. 2021, 45. [Google Scholar] [CrossRef]
- Shukla, A.; Tyagi, R.; Meena, S.; Datta, D.; Srivastava, S.K.; Khan, F. 2D- and 3D-QSAR modelling, molecular docking and in vitro evaluation studies on 18β-glycyrrhetinic acid derivatives against triple-negative breast cancer cell line. J. Biomol. Struct. Dyn. 2020, 38, 168–185. [Google Scholar] [CrossRef]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Gramatica, P.; Cassani, S.; Chirico, N. QSARINS-chem: Insubria datasets and new QSAR/QSPR models for environmental pollutants in QSARINS. J. Comput. Chem. 2014, 35, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- OECD Validation of (Q)SAR Models–OECD. Available online: https://www.oecd.org/env/ehs/riskassessment/validationofqsarmodels.htm (accessed on 17 July 2021).
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models; OECD Series on Testing and Assessment; No. 69; OECD Publishing: Paris, France, 2014. [Google Scholar] [CrossRef]
- Group, Q.E. The report from the expert group on (Quantitative) Structure-Activity Relationships [(Q)SARs] on the principles for the validation of (Q)SARs. Organ. Econ. CO-OPERATION Dev. Paris 2004, 49, 206. [Google Scholar]
- 37th Joint Meeting of the Chemicals Committee, OECD principles for the validation, for regulatory purposes, of (quantitative) structure-activity relationship models These principles were agreed by OECD member countries at the 37. Biotechnology 2004, 3–4. Available online: https://www.oecd.org/chemicalsafety/risk-assessment/37849783.pdf (accessed on 29 July 2021).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gandhi, A.; Masand, V.; Zaki, M.E.A.; Al-Hussain, S.A.; Ghorbal, A.B.; Chapolikar, A. Quantitative Structure–Activity Relationship Evaluation of MDA-MB-231 Cell Anti-Proliferative Leads. Molecules 2021, 26, 4795. https://doi.org/10.3390/molecules26164795
Gandhi A, Masand V, Zaki MEA, Al-Hussain SA, Ghorbal AB, Chapolikar A. Quantitative Structure–Activity Relationship Evaluation of MDA-MB-231 Cell Anti-Proliferative Leads. Molecules. 2021; 26(16):4795. https://doi.org/10.3390/molecules26164795
Chicago/Turabian StyleGandhi, Ajaykumar, Vijay Masand, Magdi E. A. Zaki, Sami A. Al-Hussain, Anis Ben Ghorbal, and Archana Chapolikar. 2021. "Quantitative Structure–Activity Relationship Evaluation of MDA-MB-231 Cell Anti-Proliferative Leads" Molecules 26, no. 16: 4795. https://doi.org/10.3390/molecules26164795
APA StyleGandhi, A., Masand, V., Zaki, M. E. A., Al-Hussain, S. A., Ghorbal, A. B., & Chapolikar, A. (2021). Quantitative Structure–Activity Relationship Evaluation of MDA-MB-231 Cell Anti-Proliferative Leads. Molecules, 26(16), 4795. https://doi.org/10.3390/molecules26164795