
Electrooxidation Is a Promising Approach to Functionalization of Pyrazole-Type Compounds

 and
and
Abstract
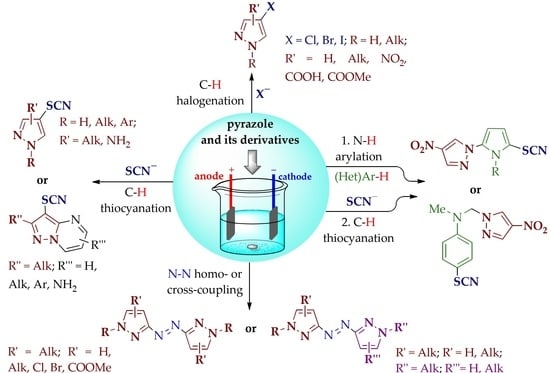
1. Introduction
2. Electrooxidative C–H Halogenation of Pyrazole and Its Substituted Derivatives
2.1. Chemical Halogenation of Pyrazoles
2.1.1. Chlorination
2.1.2. Bromination
2.1.3. Iodination
2.2. C-H An Halogenation of Pyrazoles
2.2.1. Chlorination
2.2.2. Bromination
2.2.3. Iodination
2.3. The Mechanistic Aspects of C–H (An) Halogenation of Pyrazoles
3. Electrooxidative C–H Thiocyanation of 5-Aminopyrazoles and Pyrazolo [1,5-a]pyrimidines
3.1. C–H An Thiocyanation: General Patterns and Approaches
3.2. C–H An Thiocyanation of Pyrazole Derivatives
3.2.1. CV studies and the Choice of Optimal Approach
3.2.2. Electrosynthesis
3.3. Antifungal and Antibacterial Activity of Thiocyanated Pyrazole Derivatives
4. (Electro)oxidative N–N Coupling of Aminopyrazoles
4.1. (Electro)oxidative N-N Coupling of Aminopyrazoles: Approaches and General Patterns
4.2. Synthesis of Azopyrazoles
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Godula, K.; Sames, D. C-H Bond Functionalization in Complex Organic Synthesis. Science 2006, 312, 67–72. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Morton, D. C-H Functionalization: Collaborative Methods to Redefine Chemical Logic. Angew. Chem. Int. Ed. 2014, 53, 10256–10258. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L.; Morton, D. Recent Advances in C–H Functionalization. J. Org. Chem. 2016, 81, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B. Chapter 11. Aromatic Substitution, Electrophilic. In March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 8th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 607–686. [Google Scholar]
- Chupakhin, O.N.; Postovskii, I.Y. Nucleophilic Substitution of Hydrogen in Aromatic Systems. Russ. Chem. Rev. 1976, 45, 454–468. [Google Scholar] [CrossRef]
- Chupakhin, O.N.; Charushin, V.N.; van der Plas, H.C. Nucleophilic Aromatic Substitution of Hydrogen; Academic Press: New York, NY, USA, 1994. [Google Scholar]
- Charushin, V.N.; Chupakhin, O.N. Nucleophilic aromatic substitution of hydrogen and related reactions. Mendeleev Commun. 2007, 17, 249–254. [Google Scholar] [CrossRef]
- Chupakhin, O.N.; Charushin, V.N. Recent advances in the field of nucleophilic aromatic substitution of hydrogen. Tetrahedron Lett. 2016, 57, 2665–2672. [Google Scholar] [CrossRef]
- Charushin, V.N.; Chupakhin, O.N. Nucleophilic C—H functionalization of arenes: A contribution to green chemistry. Russ. Chem. Bull. 2019, 68, 453–471. [Google Scholar] [CrossRef]
- Seebach, D. Methods of Reactivity Umpolung. Angew. Chem. Int. Ed. 1979, 18, 239–258. [Google Scholar] [CrossRef]
- Weinberg, N.L.; Weinberg, H.R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. [Google Scholar] [CrossRef]
- Organic Electrochemistry, 1st ed.; Baizer, M.M., Ed.; M. Dekker: New York, NY, USA, 1973. [Google Scholar]
- Organic Electrochemistry, 2nd ed.; Baizer, M.M., Ed.; M. Dekker: New York, NY, USA, 1983. [Google Scholar]
- Yoshida, K. Electrooxidation in Organic Chemistry. The Role of Cation Radicals as Synthetic Intermediates; Wiley: New York, NY, USA, 1984. [Google Scholar]
- Organic Electrochemistry, 3rd ed.; Lund, H., Baizer, M.M., Eds.; M. Dekker: New York, NY, USA, 1991. [Google Scholar]
- Hammerich, O.; Utley, J.H.P.; Eberson, L. Organic Electrochemistry, 4th ed.; Lund, H., Hammerich, O., Eds.; M. Dekker: New York, NY, USA, 2001. [Google Scholar]
- Shchepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; Petrosyan, V.A. Direct nucleophilic functionalization of C(sp2)–H-bonds in arenes and hetarenes by electrochemical methods. Russ. Chem. Rev. 2013, 82, 747–771. [Google Scholar] [CrossRef]
- Francke, R.; Little, R.D. Redox catalysis in organic electrosynthesis: Basic principles and recent developments. Chem. Soc. Rev. 2014, 43, 2492–2521. [Google Scholar] [CrossRef]
- Karkas, M.D. Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-L.; Fang, P.; Mei, T.-S. Recent Advances in Organic Electrochemical C–H Functionalization. Chin. J. Chem. 2018, 36, 338–352. [Google Scholar] [CrossRef]
- Petrosyan, V.A. Reactions of anodic and chemical aromatic substitution. Mendeleev Commun. 2011, 21, 115–121. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Sigacheva, V.L.; Petrosyan, V.A. New data on heteroarene thiocyanation by anodic oxidation of NH4SCN. The processes of electroinduced nucleophilic aromatic substitution of hydrogen. Tetrahedron Lett. 2014, 55, 4306–4309. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Heard, D.M.; Lennox, A.J.J. Electrode Materials in Modern Organic Electrochemistry. Angew. Chem. Int. Ed. 2020, 59, 18866–18884. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A. Electrochemical halogenation of organic compounds. Russ. J. Electrochem. 2013, 49, 497–529. [Google Scholar] [CrossRef]
- Beer, H.B. Dimensionally Stable Anodes. In Electrochemistry in Industry: New Directions; Landau, U., Yeager, E., Kortan, D., Eds.; Springer: Boston, MA, USA, 1982; pp. 19–28. [Google Scholar] [CrossRef]
- Janin, Y.L. Preparation and Chemistry of 3/5-Halogenopyrazoles. Chem. Rev. 2012, 112, 3924–3958. [Google Scholar] [CrossRef] [PubMed]
- Slomczynska, U.; Olivo, P.; Beattie, J.; Starkey, G.; Noueiry, A.; Roth, R. Compounds, Compositions and Methods for Control of Hepatitis C Viral Infections. International Patent 2010096115, 26 August 2010. [Google Scholar]
- Tanigchi, T.; Kawada, A.; Kondo, M.; Quinn, J.F.; Kanitomo, J.; Yoshikawa, M.; Fashimi, M. Pyridazinone Compounds. U.S. Patent 20100197651, 5 August 2010. [Google Scholar]
- Alberati, D.; Alvares, R.S.; Bleicher, K.; Flohr, A.; Markus, R.; Groeke, K.Z.; Koerner, M.; Kuhn, B.; Peters, J.-U.; Rudolph, M. Novel Imidazopyridines. U.S. Patent 20110071128, 24 March 2011. [Google Scholar]
- Linn, D.M.; Fong, E.H. Nicotinic Acetylcholine Agonists in the Treatment of Glaucoma and Retinal Neuropathy. International Patent 2004039366, 13 May 2004. [Google Scholar]
- Tomokazu, N. Therapeutic Agent for Hyperlipemia, Arteriosclerosis, and/or Metabolic Syndrome. Japan Patent 2006182668, 13 July 2006. [Google Scholar]
- D’orchumont, H.; Fraisse, L.; Zimmerman, A. Use of Indazolecarboxamide Derivatives for the Preparation of a Medicament that is Intended for the Treatment and Prevention of Paludism. International Patent 2005099703, 27 October 2005. [Google Scholar]
- Pennell, A.M.K.; Aggen, J.B.; Wright, J.J.; Sen, S.; McMaster, B.E.; Brian, E.; Dairaghi, J.D. 1-Aryl-4-substituted Piperazines Derivatives for Use as CCR1 Antagonists for the Treatment of Inflammation and Immune Disorders. International Patent 03105853, 24 December 2003. [Google Scholar]
- Singh, J.; Gurney, M.E.; Burgin, A.; Sandanayaka, V.; Kiselyov, A.; Motta, A.; Schultz, G.; Hategan, G. Biaryl as PDE4 inhibitors for treating inflammation. International Patent 2009067600, 28 May 2009. [Google Scholar]
- Talley, J.J.; Sprott, K.; Pearson, J.P.; Game, P.; Milene, G.T.; Schairer, W.; Yang, J.J.; Kim, C.; Barden, T.; Lundigran, R. Indole compounds. International Patent 2008019357, 14 February 2008. [Google Scholar]
- Pelcman, B.; Sanin, A.; Nilsson, P.; Groth, T.; Kromann, H. Pyrazoles Useful in the Treatment of Inflammation. International Patent 2007051981, 10 May 2007. [Google Scholar]
- Banner, D.; Helpert, H.; Mauser, H.; Mayweg, V.A.; Rogers-Evans, M. Amino oxazine Derivatives. International Patent 2011070029, 16 June 2011. [Google Scholar]
- Bamba, M.; Eiki, J.; Mitsuya, M.; Nishimura, T.; Sakai, F.; Sasaki, Y.; Watanabe, H. Novel 2-Pyridinecarboxamide Derivatives. International Patent 2004081001, 23 September 2004. [Google Scholar]
- Fix-Stenzel, S.; Judd, A.; Michaelides, M. Pyrrolopyridine and Pyrrolopyrimidine Inhibitors of Kinases. International Patent 2011143459, 17 November 2011. [Google Scholar]
- You, H.; Youn, H.-S.; Im, I.; Bae, M.-H.; Lee, S.-K.; Ko, H.; Eom, S.H.; Kim, Y.-C. Design, synthesis and X-ray crystallographic study of NAmPRTase inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2011, 46, 1153–1164. [Google Scholar] [CrossRef]
- Ahearn, S.P.; Altman, M.; Chichetti, S.; Czako, B.; Daniels, M.H.; Falcone, D.; Guerin, D.; Lipford, K.; Martinez, M.; Osimboni, E.; et al. Tyrosine kinase Inhibitors. International Patent 2011084402, 14 July 2011. [Google Scholar]
- Basso, A.D. Anti-mitotic Agent and Aurora Kinase Inhibitor Combination as Anti-cancer Treatment. International Patent 2009017701, 5 February 2009. [Google Scholar]
- Hattori, K.; Matsuda, K.; Murata, M.; Nakajima, T.; Tsutsumi, H. Substituted 3-Pyrrolidinylthio-carbapenems as Antimicrobial Agents. International Patent 9321186, 28 October 1993. [Google Scholar]
- Barrett, D.; Matsuda, H.; Matsuda, K.; Matsuya, T.; Mizuno, H.; Murano, K.; Ogino, T.; Toda, A. New Compound. International Patent 02072621, 19 September 2002. [Google Scholar]
- Kudo, N.; Furuta, S.; Taniguchi, M.; Endo, T.; Sato, K. Synthesis and Herbicidal Activity of 1,5-Diarylpyrazole Derivatives. Chem. Pharm. Bull. 1999, 47, 857–868. [Google Scholar] [CrossRef]
- Fustero, S.; Román, R.; Sanz-Cervera, J.F.; Simón-Fuentes, A.; Cuñat, A.C.; Villanova, S.; Murguía, M. Improved Regioselectivity in Pyrazole Formation through the Use of Fluorinated Alcohols as Solvents: Synthesis and Biological Activity of Fluorinated Tebufenpyrad Analogs. J. Org. Chem. 2008, 73, 3523–3529. [Google Scholar] [CrossRef]
- Higashino, Y.; Ikeda, Y.; Koike, S.; Kyomura, N.; Okui, S.; Tomita, H. Pyrazoles and Agricultural Chemicals Containing Them as Active Ingredients. International Patent 9737990, 16 October 1997. [Google Scholar]
- Auler, T.; Bojack, G.; Bretschneider, T.; Drewes, M.W.; Feucht, D.; Fischer, R.; Hills, M.; Kehne, H.; Konze, J.; Kuck, K.-H.; et al. N-Heterocyclyl Phenyl-substituted Cyclic Ketoenols. International Patent 2004111042, 23 October 2004. [Google Scholar]
- Fischer, R.; Gomibuchi, T.; Nakakura, N.; Otsu, Y.; Shibuya, K.; Wada, K.; Yoneta, Y. N1-((Pyrazol-1-ymethyl)-2-methylphenyl)-phatalamide Derivatives and Related Compounds Insecticides. International Patent 2005095351, 13 October 2005. [Google Scholar]
- Hüttel, R.; Schäfer, O.; Welzel, G. Die Chlorierung der Pyrazole. Justus Liebigs Ann. Chem. 1956, 598, 186–197. [Google Scholar] [CrossRef]
- Ohsawa, A.; Kaihoh, T.; Itoh, T.; Okada, M.; Kawabata, C.; Yamaguchi, K.; Igeta, H. Reactions of N-Aminopyrazoles with Halogenating Reagents and Synthesis of 1, 2, 3-Triazines. Chem. Pharm. Bull. 1988, 36, 3838–3848. [Google Scholar] [CrossRef][Green Version]
- Stefani, H.A.; Pereira, C.M.P.; Almeida, R.B.; Braga, R.C.; Guzen, K.P.; Cella, R. A mild and efficient method for halogenation of 3,5-dimethyl pyrazoles by ultrasound irradiation using N-halosuccinimides. Tetrahedron Lett. 2005, 46, 6833–6837. [Google Scholar] [CrossRef]
- Zhao, Z.G.; Wang, Z.X. Halogenation of Pyrazoles Using N-Halosuccinimides in CCl4 and in Water. Synthetic Commun. 2007, 37, 137–147. [Google Scholar] [CrossRef]
- Mullens, P.R. An improved synthesis of 1-methyl-1H-pyrazole-4-boronic acid pinacol ester and its corresponding lithium hydroxy ate complex: Application in Suzuki couplings. Tetrahedron Lett. 2009, 50, 6783–6786. [Google Scholar] [CrossRef]
- Khan, K.M.; Maharvi, G.M.; Choudhary, M.I.; Rahman, A.-U.; Perveen, S. A modified, economical and efficient synthesis of variably substituted pyrazolo[4,3-d]pyrimidin-7-ones. J. Het. Chem. 2005, 42, 1085–1093. [Google Scholar] [CrossRef]
- Kerr, E.R.; Mather, W.B. Electrochemical Chlorination of Hydrocarbon in Hydrochloric Acid-Acetic Acid Solytion. U.S. Patent 3692646, 19 September 1972. [Google Scholar]
- Ehlert, M.K.; Storr, A.; Thompson, R.C. Metal pyrazolate polymers. Part 3. Synthesis and study of Cu(I) and Cu(II) complexes of 4-Xdmpz (where X = H, Cl, Br, I, and CH3 for Cu(I) and X = H, Cl, Br, and CH3 for Cu(II); dmpz = 3,5-dimethylpyrazolate). Can. J. Chem. 1992, 70, 1121–1128. [Google Scholar] [CrossRef]
- Petko, K.; Sokolenko, T.; Yagupolskii, L. Chemical properties of derivatives of N-difluoromethyl-and N-2-H-tetrafluoroethylpyrazoles. Chem. Heterocycl. Compd. 2006, 42, 1177–1184. [Google Scholar] [CrossRef]
- Akopyan, G.A. Bromination of pyrazole-3(5)-carboxylic acid. Russ. J. Gen. Chem. 2007, 77, 1652–1653. [Google Scholar] [CrossRef]
- Guillou, S.; Bonhomme, F.J.; Ermolenko, M.S.; Janin, Y.L. Simple preparations of 4 and 5-iodinated pyrazoles as useful building blocks. Tetrahedron 2011, 67, 8451–8457. [Google Scholar] [CrossRef]
- Salanouve, E.; Guillou, S.; Bizouarne, M.; Bonhomme, F.J.; Janin, Y.L. 3-Methoxypyrazoles from 1,1-dimethoxyethene, few original results. Tetrahedron 2012, 68, 3165–3171. [Google Scholar] [CrossRef]
- Guillou, S.; Janin, Y.L. 5-Iodo-3-Ethoxypyrazoles: An Entry Point to New Chemical Entities. Chem. Eur. J. 2010, 16, 4669–4677. [Google Scholar] [CrossRef]
- Sinyakov, A.; Vasilevskii, S.; Shvartsberg, M. A new rearrangement of chloroethynylpyrazoles. Russ. Chem. Bull. 1977, 26, 2142–2147. [Google Scholar] [CrossRef]
- Vasilevskii, S.F.; Shvartsberg, M.S. Oxidative iodination of substituted N-methylpyrazoles. Russ. Chem. Bull. 1980, 29, 778–784. [Google Scholar] [CrossRef]
- Tret’yakov, E.V.; Vasitevsky, S.F. Nitrodeiodination of 4-iodo-l-methylpyrazoles. Russ. Chem. Bull. 1996, 45, 2581–2584. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A.; Ugrak, B.I. Electrosynthesis of 4-chloropyrazolecarboxylic acids. Russ. Chem. Bull. 2009, 58, 291–296. [Google Scholar] [CrossRef]
- Perevalov, V.P.; Manaev, Y.A.; Bezborodov, B.V.; Stepanov, B.I. Chlorination of 1,5-dimethylpyrazole. Chem. Heterocycl. Compd. 1990, 26, 301–303. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A.; Ugrak, B.I. Electrosynthesis of 4-chloro derivatives of pyrazole and alkylpyrazoles. Russ. J. Electrochem. 2008, 44, 1320–1326. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A. The Reactivity Trends in Electrochemical Chlorination and Bromination of N-Substituted and N-Unsubstituted Pyrazoles. Chem. Heterocycl. Compd. 2014, 49, 1599–1610. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A.; Ugrak, B.I. Electrosynthesis of 4-bromosubstituted pyrazole and its derivatives. Russ. J. Electrochem. 2010, 46, 123–129. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A.; Ugrak, B.I. Electrosynthesis of 4-iodopyrazole and its derivatives. Russ. Chem. Bull. 2010, 59, 1549–1555. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A. New approach to electrochemical iodination of arenes exemplified by the synthesis of 4-iodopyrazoles of different structures. Russ. Chem. Bull. 2014, 63, 360–367. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Petrosyan, V.A. Efficient iodination of structurally varying pyrazoles in heterophase medium. Russ. Chem. Bull. 2013, 62, 1044–1051. [Google Scholar] [CrossRef]
- Noszticzius, Z.; Noszticzius, E.; Schelly, Z.A. Use of ion-selective electrodes for monitoring oscillating reactions. 1. Potential response of the silver halide membrane electrodes to hypohalous acids. J. Am. Chem. Soc. 1982, 104, 6194–6199. [Google Scholar] [CrossRef]
- Kanishchev, M.I.; Korneeva, N.V.; Shevelev, S.A.; Fainzil’berg, A.A. Nitropyrazoles (review). Chem. Heterocycl. Compd. 1988, 24, 353–370. [Google Scholar] [CrossRef]
- Belen’kii, L.I.; Chuvylkin, N.D. Relationships and features of electrophilic substitution reactions in the azole series. Chem. Heterocycl. Compd. 1996, 32, 1319–1343. [Google Scholar] [CrossRef]
- Pevzner, M.S. Aromatic N-Haloazoles. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 75, pp. 1–77. [Google Scholar]
- Lyalin, B.V.; Petrosyan, V.A. Oxidation of organic compounds on NiOOH electrode. Russ. J. Electrochem. 2010, 46, 1199–1214. [Google Scholar] [CrossRef]
- Nikoofar, K. A Brief on Thiocyanation of N-Activated Arenes and N-Bearing Heteroaromatic Compounds. Chem. Sci. Trans. 2013, 3, 691–700. [Google Scholar] [CrossRef][Green Version]
- Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Recent advances in the chemistry of organic thiocyanates. Chem. Soc. Rev. 2016, 45, 494–505. [Google Scholar] [CrossRef]
- Majedi, S.; Sreerama, L.; Vessally, E.; Behmagham, F. Metal-Free Regioselective Thiocyanation of (Hetero) Aromatic C-H Bonds using Ammonium Thiocyanate: An Overview. J. Chem. Lett. 2020, 1, 25–31. [Google Scholar] [CrossRef]
- Rezayati, S.; Ramazani, A. A review on electrophilic thiocyanation of aromatic and heteroaromatic compounds. Tetrahedron 2020, 76, 131382. [Google Scholar] [CrossRef]
- Maurya, C.K.; Mazumder, A.; Gupta, P.K. Phosphorus pentasulfide mediated conversion of organic thiocyanates to thiols. Beilstein J. Org. Chem. 2017, 13, 1184–1188. [Google Scholar] [CrossRef]
- Noland, W.E.; Lanzatella, N.P.; Dickson, R.R.; Messner, M.E.; Nguyen, H.H. Access to Indoles via Diels–Alder Reactions of 5-Methylthio-2-vinylpyrroles with Maleimides. J. Heterocycl. Chem. 2013, 50, 795–808. [Google Scholar] [CrossRef]
- Maurya, C.K.; Mazumder, A.; Kumar, A.; Gupta, P.K. Synthesis of Disulfanes from Organic Thiocyanates Mediated by Sodium in Silica Gel. Synlett 2016, 27, 409–411. [Google Scholar] [CrossRef]
- Biswas, K.; Ghosh, S.; Ghosh, P.; Basu, B. Cyclic ammonium salts of dithiocarbamic acid: Stable alternative reagents for the synthesis of S-alkyl carbodithioates from organyl thiocyanates in water. J. Sulfur Chem. 2016, 37, 361–376. [Google Scholar] [CrossRef]
- Li, W.-T.; Wu, W.-H.; Tang, C.-H.; Tai, R.; Chen, S.-T. One-Pot Tandem Copper-Catalyzed Library Synthesis of 1-Thiazolyl-1,2,3-triazoles as Anticancer Agents. ACS Comb. Sci. 2011, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Baghershiroudi, M.; Safa, K.D.; Adibkia, K.; Lotfipour, F. Synthesis and antibacterial evaluation of new sulfanyltetrazole derivatives bearing piperidine dithiocarbamate moiety. Synth. Commun. 2018, 48, 323–328. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Terent’ev, A.O.; Ramenskaya, G.V.; Grammatikova, N.É.; Rodionova, G.M.; Ilovaiskii, A.I. Synthesis and Antifungal Activity of Arylthiocyanates. Pharm. Chem. J. 2013, 47, 422–425. [Google Scholar] [CrossRef]
- Fortes, M.P.; da Silva, P.B.N.; da Silva, T.G.; Kaufman, T.S.; Militão, G.C.G.; Silveira, C.C. Synthesis and preliminary evaluation of 3-thiocyanato-1H-indoles as potential anticancer agents. Eur. J. Med. Chem. 2016, 118, 21–26. [Google Scholar] [CrossRef]
- Chao, M.N.; Matiuzzi, C.E.; Storey, M.; Li, C.; Szajnman, S.H.; Docampo, R.; Moreno, S.N.J.; Rodriguez, J.B. Aryloxyethyl Thiocyanates Are Potent Growth Inhibitors of Trypanosoma cruzi and Toxoplasma gondii. ChemMedChem 2015, 10, 1094–1108. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Khodonov, V.M.; Neverov, S.V.; Grammatikova, N.É.; Petrosyan, V.A. “Metal-free” synthesis and antifungal activity of 3-thiocyanatopyrazolo[1,5-a]pyrimidines. Russ. Chem. Bull. 2021, 70, 600–604. [Google Scholar] [CrossRef]
- Yi, X.-J.; El-Idreesy, T.T.; Eldebss, T.M.A.; Farag, A.M.; Abdulla, M.M.; Hassan, S.A.; Mabkhot, Y.N. Synthesis, biological evaluation, and molecular docking studies of new pyrazol-3-one derivatives with aromatase inhibition activities. Chem. Biol. Drug Des. 2016, 88, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L. Substitution and Addition Reactions of Thiocyanogen. In Organic Reactions; John Wiley & Sons, Inc.: New York, NY, USA, 1946; Volume 3, pp. 240–266. [Google Scholar]
- Guy, R.G. Syntheses and preparative applications of thiocyanates. In Cyanates and Their Thio Derivatives; Patai, S., Ed.; John Wiley & Sons, Ltd.: New York, NY, USA, 1977; Volume 2, pp. 819–886. [Google Scholar]
- Jason, A.; Phadke, A.S.; Deshpande, M.; Agarwak, A.; Chen, D.; Gadhachanda, V.R.; Hashimoto, A.; Pais, G.; Wang, Q.; Wang, X. Aryl, Heteroaryl, and Heterocyclic Compounds for Treatment of Medical Disorders. WO2017035353A1, 2 March 2017. [Google Scholar]
- Rodríguez, R.; Camargo, P.; Sierra, C.A.; Soto, C.Y.; Cobo, J.; Nogueras, M. Iodine mediated an efficient and greener thiocyanation of aminopyrimidines by a modification of the Kaufmann’s reaction. Tetrahedron Lett. 2011, 52, 2652–2654. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, S.M.S.; Madan, C. NBS or DEAD as effective reagents in α-thiocyanation of enolizable ketones with ammonium thiocyanate. Tetr. Lett. 2011, 52, 1432–1435. [Google Scholar] [CrossRef]
- Mahajan, U.S.; Akamanchi, K.G. Facile Method for Thiocyanation of Activated Arenes Using Iodic Acid in Combination with Ammonium Thiocyanate. Synthetic Commun. 2009, 39, 2674–2682. [Google Scholar] [CrossRef]
- Khazaei, A.; Zolfigol, M.A.; Mokhlesi, M.; Panah, F.D.; Sajjadifar, S. Simple and Highly Efficient Catalytic Thiocyanation of Aromatic Compounds in Aqueous Media. Helv. Chim. Acta 2012, 95, 106–114. [Google Scholar] [CrossRef]
- Wu, J.; Wu, G.; Wu, L. Thiocyanation of Aromatic and Heteroaromatic Compounds using Ammonium Thiocyanate and I2O5. Synthetic Commun. 2008, 38, 2367–2373. [Google Scholar] [CrossRef]
- Ali, D.; Panday, A.K.; Choudhury, L.H. Hydrogen Peroxide-Mediated Rapid Room Temperature Metal-Free C(sp2)-H Thiocyanation of Amino Pyrazoles, Amino Uracils, and Enamines. J. Org. Chem. 2020, 85, 13610–13620. [Google Scholar] [CrossRef]
- Sajjadifar, S.; Louie, O. Regioselective Thiocyanation of Aromatic and Heteroaromatic Compounds by Using Boron Sulfonic Acid as a New, Efficient, and Cheap Catalyst in Water. J. Chem. 2013, 2013, 6. [Google Scholar] [CrossRef]
- Khalili, D. Highly efficient and regioselective thiocyanation of aromatic amines, anisols and activated phenols with H2O2/NH4SCN catalyzed by nanomagnetic Fe3O4. Chin. Chem. Lett. 2015, 26, 547–552. [Google Scholar] [CrossRef]
- Khazaei, A.; Zolfigol, M.A.; Mokhlesi, M.; Pirveysian, M. Citric acid as a trifunctional organocatalyst for thiocyanation of aromatic and heteroaromatic compounds in aqueous media. Can. J. Chem. 2012, 90, 427–432. [Google Scholar] [CrossRef]
- Songsichan, T.; Katrun, P.; Khaikate, O.; Soorukram, D.; Pohmakotr, M.; Reutrakul, V.; Kuhakarn, C. Thiocyanation of Pyrazoles Using KSCN/K2S2O8 Combination. SynOpen 2018, 2, 6–16. [Google Scholar] [CrossRef]
- Mete, T.B.; Khopade, T.M.; Bhat, R.G. Transition-metal-free regioselective thiocyanation of phenols, anilines and heterocycles. Tetrahedron Lett. 2017, 58, 415–418. [Google Scholar] [CrossRef]
- Karimi Zarchi, M.A.; Banihashemi, R. Thiocyanation of aromatic and heteroaromatic compounds using polymer-supported thiocyanate ion as the versatile reagent and ceric ammonium nitrate as the versatile single-electron oxidant. J. Sulfur Chem. 2016, 37, 282–295. [Google Scholar] [CrossRef]
- Pan, X.-Q.; Lei, M.-Y.; Zou, J.-P.; Zhang, W. Mn(OAc)3-promoted regioselective free radical thiocyanation of indoles and anilines. Tetrahedron Lett. 2009, 50, 347–349. [Google Scholar] [CrossRef]
- Das, B.; Kumar, A.S. Efficient Thiocyanation of Indoles Using Para-Toluene Sulfonic Acid. Synth. Commun. 2010, 40, 337–341. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, Q.; Wei, S.; Qu, J.; Wang, B. Highly Efficient and Practical Thiocyanation of Imidazopyridines Using an N-Chlorosuccinimide/NaSCN Combination. Eur. J. Org. Chem. 2016, 2016, 3373–3379. [Google Scholar] [CrossRef]
- Muniraj, N.; Dhineshkumar, J.; Prabhu, K.R. N-Iodosuccinimide Catalyzed Oxidative Selenocyanation and Thiocyanation of Electron Rich Arenes. ChemistrySelect 2016, 1, 1033–1038. [Google Scholar] [CrossRef]
- Mokhtari, B.; Azadi, R.; Rahmani-Nezhad, S. In situ-generated N-thiocyanatosuccinimide (NTS) as a highly efficient reagent for the one-pot thiocyanation or isothiocyanation of alcohols. Tetrahedron Lett. 2009, 50, 6588–6589. [Google Scholar] [CrossRef]
- Memarian, H.R.; Mohammadpoor-Baltork, I.; Nikoofar, K. DDQ-promoted thiocyanation of aromatic and heteroaromatic compounds. Can. J. Chem. 2007, 85, 930–937. [Google Scholar] [CrossRef]
- Memarian, H.R.; Mohammadpoor-Baltork, I.; Nikoofar, K. Ultrasound-assisted thiocyanation of aromatic and heteroaromatic compounds using ammonium thiocyanate and DDQ. Ultrason. Sonochem. 2008, 15, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Gitkis, A.; Becker, J.Y. Anodic thiocyanation of mono- and disubstituted aromatic compounds. Electrochim. Acta 2010, 55, 5854–5859. [Google Scholar] [CrossRef]
- Fotouhi, L.; Nikoofar, K. Electrochemical thiocyanation of nitrogen-containing aromatic and heteroaromatic compounds. Tetrahedron Lett. 2013, 54, 2903–2905. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Jiang, H.; Sun, L. A low-cost electrochemical thio- and selenocyanation strategy for electron-rich arenes under catalyst- and oxidant-free conditions. RSC Adv. 2018, 8, 22042–22045. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Yaubasarova, R.R.; Neverov, S.V.; Petrosyan, V.A. Reactivity of electrogenerated thiocyanogen in the thiocyanation of pyrazolo[1,5-a]pyrimidines. Mendeleev Commun. 2016, 26, 413–414. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Mel’nikova, E.I.; Yaubasarova, R.R.; Gorpinchenko, N.V.; Petrosyan, V.A. “Metal-free” electrooxidative C—H thiocyanation of arenes. Russ. Chem. Bull. 2019, 68, 2140–2141. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Yaubasarova, R.R.; Neverov, S.V.; Petrosyan, V.A. Electrooxidative C–H Functionalization of Heteroarenes. Thiocyanation of Pyrazolo[1,5-a]pyrimidines. Eur. J. Org. Chem. 2019, 2019, 4233–4238. [Google Scholar] [CrossRef]
- Yaubasarova, R.R.; Kokorekin, V.A.; Ramenskaya, G.V.; Petrosyan, V.A. Double electrooxidative C–H functionalization of (het)arenes with thiocyanate and 4-nitropyrazolate ions. Mendeleev Commun. 2019, 29, 334–336. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Melnikova, E.I.; Yaubasarova, R.R.; Petrosyan, V.A. Electrooxidative C–H thiocyanation of hetarenes: Voltammetric assessment of thiocyanogen reactivity. Mendeleev Commun. 2020, 30, 70–72. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Neverov, S.V.; Kuzina, V.N.; Petrosyan, V.A. A New Method for the Synthesis of 3-Thiocyanatopyrazolo[1,5-a]pyrimidines. Molecules 2020, 25, 4169. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, F. New Developments in the Study of the Structure of Parathiocyanogen: (SCN)x, An Inorganic Polymer. J. Inorg. Organomet. Polym. 1997, 7, 35–50. [Google Scholar] [CrossRef]
- Shaabani, A.; Nazeri, M.T.; Afshari, R. 5-Amino-pyrazoles: Potent reagents in organic and medicinal synthesis. Mol. Divers. 2019, 23, 751–807. [Google Scholar] [CrossRef] [PubMed]
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo[1,5-a]pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352. [Google Scholar] [CrossRef] [PubMed]
- Arias-Gómez, A.; Godoy, A.; Portilla, J. Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses as an Antitumor Scaffold. Molecules 2021, 26, 2708. [Google Scholar] [CrossRef] [PubMed]
- Sigacheva, V.L.; Kokorekin, V.A.; Strelenko, Y.A.; Neverov, S.V.; Petrosyan, V.A. Electrochemical Azolation of N-substituted Pyrroles: A New Case in SNH(An) Reactions. Mendeleev Commun. 2012, 22, 270–272. [Google Scholar] [CrossRef]
- Henton, D.R.; McCreery, R.A.; Swenton, J.S. Anodic oxidation of 1,4-dimethoxy aromatic compounds. A facile route to functionalized quinone bisketals. J. Org. Chem. 1980, 45, 369–378. [Google Scholar] [CrossRef]
- Adibi, H.; Rashidi, A.; Khodaei, M.M.; Alizadeh, A.; Majnooni, M.B.; Pakravan, N.; Abiri, R.; Nematollahi, D. Catecholthioether Derivatives: Preliminary Study of in-Vitro Antimicrobial and Antioxidant Activities. Chem. Pharm. Bull. 2011, 59, 1149–1152. [Google Scholar] [CrossRef]
- Kokorekin, V.A.; Solomatin, Y.A.; Gening, M.L.; Petrosyan, V.A. Acid-catalyzed SNH(An) thiolation of p-dihydroxybenzene. Mendeleev Commun. 2017, 27, 586–588. [Google Scholar] [CrossRef]
- Chen, C.; Niu, P.; Shen, Z.; Li, M. Electrochemical Sulfenylation of Indoles with Disulfides Mediated by Potassium Iodide. J. Electrochem. Soc. 2018, 165, G67–G74. [Google Scholar] [CrossRef]
- Petrosyan, V.A.; Burasov, A.V. Anodic azolation of 1,2- and 1,3-dimethoxybenzenes. Russ. Chem. Bull. 2010, 59, 522–527. [Google Scholar] [CrossRef]
- Feng, P.; Ma, G.; Chen, X.; Wu, X.; Lin, L.; Liu, P.; Chen, T. Electrooxidative and Regioselective C−H Azolation of Phenol and Aniline Derivatives. Angew. Chem. Int. Ed. 2019, 58, 8400–8404. [Google Scholar] [CrossRef]
- Wang, J.-H.; Lei, T.; Nan, X.-L.; Wu, H.-L.; Li, X.-B.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Regioselective Ortho Amination of an Aromatic C–H Bond by Trifluoroacetic Acid via Electrochemistry. Org. Lett. 2019, 21, 5581–5585. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P.; Eremin, D.B.; Yakukhnov, S.A.; Dilman, A.D.; Levin, V.V.; Egorov, M.P.; Karlov, S.S.; Kustov, L.M.; Tarasov, A.L.; Greish, A.A.; et al. Organic and hybrid systems: From science to practice. Mendeleev Commun. 2017, 27, 425–438. [Google Scholar] [CrossRef]
- Hunger, K. Industrial Dyes: Chemistry, Properties, Applications; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Sandborn, W.J. Rational selection of oral 5-aminosalicylate formulations and prodrugs for the treatment of ulcerative colitis. Am. J. Gastroenterol. 2002, 97, 2939–2941. [Google Scholar] [CrossRef]
- Ovchinnikov, I.V.; Kulikov, A.S.; Makhova, N.N.; Tosco, P.; Di Stilo, A.; Fruttero, R.; Gasco, A. Synthesis and vasodilating properties of N-alkylamide derivatives of 4-amino-3-furoxancarboxylic acid and related azo derivatives. Il Farmaco 2003, 58, 677–681. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Khalili, D.; Motevalli, S. Easily Prepared Azopyridines as Potent and Recyclable Reagents for Facile Esterification Reactions. An Efficient Modified Mitsunobu Reaction. J. Org. Chem. 2008, 73, 4882–4887. [Google Scholar] [CrossRef]
- Iranpoor, N.; Firouzabadi, H.; Shahin, R.; Khalili, D. 2,2′-Azobenzothiazole as a New Recyclable Oxidant for Heterogeneous Thiocyanation of Aromatic Compounds with Ammonium Thiocyanate. Synth. Commun. 2011, 42, 2040–2047. [Google Scholar] [CrossRef]
- Ovchinnikov, I.V.; Makhova, N.N.; Khmel’nitsky, L.I.; Kuz’min, V.S.; Akimova, L.N.; Pepekin, V.I. Dinitrodiazenofuroxan as a new energetic explosive. Dokl. Chem. 1998, 359, 67–70. [Google Scholar]
- Veauthier, J.M.; Chavez, D.E.; Tappan, B.C.; Parrish, D.A. Synthesis and Characterization of Furazan Energetics ADAAF and DOATF. J. Energ. Mater. 2010, 28, 229–249. [Google Scholar] [CrossRef]
- Firouzabadi, H.; Mostafavipoor, Z. Barium Manganate. A Versatile Oxidant in Organic Synthesis. Bull. Chem. Soc. Jpn. 1983, 56, 914–917. [Google Scholar] [CrossRef]
- Birchall, J.M.; Haszeldine, R.N.; Kemp, J.E.G. Polyfluoroarenes. Part X. Polyfluoroaromatic azo-compounds. J. Chem. Soc. C 1970, 449–455. [Google Scholar] [CrossRef]
- Farhadi, S.; Zaringhadama, P.; Sahamiehb, R.Z. Photo-assisted Oxidation of Anilines and Other Primary Aromatic Amines to Azo Compounds Using Mercury (II) Oxide as a Photo-Oxidant. Acta Chim. Slov. 2007, 54, 647–653. [Google Scholar]
- Huang, H.; Sommerfeld, D.; Dunn, B.C.; Lloyd, C.R.; Eyring, E.M. Ferrate(VI) oxidation of aniline. J. Chem. Soc. Dalton Trans. 2001, 1301–1305. [Google Scholar] [CrossRef]
- Chavez, D.E.; Parrish, D.A.; Leonard, P. The Synthesis and Characterization of a New Furazan Heterocyclic System. Synlett 2012, 23, 2126–2128. [Google Scholar] [CrossRef]
- Okumura, S.; Lin, C.-H.; Takeda, Y.; Minakata, S. Oxidative Dimerization of (Hetero)aromatic Amines Utilizing t-BuOI Leading to (Hetero)aromatic Azo Compounds: Scope and Mechanistic Studies. J. Org. Chem. 2013, 78, 12090–12105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jiao, N. Copper-Catalyzed Aerobic Oxidative Dehydrogenative Coupling of Anilines Leading to Aromatic Azo Compounds using Dioxygen as an Oxidant. Angew. Chem. 2010, 122, 6310–6313. [Google Scholar] [CrossRef]
- Jiang, B.; Ning, Y.; Fan, W.; Tu, S.-J.; Li, G. Oxidative Dehydrogenative Couplings of Pyrazol-5-amines Selectively Forming Azopyrroles. J. Org. Chem. 2014, 79, 4018–4024. [Google Scholar] [CrossRef]
- Takeda, Y.; Okumura, S.; Minakata, S. Oxidative Dimerization of Aromatic Amines using tBuOI: Entry to Unsymmetric Aromatic Azo Compounds. Angew. Chem. Int. Ed. 2012, 51, 7804–7808. [Google Scholar] [CrossRef]
- Pandit, R.P.; Lee, Y.R. Construction of Multifunctionalized Azopyrazoles by Silver- Catalyzed Cascade Reaction of Diazo Compounds. Adv. Synth. Catal. 2015, 357, 2657–2664. [Google Scholar] [CrossRef]
- Wawzonek, S.; McIntyre, T. Electrolytic oxidation of aromatic amines. J. Electrochem. Soc. 1967, 114, 1025. [Google Scholar] [CrossRef]
- Wawzonek, S.; McIntyre, T. Electrolytic preparation of azobenzenes. J. Electrochem. Soc. 1972, 119, 1350. [Google Scholar] [CrossRef]
- Schäfer, H.-J. Oxidation of organic compounds at the nickel hydroxide electrode. In Electrochemistry I. Top. Curr. Chem; Steckhan, E., Ed.; Springer: Berlin, Germany; New York, NY, USA, 1987; Volume 142, pp. 101–129. [Google Scholar]
- Lyalin, B.V.; Sigacheva, V.L.; Kokorekin, V.A.; Petrosyan, V.A. A new synthesis of azopyrazoles by oxidation of C-aminopyrazoles on a NiO(OH) electrode. Mendeleev Commun. 2015, 25, 479–481. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Sigacheva, V.L.; Kokorekin, V.A.; Petrosyan, V.A. Electrosynthesis of azopyrazoles via the oxidation of N-alkylaminopyrazoles on a NiO(OH) anode in aqueous alkali—A green method for N-N homocoupling. Tetrahedron Lett. 2018, 59, 2741–2744. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Sigacheva, V.L.; Petrosyan, V.A. Electrocatalytic N—N cross-coupling of N-alkyl-3-aminopyrazoles at a nickel anode. Russ. Chem. Bull. 2020, 69, 2020–2021. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Sigacheva, V.L.; Kokorekin, V.A.; Dutova, T.Y.; Rodionova, G.M.; Petrosyan, V.A. Oxidative transformation of N-substituted 3-aminopyrazoles to azopyrazoles using electrogenerated bromine as a mediator. Russ. Chem. Bull. 2018, 67, 510–516. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Sigacheva, V.L.; Kokorekin, V.A.; Petrosyan, V.A. Oxidative conversion of N-substituted 3-aminopyrazoles to azopyrazoles using electrogenerated NaOCl as the mediator. Arkivoc 2017, 2017, 55–62. [Google Scholar] [CrossRef]
- Lyalin, B.V.; Sigacheva, V.L.; Ugrak, B.I.; Petrosyan, V.A. Oxidative N—N coupling of N-alkyl-3-aminopyrazoles to azopyrazoles in aqueous solutions of NaOCl and NaOBr. Russ. Chem. Bull. 2021, 70, 164–170. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Az‒H, Az‒Cl and other Products (Yield, %) [70,71] | Entry | Az‒H, Az‒Cl (Yield, %) [68,70,71] | ||||
|---|---|---|---|---|---|---|---|
| 1 |  1a |  1b (46 2) |  1b-1b′ (8 2) | 8 |  8a |  8b (41 2) | |
| 2 |  2a |  2b (71 3) | 9 |  9a |  9b (64 3) | ||
| 3 |  3a |  3b (34 2) | 10 |  10a |  10b (92 3) | ||
| 4 |  4a |  4b (70 3) | 11 |  11a |  11b (69 3) | ||
| 5 |  5a |  5b (15 2) |  5b’ (35 2) | 12 |  12a |  12b (93 3) | |
| 6 |  6a |  6b (47 2) |  6b’ (13 2) |  6b’’ (4)2 | 13 |  13a |  13b (84 3) |
| 7 |  7a |  7b (8 2) | 14 |  14a |  14b (4 2) | ||
| Entry | Az‒H, Az‒Br and Other Products (Yield, %) [71,72] | Entry | Az‒H, Az‒Br (Yield, %) [71,72] | ||||
|---|---|---|---|---|---|---|---|
| 1 |  1a |  1c (70 2) | 8 |  8a |  8c (89 2) | ||
| 2 |  2a |  2c (76 3) |  2c’ (5 3) | 9 |  9a |  9c (15 3) | |
| 3 |  3a |  3c (66 3) | 10 |  10a |  10c (68 3) | ||
| 4 |  4a |  4c (94 2) | 11 |  11a |  11c (78 2) | ||
| 5 |  5a |  5c (55 3) |  5c’ (26 3) | 12 |  12a |  12c (84 2) | |
| 6 |  6a |  6c (88 2) | 13 |  13a |  13c (84 2) | ||
| 7 |  7a |  7c (0 5) | 14 |  14a |  14c (0 3) | ||
| Entry | Az‒H, Az‒I (Yield, %) [73,74] | Entry | Az‒H, Az‒I (Yield, %) [73,74] | ||
|---|---|---|---|---|---|
| 1 |  1a |  1d (57 1,3, 93 2,3) | 8 |  8a |  8d (2 1,4, 82 2,3) |
| 2 |  2a |  2d (5 1,4, 79 2,3) | 9 |  9a |  9d (0 1,4) |
| 3 |  3a |  3d (71 1,3) | 10 |  10a |  10d (30 1,4, 86 2,3) |
| 4 |  4a |  4d (86 1,3, 93 2,3) | 11 |  11a |  11d (0 1,4, 74 2,3) |
| 5 |  5a |  5d (35 1,4) | 12 |  12a |  12d (0 1,4, 78 2,3) |
| 6 |  6a |  6d (42 1,5) | 13 |  14a |  14d (0 1,5, 79 2,3) |
| 7 |  7a |  7d (0 1,5) | 14 |  15a |  15d (40 1,4) |
| Entry | Az–H, Az–SCN (Yield, %) [22,122,123,126] | Entry | Az–H, Az–SCN (Yield, %) [22,122,123,126] | ||
|---|---|---|---|---|---|
| 1 |  1e |  1f (83 2,5,6, 72 2,4,6, 74 2,5,7, 69 3,5,7) | 6 |  6e |  6f (86 2,4,6) |
| 2 |  2e |  2f (87 2,5,6, 78 2,5,7, 71 3,5,7) | 7 |  7e |  7f (83 2,4,6, 80 2,4,7, 75 2,5,7, 77 3,4,7, 71 3,5,7) |
| 3 |  3e |  3f (65 2,4,6, 57 3,4,6) | 8 |  8e |  8f (85 2,4,6, 82 2,5,6) |
| 4 |  4e |  4f (75 2,4,6, 68 3,4,6) | 9 |  9e |  9f (75 2,4,6, 66 2,5,6) |
| 5 |  5e |  5f (89 2,4,6, 64 2,5,6) | |||
| Entry | Az–H (Epox, V2), Az–SCN (Yield, %) [122,123] | Entry | Az–H (Epox, V2), Az–SCN (Yield, %) [122,123] | ||
|---|---|---|---|---|---|
| 1 |  10e (1.75) |  10f (81 3,5, 65 4,5, 60 4,6) | 4 |  13e (1.85) |  13f (80 3,5, 62 4,5, 55 4,6) |
| 2 |  11e (1.77) |  11f (793,5, 604,5) | 5 |  14e (1.85) |  14f (73 3,5, 60 4,5) |
| 3 |  12e (1.79) |  12f (77 3,5, 60 4,5, 52 4,6) | 6 |  15e (1.88) |  15f (69 3,5, 63 4,5, 47 4,6) |
| Entry | Az–NH2, Az–N=N–Az and Other Products (Yield, %) [161,163,165] | Entry | Az–NH2, Az–N=N–Az and Other Products (Yield, %) [161,163,165] | ||
|---|---|---|---|---|---|
| 1 |  1k |  1k-1k (82 2, 34 4, 72 5, 40 6, 75 7) | 7 |  5k |  5k-5k (86 3) |
 1k′ (593, 224, 15) |  1k′-1k′ (283, 48 4) | 8 |  6k |  6k-6k (792, 2 6) | |
 1k″ (76, 57) |  1k″-1k″ (40 6, 14 7) |  6k′ (3 3) |  6k′-6k′ (80 3) | ||
| 2 |  1k′ |  1k′-1k′ (77 2, 62 4) |  6k″ (6) |  6k″-6k″ (79 6) | |
| 3 |  1k″ |  1k″-1k″ (87 2) | 9 |  7k |  7k-7k (55 2) |
| 4 |  2k |  2k-2k (712, 735) | 10 |  8k |  8k-8k (52 2) |
 2k′ (25) |  2k′-2k′ (6 5) | 11 |  9k |  9k-9k8 (67 2) | |
| 5 |  3k |  3k-3k (86 2) | 12 |  10k |  10k-10k (93 4, 86 6) |
| 6 |  4k |  4k-4k (88 2, 62 3, 70 4) | |||
| Entry | Az1–NH2 | H2N–Az2 | Az1–N=N–Az2 | Yield, % |
|---|---|---|---|---|
| 1 |  1k 1k |  2k 2k |  1k-2k 1k-2k | 50 (1k-2k) 36 (1k-1k) 37(2k-2k) |
| 2 |  4k 4k |  5k 5k |  4k-5k 4k-5k | 48 (4k-5k) 29 (1k-1k) 23 (2k-2k) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyalin, B.V.; Sigacheva, V.L.; Kudinova, A.S.; Neverov, S.V.; Kokorekin, V.A.; Petrosyan, V.A. Electrooxidation Is a Promising Approach to Functionalization of Pyrazole-Type Compounds. Molecules 2021, 26, 4749. https://doi.org/10.3390/molecules26164749
Lyalin BV, Sigacheva VL, Kudinova AS, Neverov SV, Kokorekin VA, Petrosyan VA. Electrooxidation Is a Promising Approach to Functionalization of Pyrazole-Type Compounds. Molecules. 2021; 26(16):4749. https://doi.org/10.3390/molecules26164749
Chicago/Turabian StyleLyalin, Boris V., Vera L. Sigacheva, Anastasia S. Kudinova, Sergey V. Neverov, Vladimir A. Kokorekin, and Vladimir A. Petrosyan. 2021. "Electrooxidation Is a Promising Approach to Functionalization of Pyrazole-Type Compounds" Molecules 26, no. 16: 4749. https://doi.org/10.3390/molecules26164749
APA StyleLyalin, B. V., Sigacheva, V. L., Kudinova, A. S., Neverov, S. V., Kokorekin, V. A., & Petrosyan, V. A. (2021). Electrooxidation Is a Promising Approach to Functionalization of Pyrazole-Type Compounds. Molecules, 26(16), 4749. https://doi.org/10.3390/molecules26164749