
The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches



Abstract
:1. Introduction
2. Literature Search Methods
2.1. Peer-Reviewed Publications
2.2. International Patents
3. The Therapeutic Potential of Celastrol in Neurodegenerative Disorders
4. The Therapeutic Potential of Celastrol in Neurodegenerative Disorders Induced by Cadmium
5. The Therapeutic Potential of Celastrol in Neuropsychiatric Disorders
6. The Therapeutic Potential of Celastrol in Cerebral Ischemia, Ischemic Stroke, and Traumatic Brain Injury
7. The Therapeutic Potential of Celastrol in Epilepsy
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gunatilaka, A.A.L. Triterpenoid Quinonemethides and Related Compounds (Celastroloids). Prog. Chem. Org. Nat. Prod. 1996, 67, 1–123. [Google Scholar]
- Lange, B.M.; Fischedick, J.T.; Lange, M.F.; Srividya, N.; Samec, D.; Poirier, B.C. Integrative Approaches for the Identification and Localization of Specialized Metabolites in Tripterygium Roots. Plant Physiol. 2017, 173, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, N.L.; Miettinen, K.; Zhao, Y.; Ignea, C.; Andreadelli, A.; Raadam, M.H.; Makris, A.M.; Moller, B.L.; Staerk, D.; Bak, S.; et al. Integrating pathway elucidation with yeast engineering to produce polpunonic acid the precursor of the anti-obesity agent celastrol. Microb. Cell Factories 2020, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Abu Bakar, M.H.; Sarmidi, M.R.; Tan, J.S.; Mohamad Rosdi, M.N. Celastrol attenuates mitochondrial dysfunction and inflammation in palmitate-mediated insulin resistance in C3A hepatocytes. Eur. J. Pharmacol. 2017, 799, 73–83. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, Q.; Zhang, X.; Zhang, H.; Liu, Y.; Wu, X.; Li, M.; Li, X.; Zhang, J.; Ruan, X.; et al. Celastrol ameliorates inflammation through inhibition of NLRP3 inflammasome activation. Oncotarget 2017, 8, 67300–67314. [Google Scholar] [CrossRef] [Green Version]
- Klaic, L.; Morimoto, R.I.; Silverman, R.B. Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascao, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A Spectrum of Treatment Opportunities in Chronic Diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.L.; Sun, Y.; Dong, Y.; Xiao, Y.L.; Hu, D.W.; Shi, Y.P.; Zhu, Q.L.; Dai, H.; Zhang, N.Z. A prospective, controlled, double-blind, cross-over study of tripterygium wilfodii hook F in treatment of rheumatoid arthritis. Chin. Med. J. 1989, 102, 327–332. [Google Scholar]
- Ji, S.M.; Wang, Q.W.; Chen, J.S.; Sha, G.Z.; Liu, Z.H.; Li, L.S. Clinical trial of Tripterygium Wilfordii Hook F. in human kidney transplantation in China. Transplant. Proc. 2006, 38, 1274–1279. [Google Scholar] [CrossRef]
- Brinker, A.M.; Ma, J.; Lipsky, P.E.; Raskin, I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae). Phytochemistry 2007, 68, 732–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, A.; West, J.D.; Klaic, L.; Westerheide, S.D.; Silverman, R.B.; Morimoto, R.I.; Morano, K.A. Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell 2008, 19, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Klaic, L.; Trippier, P.C.; Mishra, R.K.; Morimoto, R.I.; Silverman, R.B. Remarkable stereospecific conjugate additions to the Hsp90 inhibitor celastrol. J. Am. Chem. Soc. 2011, 133, 19634–19637. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine”, is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreeramulu, S.; Gande, S.L.; Gobel, M.; Schwalbe, H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. 2009, 48, 5853–5855. [Google Scholar] [CrossRef]
- Wu, C. Heat shock transcription factors: Structure and regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar] [CrossRef]
- Lee, J.H.; Koo, T.H.; Yoon, H.; Jung, H.S.; Jin, H.Z.; Lee, K.; Hong, Y.S.; Lee, J.J. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem. Pharmacol. 2006, 72, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Jaquet, V.; Marcoux, J.; Forest, E.; Leidal, K.G.; McCormick, S.; Westermaier, Y.; Perozzo, R.; Plastre, O.; Fioraso-Cartier, L.; Diebold, B.; et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br. J. Pharmacol. 2011, 164, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat. Cell Biol. 2004, 6, 1054–1061. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.M.; Brown, I.R. Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones 2007, 12, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Cleren, C.; Calingasan, N.Y.; Chen, J.; Beal, M.F. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 2005, 94, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gines, S.; MacDonald, M.E.; Gusella, J.F. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci. 2005, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Kiaei, M.; Kipiani, K.; Petri, S.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Celastrol blocks neuronal cell death and extends life in transgenic mouse model of amyotrophic lateral sclerosis. Neuro Degener. Dis. 2005, 2, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Lu, A.; Zhang, L.; Tang, Y.; Zhu, H.; Xu, H.; Feng, Y.; Han, C.; Zhou, G.; Rigby, A.C.; et al. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappa B survival signaling. Genes Dev. 2004, 18, 1466–1481. [Google Scholar] [CrossRef] [Green Version]
- Weiss, Y.G.; Bromberg, Z.; Raj, N.; Raphael, J.; Goloubinoff, P.; Ben-Neriah, Y.; Deutschman, C.S. Enhanced heat shock protein 70 expression alters proteasomal degradation of IkappaB kinase in experimental acute respiratory distress syndrome. Crit. Care Med. 2007, 35, 2128–2138. [Google Scholar] [CrossRef]
- Chen, H.; Wu, Y.; Zhang, Y.; Jin, L.; Luo, L.; Xue, B.; Lu, C.; Zhang, X.; Yin, Z. Hsp70 inhibits lipopolysaccharide-induced NF-kappaB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006, 580, 3145–3152. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.; Alvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Paris, D.; Ganey, N.J.; Laporte, V.; Patel, N.S.; Beaulieu-Abdelahad, D.; Bachmeier, C.; March, A.; Ait-Ghezala, G.; Mullan, M.J. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J. Neuroinflamm. 2010, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, H.; Lobo, N.; Guo, X.; Gentleman, S.M.; Ma, D. Celastrol enhances cell viability and inhibits amyloid-beta production induced by lipopolysaccharide in vitro. J. Alzheimer Dis. JAD 2014, 41, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Wang, Y.; Peng, B.; Zhang, X.; Zhang, D.; Xu, L. Effects of celastrol on Tau hyperphosphorylation and expression of HSF-1 and HSP70 in SH-SY5Y neuroblastoma cells induced by amyloid-beta peptides. Biotechnol. Appl. Biochem. 2018, 65, 390–396. [Google Scholar] [CrossRef]
- Konieczny, J.; Jantas, D.; Lenda, T.; Domin, H.; Czarnecka, A.; Kuter, K.; Smialowska, M.; Lason, W.; Lorenc-Koci, E. Lack of neuroprotective effect of celastrol under conditions of proteasome inhibition by lactacystin in in vitro and in vivo studies: Implications for Parkinson’s disease. Neurotox. Res. 2014, 26, 255–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.S.; Kim, H.; Lee, H.J.; Sapkota, K.; Park, S.E.; Kim, S.; Kim, S.J. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem. Res. 2014, 39, 84–96. [Google Scholar] [CrossRef]
- Faust, K.; Gehrke, S.; Yang, Y.; Yang, L.; Beal, M.F.; Lu, B. Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2009, 10, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.W.; Lin, C.C.; Chen, Y.H.; Yang, H.B.; Hung, S.Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy. Antioxidants 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.H.; Liu, S.B.; Zhang, H.Y.; Zhou, F.H.; Liu, Y.X.; Lu, Q.; Yang, L. Antioxidant effects of celastrol against hydrogen peroxide-induced oxidative stress in the cell model of amyotrophic lateral sclerosis. Sheng Li Xue Bao Acta Physiol. Sin. 2017, 69, 751–758. [Google Scholar]
- Kalmar, B.; Greensmith, L. Activation of the heat shock response in a primary cellular model of motoneuron neurodegeneration-evidence for neuroprotective and neurotoxic effects. Cell. Mol. Biol. Lett. 2009, 14, 319–335. [Google Scholar] [CrossRef]
- Brown, I.R. Heat shock proteins and protection of the nervous system. Ann. N. Y. Acad. Sci. 2007, 1113, 147–158. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Sarge, K.D. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J. Mol. Med. 2007, 85, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Abdin, A.A.; Hasby, E.A. Modulatory effect of celastrol on Th1/Th2 cytokines profile, TLR2 and CD3+ T-lymphocyte expression in a relapsing-remitting model of multiple sclerosis in rats. Eur. J. Pharmacol. 2014, 742, 102–112. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, L.; Xu, L.M.; Cao, F.F.; Peng, B.; Zhang, X.; Shen, Y.F.; Uzan, G.; Zhang, D.H. Celastrol Ameliorates EAE Induction by Suppressing Pathogenic T Cell Responses in the Peripheral and Central Nervous Systems. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2015, 10, 506–516. [Google Scholar] [CrossRef]
- Yang, H.; Liu, C.; Jiang, J.; Wang, Y.; Zhang, X. Celastrol Attenuates Multiple Sclerosis and Optic Neuritis in an Experimental Autoimmune Encephalomyelitis Model. Front. Pharmacol. 2017, 8, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesha, S.H.; Moudgil, K.D. Celastrol suppresses experimental autoimmune encephalomyelitis via MAPK/SGK1-regulated mediators of autoimmune pathology. Inflamm. Res. Off. J. Eur. Histamine Res. 2019, 68, 285–296. [Google Scholar] [CrossRef]
- Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.M.; Aschner, M.; Marreilha Dos Santos, A.P. Neurotoxicity of Metal Mixtures. Adv. Neurobiol. 2017, 18, 227–265. [Google Scholar]
- Chen, S.; Gu, C.; Xu, C.; Zhang, J.; Xu, Y.; Ren, Q.; Guo, M.; Huang, S.; Chen, L. Celastrol prevents cadmium-induced neuronal cell death via targeting JNK and PTEN-Akt/mTOR network. J. Neurochem. 2014, 128, 256–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, X.; Gu, C.; Zhang, H.; Zhang, R.; Dong, X.; Liu, C.; Hu, X.; Ji, X.; Huang, S.; et al. Celastrol ameliorates Cd-induced neuronal apoptosis by targeting NOX2-derived ROS-dependent PP5-JNK signaling pathway. J. Neurochem. 2017, 141, 48–62. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, Y.; Dong, X.; Liu, B.; Zhang, N.; Wang, X.; Liu, L.; Xu, C.; Huang, S.; Chen, L. Celastrol Attenuates Cadmium-Induced Neuronal Apoptosis via Inhibiting Ca(2+) -CaMKII-Dependent Akt/mTOR Pathway. J. Cell. Physiol. 2017, 232, 2145–2157. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, N.; Zhang, H.; Liu, C.; Dong, X.; Wang, X.; Zhu, Y.; Xu, C.; Liu, L.; Yang, S.; et al. Celastrol prevents cadmium-induced neuronal cell death by blocking reactive oxygen species-mediated mammalian target of rapamycin pathway. Br. J. Pharmacol. 2017, 174, 82–100. [Google Scholar] [CrossRef]
- Colaianna, M.; Schiavone, S.; Zotti, M.; Tucci, P.; Morgese, M.G.; Backdahl, L.; Holmdahl, R.; Krause, K.H.; Cuomo, V.; Trabace, L. Neuroendocrine profile in a rat model of psychosocial stress: Relation to oxidative stress. Antioxid. Redox Signal. 2013, 18, 1385–1399. [Google Scholar] [CrossRef] [Green Version]
- Schiavone, S.; Trabace, L. Inflammation, Stress Response, and Redox Dysregulation Biomarkers: Clinical Outcomes and Pharmacological Implications for Psychosis. Front. Psychiatry 2017, 8, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, S.; Tucci, P.; Trabace, L.; Morgese, M.G. Early Celastrol Administration Prevents Ketamine-Induced Psychotic-Like Behavioral Dysfunctions, Oxidative Stress and IL-10 Reduction in The Cerebellum of Adult Mice. Molecules 2019, 24, 3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bove, M.; Tucci, P.; Dimonte, S.; Trabace, L.; Schiavone, S.; Morgese, M.G. Postnatal Antioxidant and Anti-inflammatory Treatments Prevent Early Ketamine-Induced Cortical Dysfunctions in Adult Mice. Front. Neurosci. 2020, 14, 590088. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, J.; Zhu, Y.; Li, J.; Chi, H.; Tian, C.; Meng, Y.; Liu, Y.; Wang, J.; Lin, N. Celastrol alleviates comorbid obesity and depression by directly binding amygdala HnRNPA1 in a mouse model. Clin. Transl. Med. 2021, 11, e394. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Okimura, K.; Shen, J.; Guh, Y.-J.; Tamai, T.K.; Shimada, A.; Minou, S.; Okushi, Y.; Shimmura, T.; Furukawa, Y.; et al. Seasonal changes in NRF2 antioxidant pathway regulates winter depression-like behavior. Proc. Natl. Acad. Sci. USA 2020, 117, 9594–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-de-Saavedra, M.D.; Budni, J.; Cunha, M.P.; Gómez-Rangel, V.; Lorrio, S.; del Barrio, L.; Lastres-Becker, I.; Parada, E.; Tordera, R.M.; Rodrigues, A.L.S.; et al. Nrf2 participates in depressive disorders through an anti-inflammatory mechanism. Psychoneuroendocrinology 2013, 38, 2010–2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaianna, M.; Tucci, P.; Zotti, M.; Morgese, M.G.; Schiavone, S.; Govoni, S.; Cuomo, V.; Trabace, L. Soluble beta amyloid(1-42): A critical player in producing behavioural and biochemical changes evoking depressive-related state? Br. J. Pharmacol. 2010, 159, 1704–1715. [Google Scholar] [CrossRef] [Green Version]
- Morgese, M.G.; Schiavone, S.; Mhillaj, E.; Bove, M.; Tucci, P.; Trabace, L. N-3 PUFA diet enrichment prevents amyloid beta-induced depressive-like phenotype. Pharmacol. Res. 2018, 129, 526–534. [Google Scholar] [CrossRef]
- Morgese, M.G.; Schiavone, S.; Bove, M.; Colia, A.L.; Dimonte, S.; Tucci, P.; Trabace, L. N-3 PUFA Prevent Oxidative Stress in a Rat Model of Beta-Amyloid-Induced Toxicity. Pharmaceuticals 2021, 14, 339. [Google Scholar] [CrossRef]
- Morgese, M.G.; Bove, M.; Francavilla, M.; Schiavone, S.; Dimonte, S.; Colia, A.L.; Bevilacqua, M.; Trabace, L.; Tucci, P. Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model. Biomolecules 2021, 11, 686. [Google Scholar] [CrossRef]
- Jiang, M.; Liu, X.; Zhang, D.; Wang, Y.; Hu, X.; Xu, F.; Jin, M.; Cao, F.; Xu, L. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an IL-33/ST2 axis-mediated microglia/macrophage M2 polarization. J. Neuroinflamm. 2018, 15, 78. [Google Scholar] [CrossRef]
- Li, Y.; He, D.; Zhang, X.; Liu, Z.; Zhang, X.; Dong, L.; Xing, Y.; Wang, C.; Qiao, H.; Zhu, C.; et al. Protective effect of celastrol in rat cerebral ischemia model: Down-regulating p-JNK, p-c-Jun and NF-kappaB. Brain Res. 2012, 1464, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, C.; Jin, X.P.; Weng, S.X.; Fan, L.L.; Zheng, Z.; Li, W.L.; Wang, F.; Wang, W.F.; Hu, X.F.; et al. Celastrol may have an anti-atherosclerosis effect in a rabbit experimental carotid atherosclerosis model. Int. J. Clin. Exp. Med. 2014, 7, 1684–1691. [Google Scholar] [PubMed]
- Kim, J.Y.; Kim, N.; Zheng, Z.; Lee, J.E.; Yenari, M.A. The 70 kDa heat shock protein protects against experimental traumatic brain injury. Neurobiol. Dis. 2013, 58, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Eroglu, B.; Kimbler, D.E.; Pang, J.; Choi, J.; Moskophidis, D.; Yanasak, N.; Dhandapani, K.M.; Mivechi, N.F. Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J. Neurochem. 2014, 130, 626–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef]
- Malkov, A.; Ivanov, A.I.; Latyshkova, A.; Bregestovski, P.; Zilberter, M.; Zilberter, Y. Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models. Ann. Neurol. 2019, 85, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and Inflammation in Epilepsy. Cold Spring Harb. Perspect. Med. 2015, 6, a022699. [Google Scholar] [CrossRef] [Green Version]
- Terrone, G.; Salamone, A.; Vezzani, A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr. Pharm. Des. 2017, 23, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Shandra, O.; Wang, Y.; Mowrey, W. Effects of Celastrol and Edaravone in the Multiple-Hit Rat Model of Infantile Spasms; American Epilepsy Society: Philadelphia, PA, USA, 2015. [Google Scholar]
- Connolly, M.B. Dravet Syndrome: Diagnosis and Long-Term Course. Can. J. Neurol. Sci. J. Can. Des Sci. Neurol. 2016, 43 (Suppl. 3), S3–S8. [Google Scholar] [CrossRef] [Green Version]
- von Ruden, E.L.; Wolf, F.; Gualtieri, F.; Keck, M.; Hunt, C.R.; Pandita, T.K.; Potschka, H. Genetic and Pharmacological Targeting of Heat Shock Protein 70 in the Mouse Amygdala-Kindling Model. ACS Chem. Neurosci. 2019, 10, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Traynor, B.J.; Bruijn, L.; Conwit, R.; Beal, F.; O’Neill, G.; Fagan, S.C.; Cudkowicz, M.E. Neuroprotective agents for clinical trials in ALS: A systematic assessment. Neurology 2006, 67, 20–27. [Google Scholar] [CrossRef]
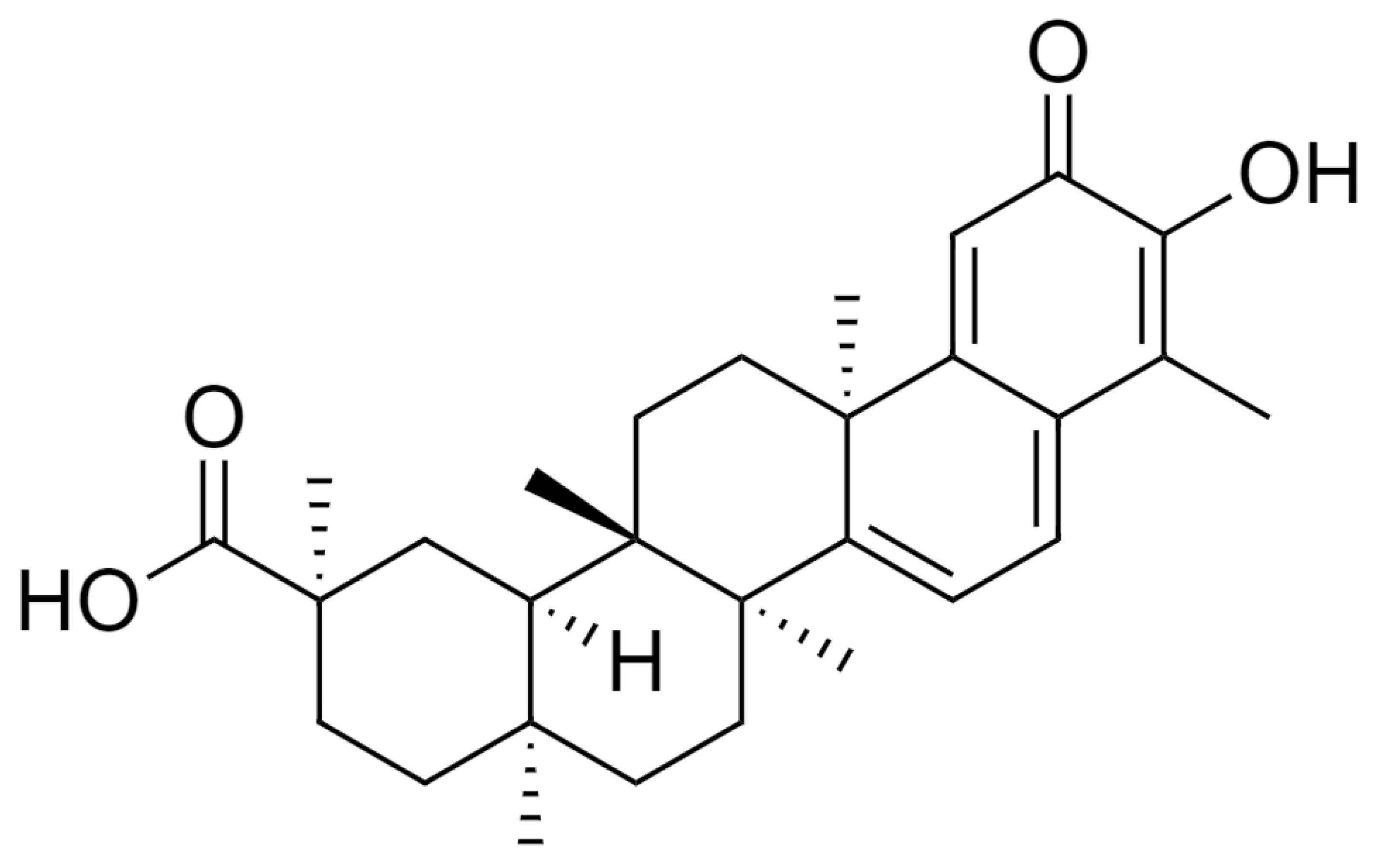
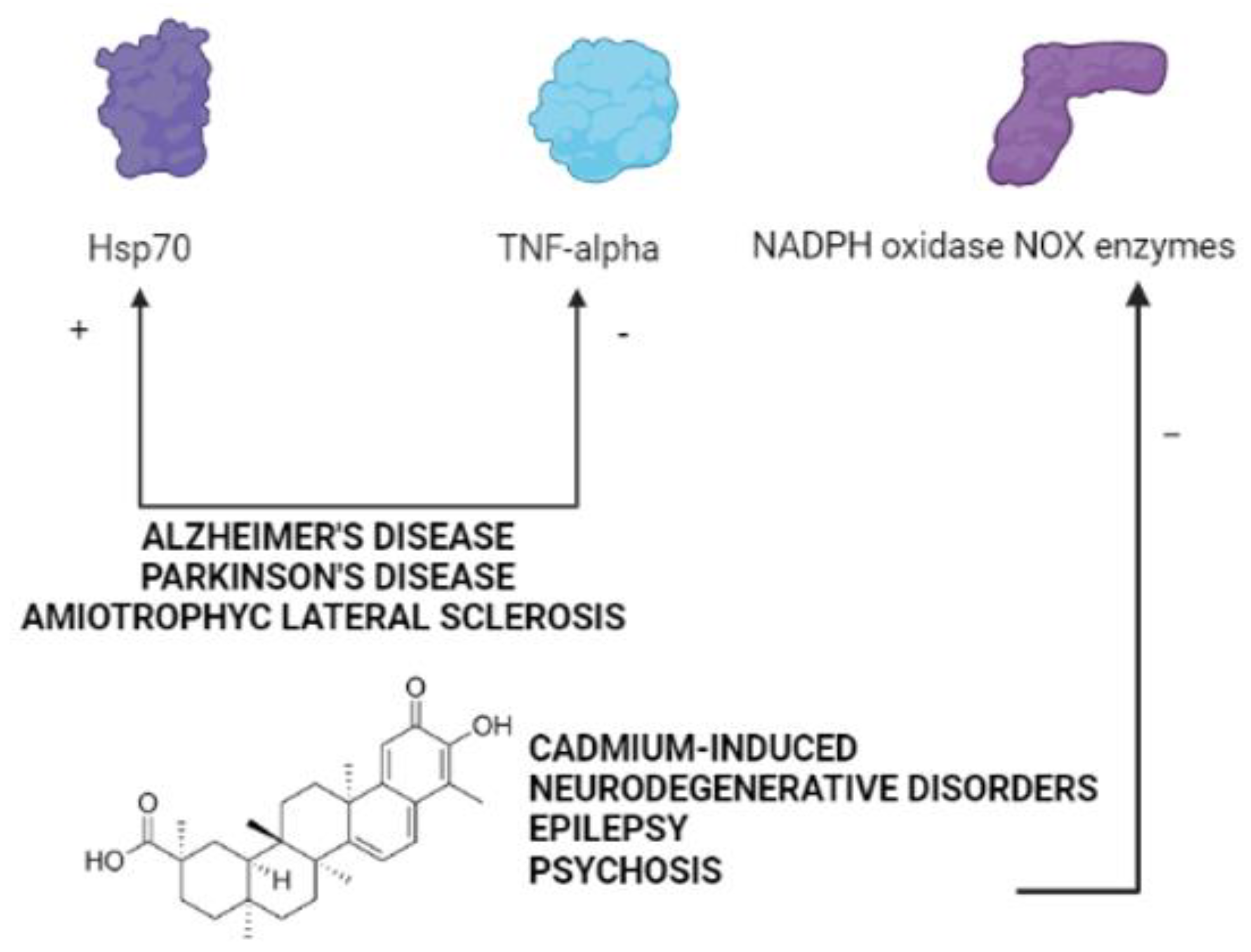

{kind=link}
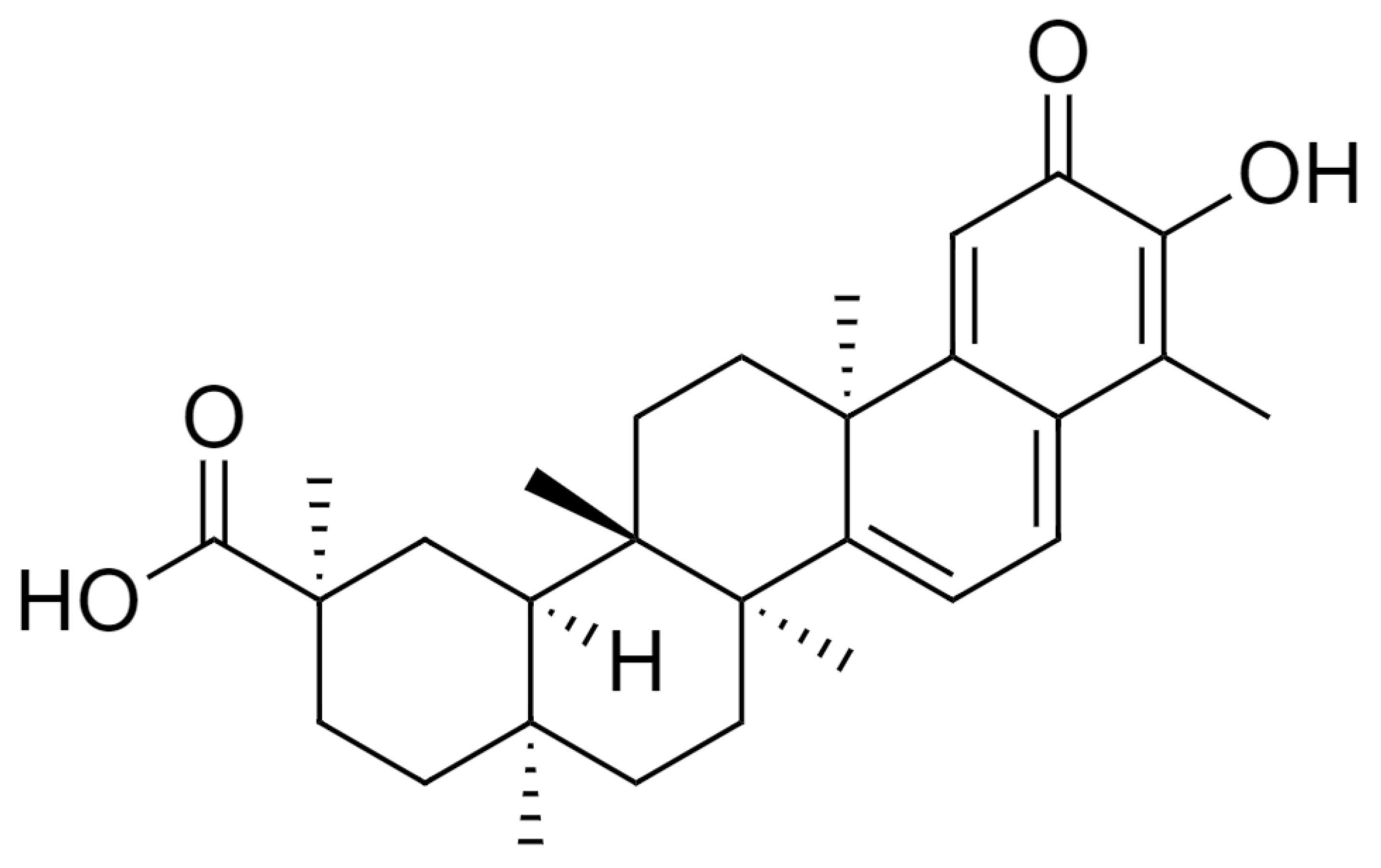
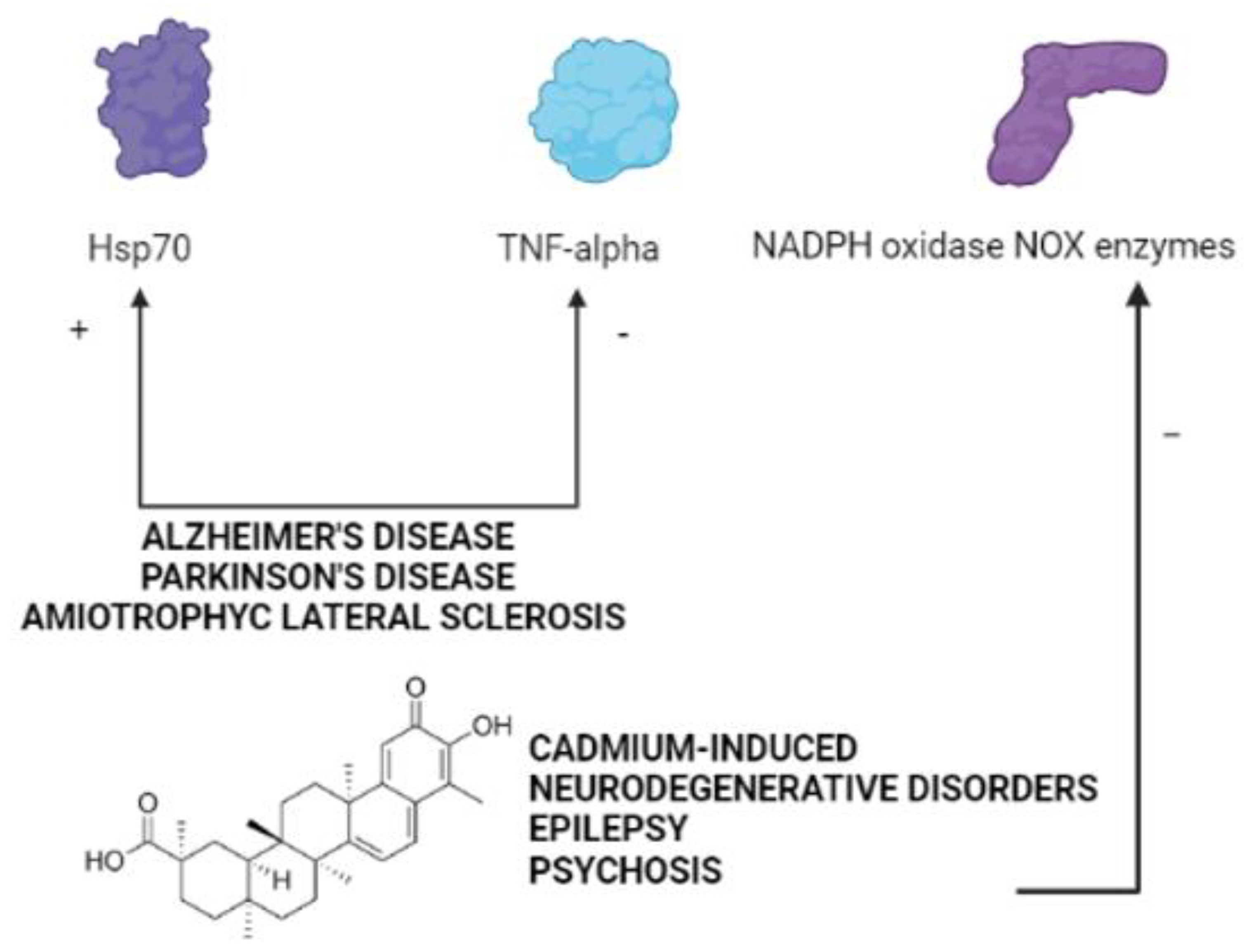{kind=link}
| Neurodegenerative Disease | In Vitro/In Vivo | Celastrol Effects | Doses/IC50 | References |
|---|---|---|---|---|
| Alzheimer’s disease | In vitro - Human monocytes and macrophages - endothelial cells | - Suppression of IL-1βproduction | 30 nM | [31] |
| - Suppression of TNF-α production | 70 nM | |||
| - Decrease of the production of induced but not constitutive nitric oxide | 50 nM IC50 = 100 nM | |||
| In vivo - LPS rat model | - Improvement of memory, learning and performance in psychomotor activity tests | 7 µg/kg i.p. | ||
| In vitro - Stable NF-κB luciferase reporter cell line of HEK293 cells - 7 W CHO cells overexpressing wild-type human APP | - Prevention of NF-κB activation | IC50 < 1 μM | [32] | |
| - Inhibition of BACE-1 expression | 5 μM | |||
| In vivo - Transgenic mice overexpressing the human APP695sw mutation and the presenilin-1 mutation M146L (Tg PS1/APPsw) | - Reduction of APP beta-cleavage with consequently inhibition of Aß1–40 and Aß1–42 production- Decrease of both soluble and insoluble Aß1–38, Aß1–40 and Aß1–42 levels - Reduction of Aß plaque burden -Microglia activation | 2.5 mg/kg/day s.c. long-lasting | ||
| In vitro - H4 human neuroglioma cells transfected to overexpress human full length APP | - Reduction of Aß production induced by LPS - Increase of Hsp70 and Bcl-2 expression - Decrease of NF-κB activity - Induction of GSK-3β posphorylation at tyrosine 216 - Reduction of COX2 expression - Decrease of Aß accumulation | 1, 10, and 100 nM (dose-dependently) | [33] | |
| In vitro - SH-SY5Y cells treated with Aβ1-42 | - Inhibition of Tau hyperphosphorylation and Hsp90 expression, induced by Aβ1–42 - No effects on the decreased HSP70 and HSF-1 expression, Tau ubiquitination, and HSP70/Tau- HSP70/CHIP interaction induced by Aβ1–42 | 600 nmol/L | [34] | |
| Parkinson’s disease | In vitro - Mouse primary cortical neurons and neuroblastoma SH-SY5Y cells incubated with lactacystin | - Absence of neuroprotective effects under conditions of the ubiquitin-proteasome system inhibition | 1 µM (co-treatment) 0.01 an 0.1 µM (pre-treatment) | [35] |
| - Reduction of cell viability and enhancement of cell death at high concentrations | 1 and 2.5 µM | |||
| In vivo - Lactacystin rat model | - No effects on the decrease of levels of dopamine and its metabolites - Absence of neuroprotective effects on dopaminergic neurons of the substantia nigra | 0.3, 1 or 3 mg/kg/1 mL i.p. | ||
| - Potentiation of the decrease in the levels of dopamine and its metabolites in the lesioned striatum - Acceleration of the total dopamine metabolism - Enhanced oxidative stress - Decrease in the number and/or density of dopaminergic neurons in the substantia nigra | 3 mg/kg/1 mL i.p. | |||
| In vitro - Human dopaminergic neuronal cell line (SH-SY5Y) treated with rotenone | - Protection from cell-injury induced and death induced by rotenone - Prevention of free radical production - Prevention of mitochondria membrane potential - Inhibition of cytochrome c release - Inhibition of Bax/Bcl-2 changes - Inhibition of caspase-9/3 activation - Inhibition of the activation of the p38 mitogen-activated protein kinase | 2.5 nM | [36] | |
| In vivo - MPTP-treated mice | - Attenuation (48%) of the loss of dopaminergic neurons of the substantia nigra - Reduction of dopamine concentration depletion - Induction of Hsp70 expression in dopaminergic neurons - Decrease of TNF-α and NF-κB immunostaining - Reduction of astrogliosis | 3 mg/kg i.p. | [25] | |
| In vivo - Drosophila DJ-1A model | - Neuroprotective effects on dopaminergic neurons | 5 and 20 µg | [37] | |
| In vitro - Dopaminergic neuronal cell line (SH-SY5Y) treated with treated with MPP+ | - Reduction of the MPP+-induced dopaminergic neuronal death, mitochondrial membrane depolarization, and ATP reduction | 0.1–3 μM celastrol (dose dependently) | [38] | |
| In vivo - MPTP-treated mice | - Suppression of motor symptoms and neurodegeneration in the substantia nigra and striatum - Enhancement of mitophagy in the striatum | 3 mg/kg/day i.p. for 3 days | ||
| Amyotrophic lateral sclerosis | In vitro -SOD1G93A transfected NSC34 cells | - Attenuation of H2O2-induced cell death - Decrease of MDA levels -Enhanced GCLC and GST mRNA expressions -Induction of ERK1/2 and Akt | 50 nmol/L | [39] |
| In vitro - Primary motoneuron cultures treated with staurosporin or H2O2 | - Induction of Hsp70 - Absence of neuroprotective effects - Neurotoxic effects and induction of cell death - Induction of the apoptotic cell death cascade | 0.3 and 3 μM | [40] | |
| In vitro - Differentiated neurons | - Neuroprotective effects via induced Hsp70 expression | 0.75 μM | [24,41] | |
| In vivo - G93A SOD1 transgenic mouse model | 2 mg/kg and 8 mg/kg p.o. | |||
| In vivo - G93A SOD1 transgenic mouse model | - Improvement of weight loss and motor performance - Delay of the onset of the disease - Increase (30%) in the neuronal number in the lumbar spinal cord - Decrease of TNF-α, iNOS, CD40, and GFAP immunoreactivity in the lumbar spinal cord - Increase of Hsp70 immunoreactivity in lumbar spinal cord neurons | 2 mg/kg and 8 mg/kg p.o. | [27] | |
| Huntington’s disease | In vivo 3-nitropropionic acid rat model | - Decrease of the lesion volume in the striatum | 3 mg/kg i.p. | [25] |
| In vitro - Cell lines expressing mutant polyglutamine protein | - Reduction of the cell killing | 0.4, 0.8 and 1.6 μM | [42] | |
| In vitro - Striatal cell line from the HdhQ111/Q111 knock-in mouse | - Inhibition of mutant huntingtin aggregation - Reverse of the abnormal cellular localization of full-length mutant huntingtin in mutant HdhQ111/Q111 striatal cells | 0.25 μM | [26] |
| Pathological Condition | In Vitro/In Vivo | Celastrol Effects | Doses or IC50 | References |
|---|---|---|---|---|
| Cerebral Ischemia and Ischemic Stroke | In vitro - Co-cultures of neurons and microglial cells, as well as neuron cultures, with or without 3 h-lasting oxygen glucose deprivation | - Protection against neuronal cell death caused by oxygen glucose deprivation in neuron-microglia co-cultures but not in neuronal cultures - Induction of M2 microglia phenotype development | 0.5 and 1 μM | [65] |
| In vivo - Murine model of middle cerebral artery occlusion | - Reduction of the infarct volume - Prevention of neuronal death - Protection against cerebral ischemia-induced neurological dysfunction - Improvement of sensorimotor functions - Promotion of M2 microglia polarization | 1 mg/kg i.p. | ||
| In vivo - Murine model of middle cerebral artery occlusion | - Reduction of brain water content - Reduction of p-JNK, p-c-Jun and NF-κB expression | 2–3 mg/kg i.p. | [66] | |
| In vivo - Rabbit model of carotid atherosclerosis | - Decrease of both plaque and arterial wall cross-section areas - Decrease of VEGF expression. | 1 mg/kg/day and 3.5 mL/kg/day gavage | [67] | |
| Traumatic brain injury | In vivo - TBI mouse model | - Improvement of TBI-induced neuronal death and behavioral alterations | 1 mg/kg i.p. | [68,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiavone, S.; Morgese, M.G.; Tucci, P.; Trabace, L. The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches. Molecules 2021, 26, 4700. https://doi.org/10.3390/molecules26154700
Schiavone S, Morgese MG, Tucci P, Trabace L. The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches. Molecules. 2021; 26(15):4700. https://doi.org/10.3390/molecules26154700
Chicago/Turabian StyleSchiavone, Stefania, Maria Grazia Morgese, Paolo Tucci, and Luigia Trabace. 2021. "The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches" Molecules 26, no. 15: 4700. https://doi.org/10.3390/molecules26154700
APA StyleSchiavone, S., Morgese, M. G., Tucci, P., & Trabace, L. (2021). The Therapeutic Potential of Celastrol in Central Nervous System Disorders: Highlights from In Vitro and In Vivo Approaches. Molecules, 26(15), 4700. https://doi.org/10.3390/molecules26154700