
N-Terminomics Strategies for Protease Substrates Profiling


Abstract
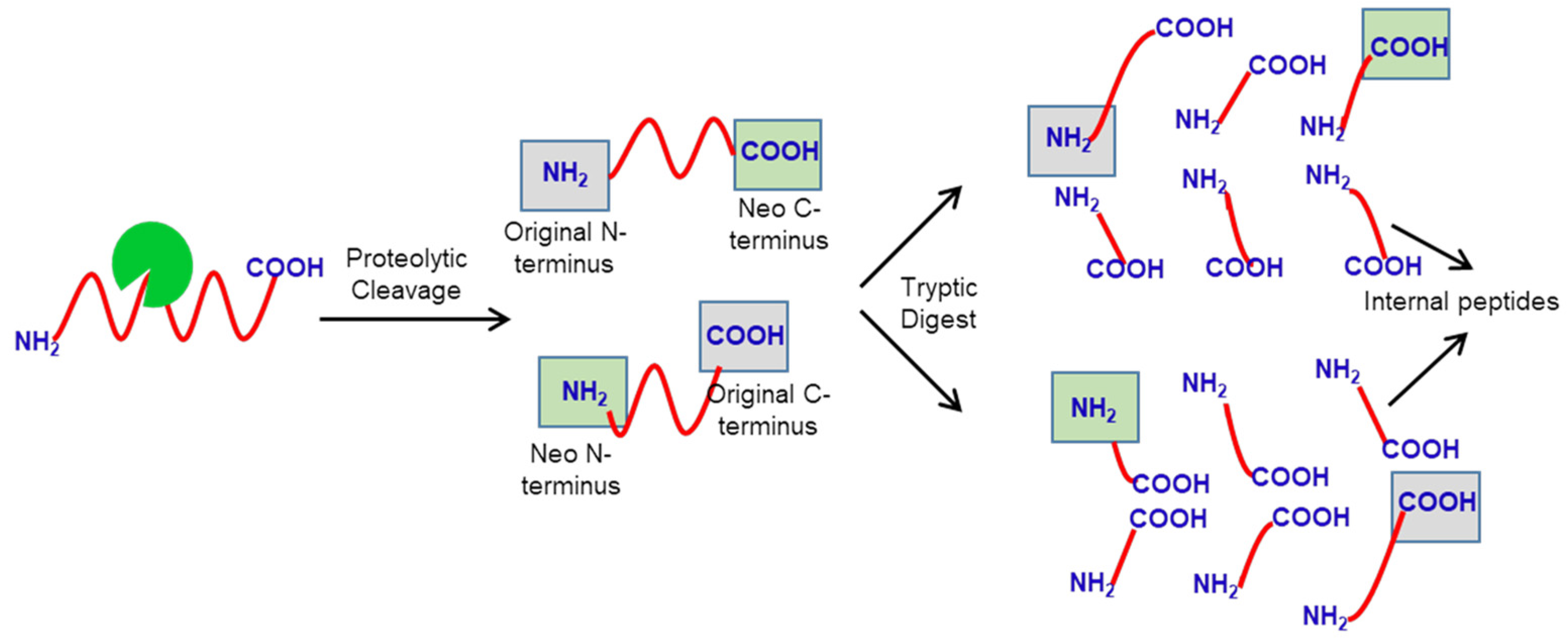
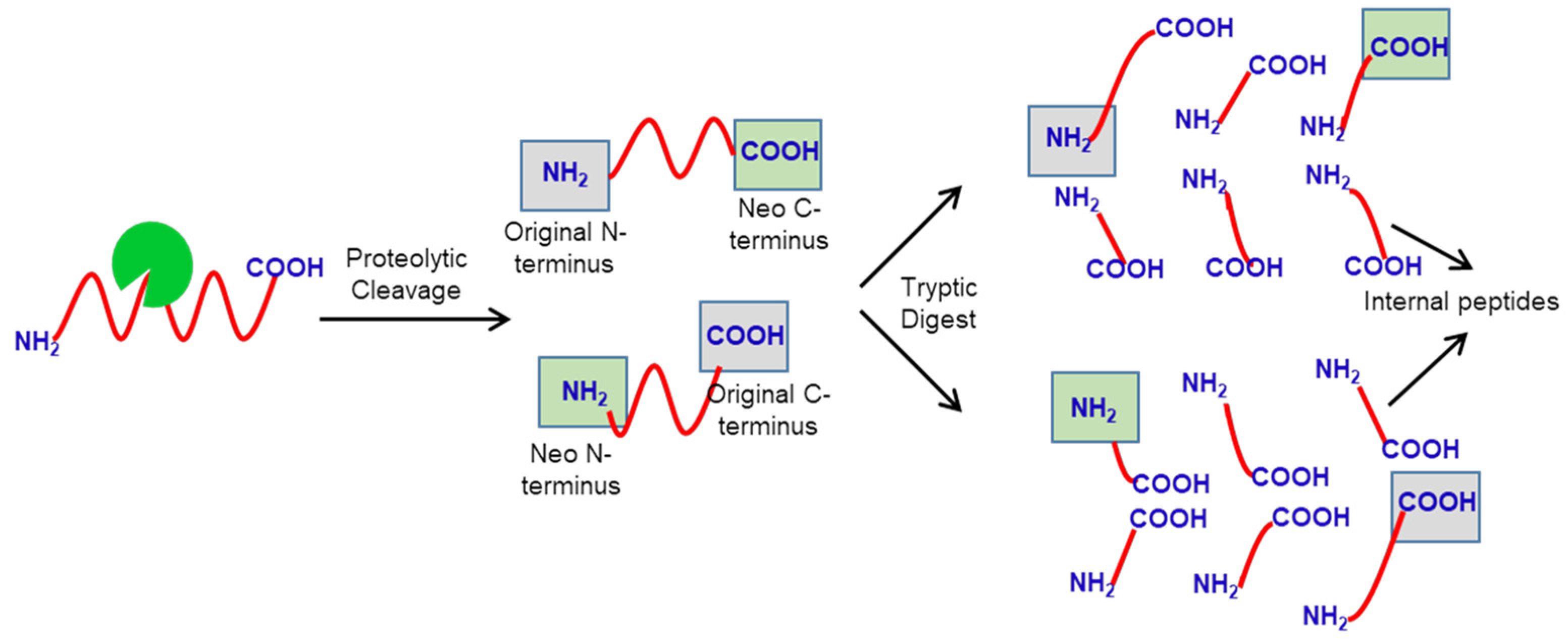
:1. Introduction
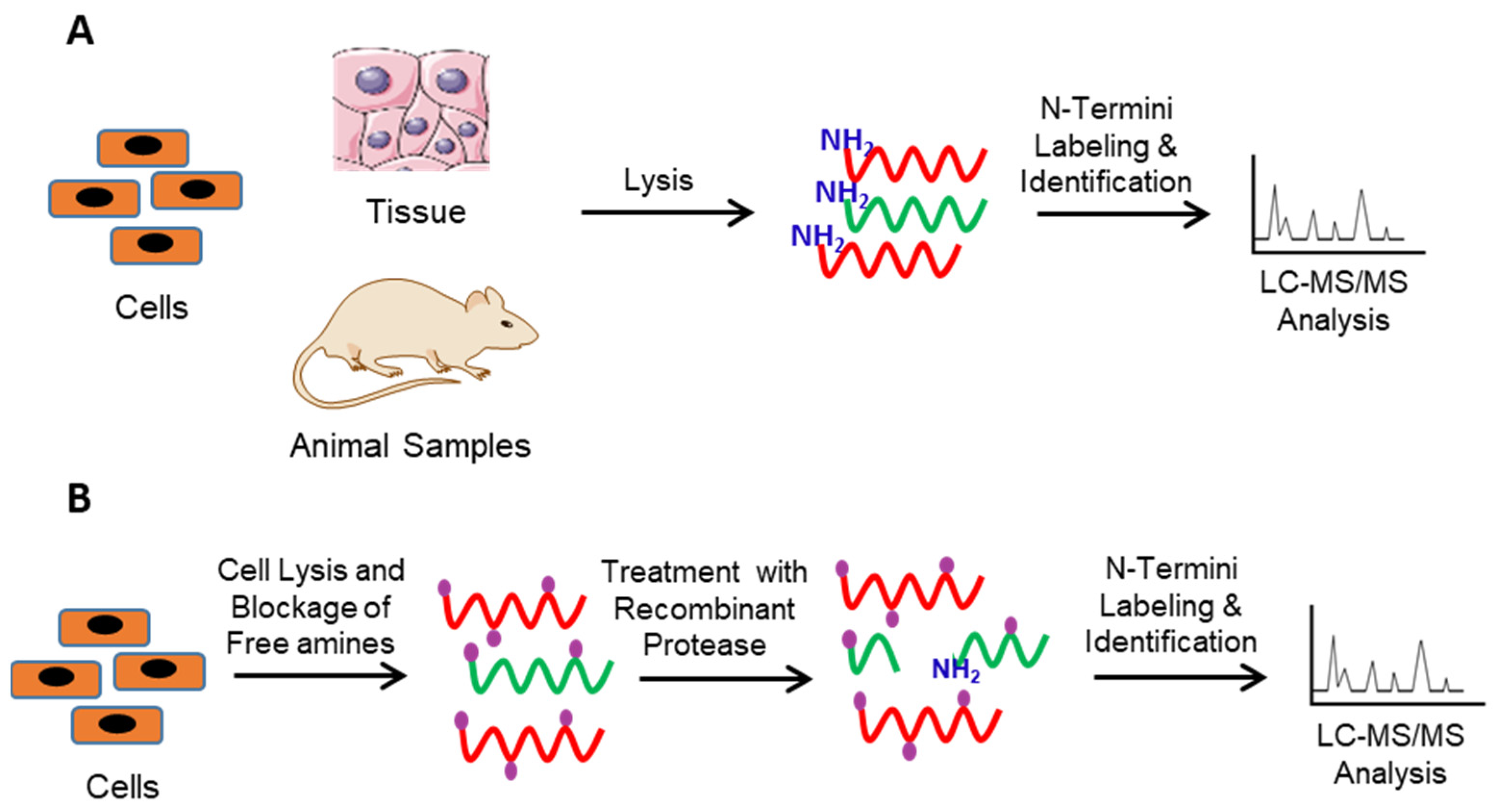
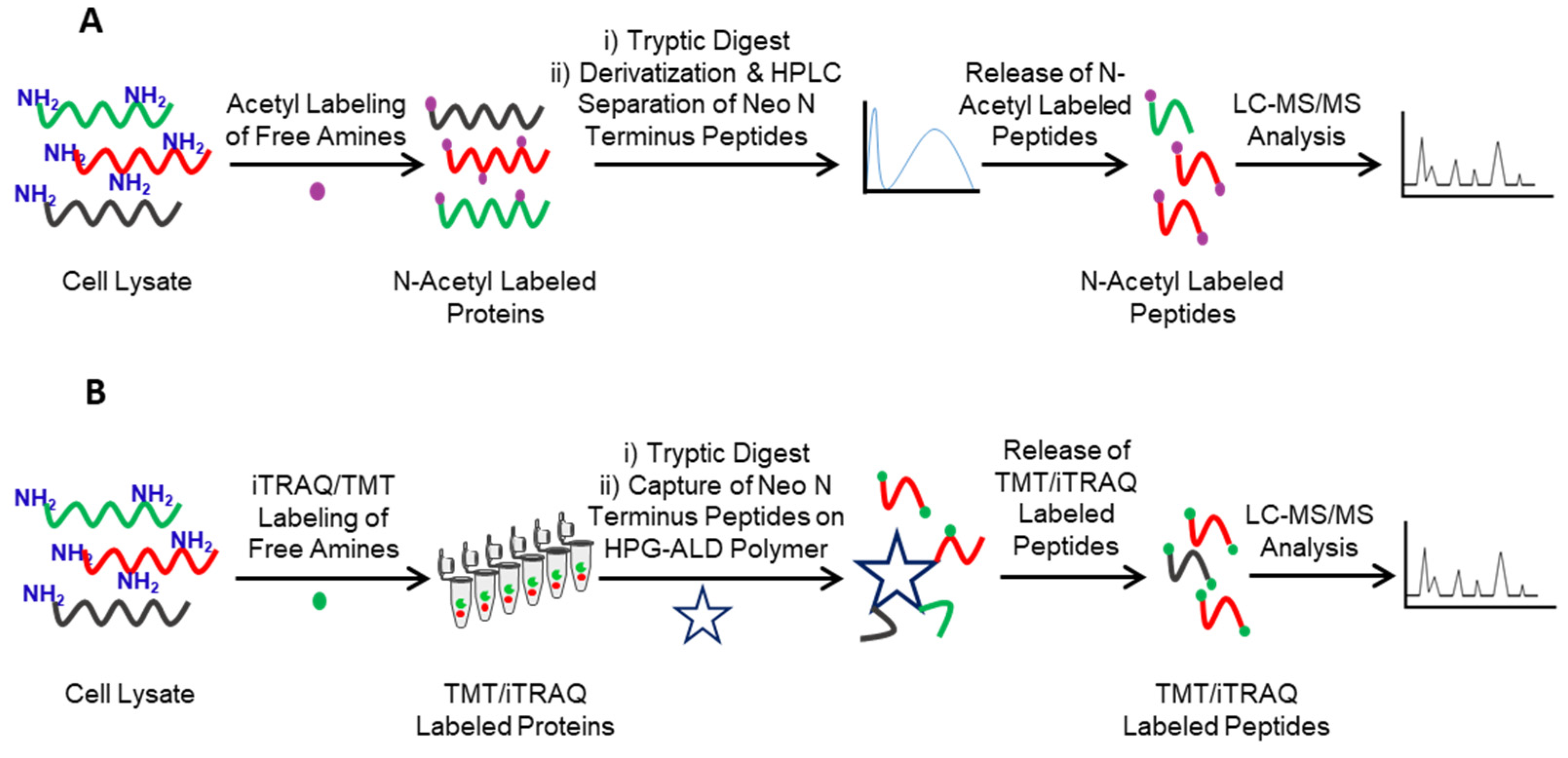
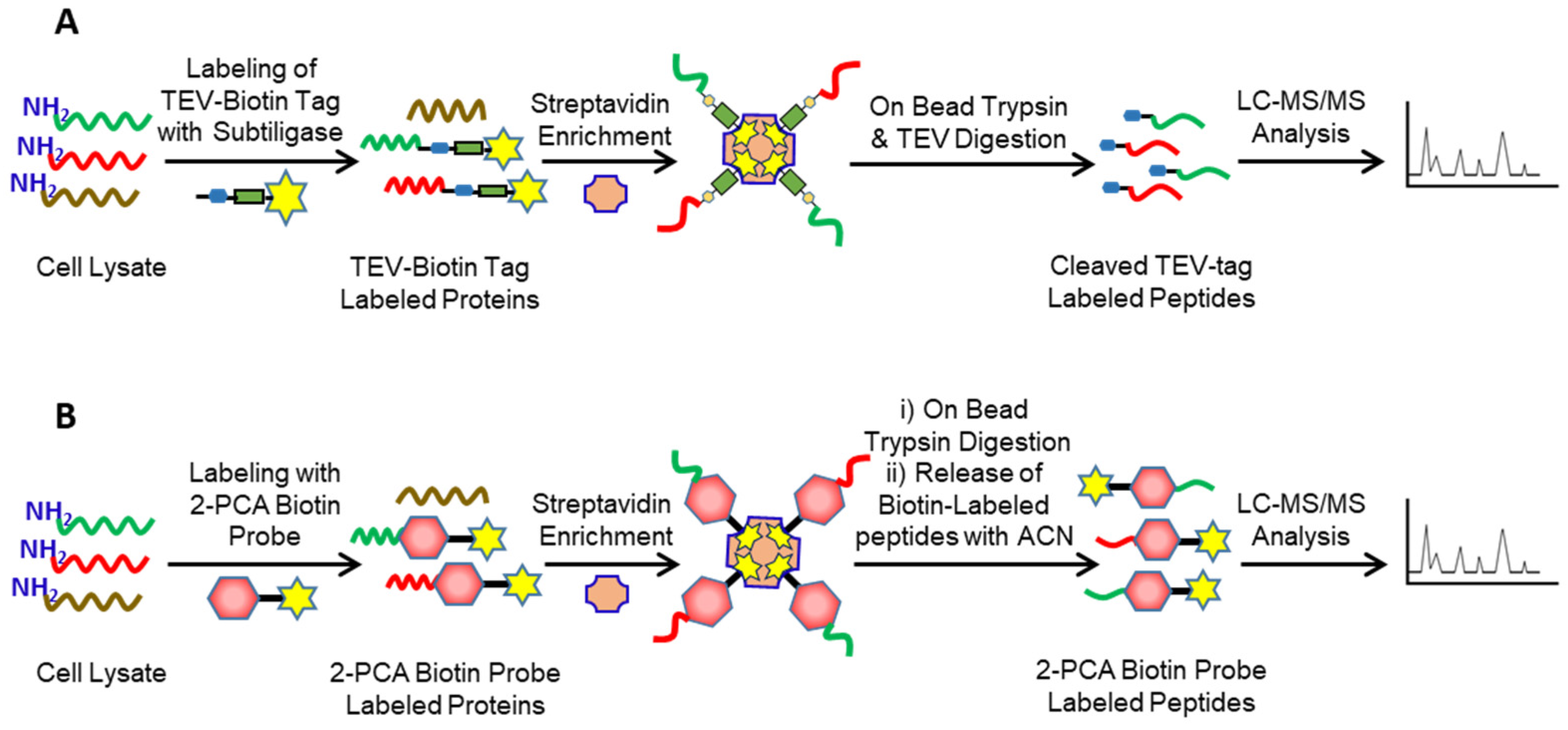
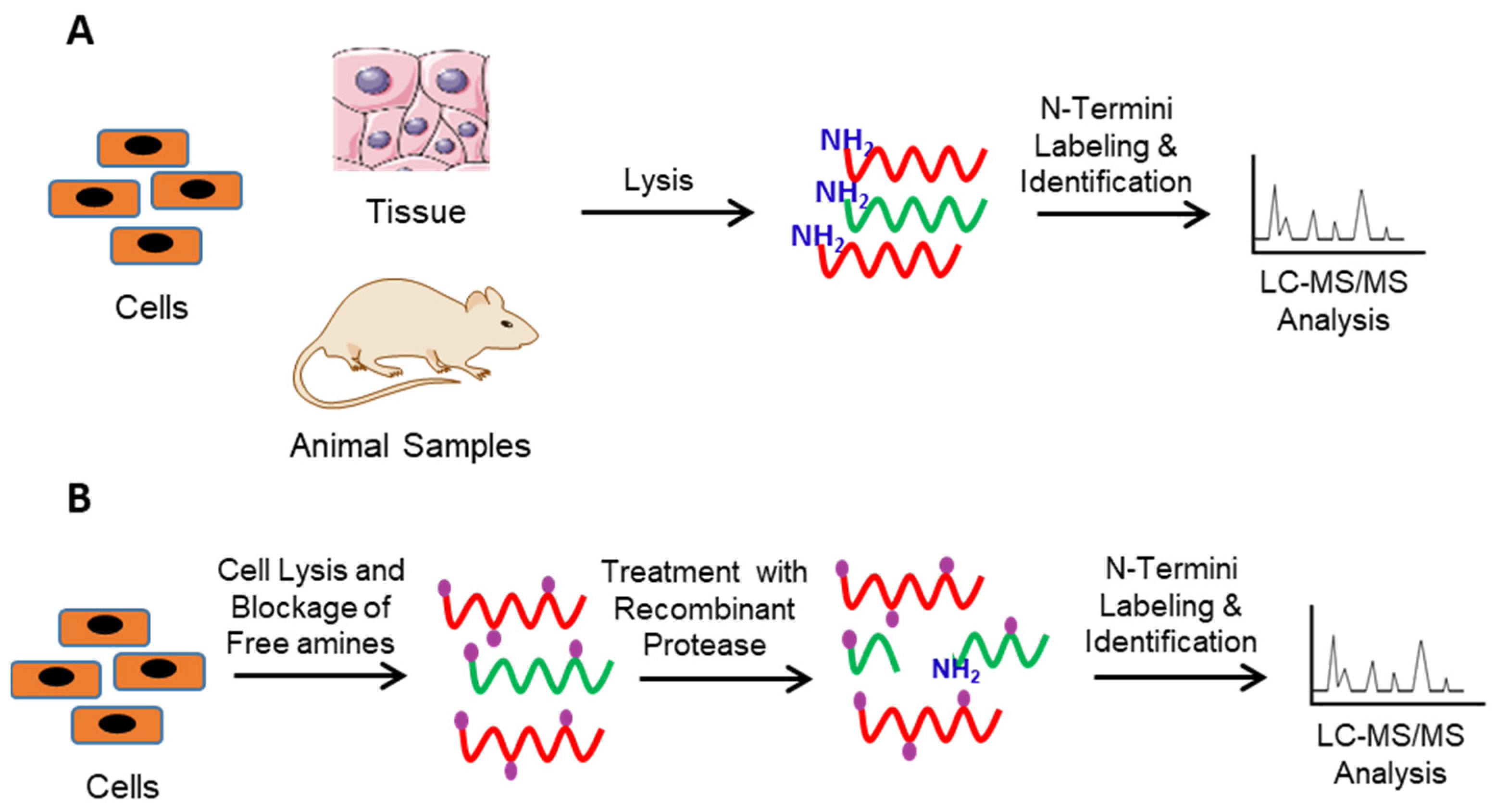
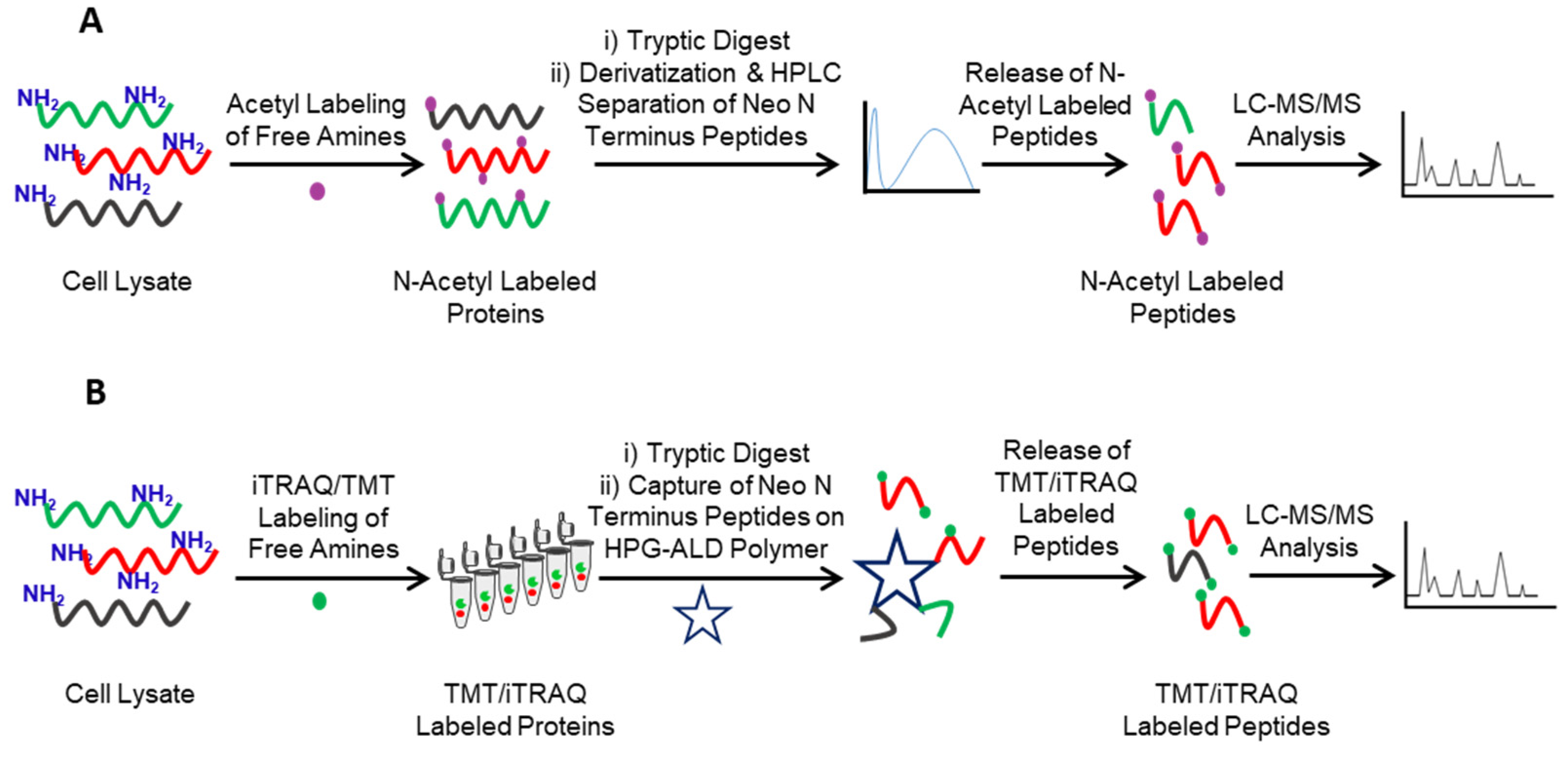
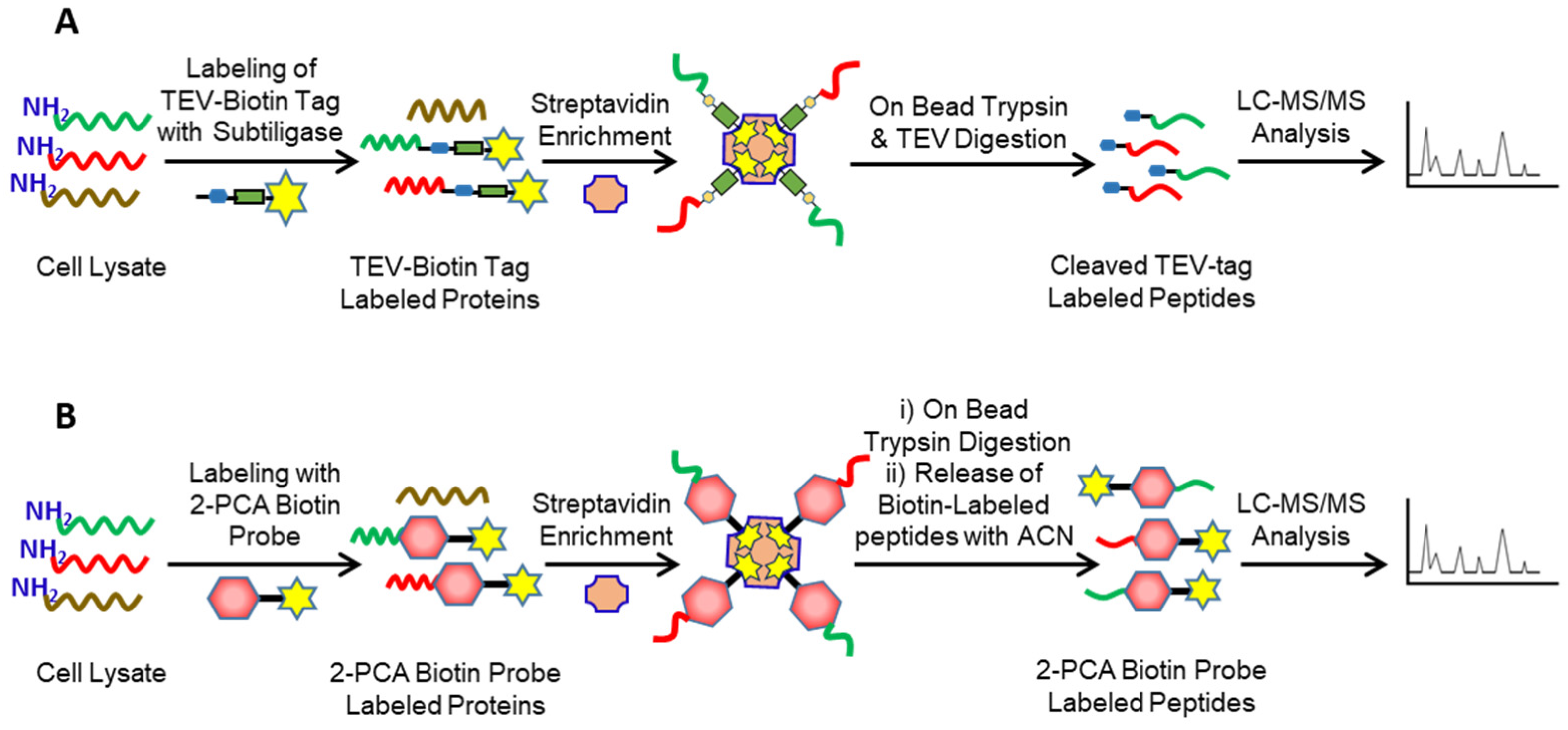
2. Identification of Protease Substrates Using N-Terminomics Methods
3. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gurumallesh, P.; Alagu, K.; Ramakrishnan, B.; Muthusamy, S. A systematic reconsideration on proteases. Int. J. Biol. Macromol. 2019, 128, 254–267. [Google Scholar] [CrossRef]
- Quesada, V.; Ordóñez, G.R.; Sánchez, L.M.; Puente, X.S.; López-Otín, C. The Degradome database: Mammalian proteases and diseases of proteolysis. Nucleic Acids Res. 2008, 37 (Suppl. 1), D239–D243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, N.D.; Morton, F.R.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2006, 34 (Suppl. 1), D270–D272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [Green Version]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Scher, W. The role of extracellular proteases in cell proliferation and differentiation. Lab. Investig. J. Tech. Methods Pathol. 1987, 57, 607–633. [Google Scholar]
- Sukharev, S.A.; Pleshakova, O.V.; Sadovnikov, V.B. Role of proteases in activation of apoptosis. Cell Death Differ. 1997, 4, 457–462. [Google Scholar] [CrossRef]
- Ruggiano, A.; Ramadan, K. DNA–protein crosslink proteases in genome stability. Commun. Biol. 2021, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T. Membrane proteases: Roles in tissue remodeling and tumour invasion. Curr. Opin. Cell Biol. 1992, 4, 802–809. [Google Scholar] [CrossRef]
- Walsh, P.N.; Ahmad, S.S. Proteases in blood clotting. Essays Biochem. 2002, 38, 95–111. [Google Scholar]
- Singh, N.; Bhattacharyya, D. Proteases in Wound Healing and Immunity. In Proteases in Human Diseases; Chakraborti, S., Chakraborti, T., Dhalla, N.S., Eds.; Springer: Singapore, 2017; pp. 147–170. [Google Scholar]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef]
- Hasanbasic, S.; Jahic, A.; Karahmet, E.; Sejranic, A.; Prnjavorac, B. The role of cysteine protease in Alzheimer disease. Mater. Socio-Med. 2016, 28, 235. [Google Scholar] [CrossRef] [Green Version]
- Suchiva, P.; Takai, T.; Kamijo, S.; Maruyama, N.; Yokomizo, T.; Sugimoto, Y.; Okumura, K.; Ikeda, S.; Ogawa, H. Inhibition of Both Cyclooxygenase-1 and-2 Promotes Epicutaneous Th2 and Th17 Sensitization and Allergic Airway Inflammation on Subsequent Airway Exposure to Protease Allergen in Mice. Int. Arch. Allergy Immunol. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, F.; Mariani, G. Biochemical, molecular and clinical aspects of coagulation factor VII and its role in hemostasis and thrombosis. Haematologica 2021, 106, 351. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Nair, S. Proteases in cardiometabolic diseases: Pathophysiology, molecular mechanisms and clinical applications. Biochim. Biophys. Acta 2015, 1852, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overall, C.M.; Lopez-Otin, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Ruf, W. Emerging insights in tissue factor-dependent signaling events. Semin. Thromb. Hemost. 2006, 32, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Parks, W.C.; Heinecke, J.W. Activation and silencing of matrix metalloproteinases. Semin. Cell Dev. Biol. 2008, 19, 2–13. [Google Scholar] [CrossRef]
- Bode, W.; Huber, R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta 2000, 1477, 241–252. [Google Scholar] [CrossRef]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef]
- Mo, X.Y.; Cascio, P.; Lemerise, K.; Goldberg, A.L.; Rock, K. Distinct Proteolytic Processes Generate the C and N Termini of MHC Class I-Binding Peptides. J. Immunol. 1999, 163, 5851–5859. [Google Scholar]
- Khan, A.R.; James, M.N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998, 7, 815–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisin, A.; Monville, C.; Plancheron, A.; Béré, E.; Gaillard, A.; Leveziel, N. Cathepsin B pH-Dependent Activity Is Involved in Lysosomal Dysregulation in Atrophic Age-Related Macular Degeneration. Oxid. Med. Cell. Longev. 2019, 2019, 5637075. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Edrington, T.C.; Storrs, S.B.; Crowley, K.S.; Ward, J.M.; Lee, T.C.; Liu, Z.L.; Li, B.; Glenn, K.C. Analyzing pepsin degradation assay conditions used for allergenicity assessments to ensure that pepsin susceptible and pepsin resistant dietary proteins are distinguishable. PLoS ONE 2017, 12, e0171926. [Google Scholar] [CrossRef] [Green Version]
- Allgayer, H.; Lengyel, E.; Boyd, D.D. Transcriptional Control of Proteases. In Proteases and Their Inhibitors in Cancer Metastasis; Foidart, J.-M., Muschel, R.J., Eds.; Springer: Dordrecht, The Netherlands, 2002; pp. 151–168. [Google Scholar]
- Klein, T.; Eckhard, U.; Dufour, A.; Solis, N.; Overall, C.M. Proteolytic Cleavage Mechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018, 118, 1137–1168. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.S. An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 2011, 80, 283–293. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-end rule pathway of protein degradation. Genes Cells 1997, 2, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.Y.; Araya, L.E.; Julien, O. Protease substrate identification using N-terminomics. ACS Chem. Biol. 2019, 14, 2361–2371. [Google Scholar] [CrossRef]
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass spectrometry-based proteomics: Existing capabilities and future directions. Chem. Soc. Rev. 2012, 41, 3912–3928. [Google Scholar] [CrossRef] [Green Version]
- Timmer, J.C.; Salvesen, G.S. N-terminomics: A high-content screen for protease substrates and their cleavage sites. Methods Mol. Biol. 2011, 753, 243–255. [Google Scholar]
- Tanco, S.; Gevaert, K.; Van Damme, P. C-terminomics: Targeted analysis of natural and posttranslationally modified protein and peptide C-termini. Proteomics 2015, 15, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Wiita, A.P.; Seaman, J.E.; Wells, J.A. Global analysis of cellular proteolysis by selective enzymatic labeling of protein N-termini. Methods Enzymol. 2014, 544, 327–358. [Google Scholar] [PubMed] [Green Version]
- Löffek, S.; Schilling, O.; Franzke, C.W. Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, C.; Ghesquière, B.; Impens, F.; Gevaert, K.; Kumar, J.D.; Cash, N.; Kandola, S.; Hegyi, P.; Wang, T.C.; Dockray, G.J.; et al. Mapping proteolytic processing in the secretome of gastric cancer-associated myofibroblasts reveals activation of MMP-1, MMP-2, and MMP-3. J. Proteome Res. 2013, 12, 3413–3422. [Google Scholar] [CrossRef]
- Prudova, A.; Gocheva, V.; Auf dem Keller, U.; Eckhard, U.; Olson, O.C.; Akkari, L.; Butler, G.S.; Fortelny, N.; Lange, P.F.; Mark, J.C. TAILS N-terminomics and proteomics show protein degradation dominates over proteolytic processing by cathepsins in pancreatic tumors. Cell Rep. 2016, 16, 1762–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colige, A.; Monseur, C.; Crawley, J.T.B.; Santamaria, S.; de Groot, R. Proteomic discovery of substrates of the cardiovascular protease ADAMTS7. J. Biol. Chem. 2019, 294, 8037–8045. [Google Scholar] [CrossRef] [Green Version]
- Prudova, A.; Auf dem Keller, U.; Butler, G.S.; Overall, C.M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteom. 2010, 9, 894–911. [Google Scholar] [CrossRef] [Green Version]
- Plasman, K.; Demol, H.; Bird, P.I.; Gevaert, K.; Van Damme, P. Substrate specificities of the granzyme tryptases A and K. J. Proteome Res. 2014, 13, 6067–6077. [Google Scholar] [CrossRef]
- Crawford, E.D.; Wells, J.A. Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 2011, 80, 1055–1087. [Google Scholar] [CrossRef]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Martens, L.; Van Damme, J.; Hugelier, K.; Staes, A.; Vandekerckhove, J.; Gevaert, K. Caspase-specific and nonspecific in vivo protein processing during Fas-induced apoptosis. Nat. Methods 2005, 2, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Wejda, M.; Impens, F.; Takahashi, N.; Van Damme, P.; Gevaert, K.; Vandenabeele, P. Degradomics reveals that cleavage specificity profiles of caspase-2 and effector caspases are alike. J. Biol. Chem. 2012, 287, 33983–33995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impens, F.; Timmerman, E.; Staes, A.; Moens, K.; Arien, K.K.; Verhasselt, B.; Vandekerckhove, J.; Gevaert, K. A catalogue of putative HIV-1 protease host cell substrates. Biol. Chem. 2012, 393, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Declercq, W.; Kalai, M.; Saelens, X.; Vandenabeele, P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 2002, 9, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Rano, T.A.; Peterson, E.P.; Rasper, D.M.; Timkey, T.; Garcia-Calvo, M.; Houtzager, V.M.; Nordstrom, P.A.; Roy, S.; Vaillancourt, J.P.; et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J. Biol. Chem. 1997, 272, 17907–17911. [Google Scholar] [CrossRef] [Green Version]
- Stennicke, H.R.; Renatus, M.; Meldal, M.; Salvesen, G.S. Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 2000, 350 Pt 2, 563–568. [Google Scholar] [CrossRef]
- Seaman, J.E.; Julien, O.; Lee, P.S.; Rettenmaier, T.J.; Thomsen, N.D.; Wells, J.A. Cacidases: Caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ. 2016, 23, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Agard, N.J.; Mahrus, S.; Trinidad, J.C.; Lynn, A.; Burlingame, A.L.; Wells, J.A. Global kinetic analysis of proteolysis via quantitative targeted proteomics. Proc. Natl. Acad. Sci. USA 2012, 109, 1913–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.E.; MacPherson, D.J.; Wu, P.; Julien, O.; Wells, J.A.; Hardy, J.A. Reprogramming Caspase-7 Specificity by Regio-Specific Mutations and Selection Provides Alternate Solutions for Substrate Recognition. ACS Chem. Biol. 2016, 11, 1603–1612. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.E.; Kumar, A.; Wells, J.A.; Hobman, T.C.; Julien, O.; Hardy, J.A. The Unique Cofactor Region of Zika Virus NS2B-NS3 Protease Facilitates Cleavage of Key Host Proteins. ACS Chem. Biol. 2018, 13, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Weeks, A.M.; Byrnes, J.R.; Lui, I.; Wells, J.A. Mapping proteolytic neo-N termini at the surface of living cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2018809118. [Google Scholar] [CrossRef]
- Wildes, D.; Wells, J.A. Sampling the N-terminal proteome of human blood. Proc. Natl. Acad. Sci. USA 2010, 107, 4561–4566. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, M.; Hayashi, A.; Takeuchi, M.; Dobashi, M.; Mori, Y.; Shinoda, H.; Aizawa, T.; Demura, M.; Kawano, K. Unfolding and aggregation of transthyretin by the truncation of 50 N-terminal amino acids. Proteins 2008, 72, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Janowski, R.; Abrahamson, M.; Grubb, A.; Jaskolski, M. Domain swapping in N-truncated human cystatin C. J. Mol. Biol. 2004, 341, 151–160. [Google Scholar] [CrossRef]
- Liz, M.A.; Gomes, C.M.; Saraiva, M.J.; Sousa, M.M. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. J. Lipid Res. 2007, 48, 2385–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, J.; Nazarian, A.; Lawlor, K.; Robbins, R.J.; Tempst, P. A sequence-specific exopeptidase activity test (SSEAT) for “functional” biomarker discovery. Mol. Cell. Proteom. 2008, 7, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.S.; Goldberg, R.M.; Sargent, D.J.; Meyerhardt, J.A.; Wolpin, B.M.; Green, E.M.; Pitot, H.C.; Pollak, M. Plasma insulin-like growth factors, insulin-like binding protein-3, and outcome in metastatic colorectal cancer: Results from intergroup trial N9741. Clin. Cancer Res. 2008, 14, 8263–8269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.M.; Brecht, W.J.; Xu, Q.; Tesseur, I.; Kekonius, L.; Wyss-Coray, T.; Fish, J.D.; Masliah, E.; Hopkins, P.C.; Scearce-Levie, K. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10966–10971. [Google Scholar] [CrossRef] [Green Version]
- Alcaraz, L.B.; Mallavialle, A.; David, T.; Derocq, D.; Delolme, F.; Dieryckx, C.; Mollevi, C.; Boissiere-Michot, F.; Simony-Lafontaine, J.; Du Manoir, S.; et al. A 9-kDa matricellular SPARC fragment released by cathepsin D exhibits pro-tumor activity in the triple-negative breast cancer microenvironment. Theranostics 2021, 11, 6173–6192. [Google Scholar] [CrossRef]
- Gordon, M.H.; Anowai, A.; Young, D.; Das, N.; Campden, R.I.; Sekhon, H.; Myers, Z.; Mainoli, B.; Chopra, S.; Thuy-Boun, P.S.; et al. N-Terminomics/TAILS Profiling of Proteases and Their Substrates in Ulcerative Colitis. ACS Chem. Biol. 2019, 14, 2471–2483. [Google Scholar] [CrossRef] [PubMed]
- Mallia-Milanes, B.; Dufour, A.; Philp, C.; Solis, N.; Klein, T.; Fischer, M.; Bolton, C.E.; Shapiro, S.; Overall, C.M.; Johnson, S.R. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 315, L1003–L1014. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Fung, S.Y.; Renner, F.; Blank, M.A.; Dufour, A.; Kang, S.; Bolger-Munro, M.; Scurll, J.M.; Priatel, J.J.; Schweigler, P.; et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-kappaB signalling. Nat. Commun. 2015, 6, 8777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.H.; Indarto, D.; Doucet, A.; Pogson, L.D.; Pitman, M.R.; McNicholas, K.; Menz, R.I.; Overall, C.M.; Abbott, C.A. Identifying Natural Substrates for Dipeptidyl Peptidases 8 and 9 Using Terminal Amine Isotopic Labeling of Substrates (TAILS) Reveals in Vivo Roles in Cellular Homeostasis and Energy Metabolism. J. Biol. Chem. 2013, 288, 13936–13949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferson, T.; Auf dem Keller, U.; Bellac, C.; Metz, V.V.; Broder, C.; Hedrich, J.; Ohler, A.; Maier, W.; Magdolen, V.; Sterchi, E. The substrate degradome of meprin metalloproteases reveals an unexpected proteolytic link between meprin β and ADAM10. Cell. Mol. Life Sci. 2013, 70, 309–333. [Google Scholar] [CrossRef] [Green Version]
- Zelanis, A.; Oliveira, A.K.; Prudova, A.; Huesgen, P.F.; Tashima, A.K.; Kizhakkedathu, J.; Overall, C.M.; Serrano, S.M. Deep profiling of the cleavage specificity and human substrates of snake venom metalloprotease HF3 by proteomic identification of cleavage site specificity (PICS) using proteome derived peptide libraries and terminal amine isotopic labeling of substrates (TAILS) N-terminomics. J. Proteome Res. 2019, 18, 3419–3428. [Google Scholar]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25): Chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef] [Green Version]
- Bellac, C.L.; Dufour, A.; Krisinger, M.J.; Loonchanta, A.; Starr, A.E.; Auf dem Keller, U.; Lange, P.F.; Goebeler, V.; Kappelhoff, R.; Butler, G.S. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 2014, 9, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, T.; Čaušević, M.; auf dem Keller, U.; Schilling, O.; Isbert, S.; Geyer, R.; Maier, W.; Tschickardt, S.; Jumpertz, T.; Weggen, S.; et al. Metalloprotease Meprin β Generates Nontoxic N-terminal Amyloid Precursor Protein Fragments in Vivo. J. Biol. Chem. 2011, 286, 27741–27750. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.C.; Thejoe, M.; Klein, T.; Serapio-Palacios, A.; Santos, A.S.; von Krosigk, N.; Kizhakkedathu, J.N.; Stoynov, N.; Foster, L.J.; Overall, C.M. Master Sculptor at Work: Enteropathogenic Escherichia coli Infection Uniquely Modifies Mitochondrial Proteolysis during Its Control of Human Cell Death. Msystems 2020, 5, e00283-20. [Google Scholar] [CrossRef] [PubMed]
- Jagdeo, J.M.; Dufour, A.; Fung, G.; Luo, H.; Kleifeld, O.; Overall, C.M.; Jan, E. Heterogeneous Nuclear Ribonucleoprotein M Facilitates Enterovirus Infection. J. Virol. 2015, 89, 7064–7078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griswold, A.R.; Cifani, P.; Rao, S.D.; Axelrod, A.J.; Miele, M.M.; Hendrickson, R.C.; Kentsis, A.; Bachovchin, D.A. A chemical strategy for protease substrate profiling. Cell Chem. Biol. 2019, 26, 901–907.e6. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, K.; Van Damme, J.; Goethals, M.; Thomas, G.R.; Hoorelbeke, B.; Demol, H.; Martens, L.; Puype, M.; Staes, A.; Vandekerckhove, J. Chromatographic isolation of methionine-containing peptides for gel-free proteome analysis: Identification of more than 800 Escherichia coli proteins. Mol. Cell. Proteom. 2002, 1, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, P.; Van Damme, J.; Demol, H.; Staes, A.; Vandekerckhove, J.; Gevaert, K. A review of COFRADIC techniques targeting protein N-terminal acetylation. BMC Proc. 2009, 3 (Suppl. 6), S6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrar, A.; Dissmeyer, N.; Huesgen, P.F. New beginnings and new ends: Methods for large-scale characterization of protein termini and their use in plant biology. J. Exp. Bot. 2019, 70, 2021–2038. [Google Scholar] [CrossRef] [Green Version]
- Venne, A.S.; Solari, F.A.; Faden, F.; Paretti, T.; Dissmeyer, N.; Zahedi, R.P. An improved workflow for quantitative N-terminal charge-based fractional diagonal chromatography (ChaFRADIC) to study proteolytic events in Arabidopsis thaliana. Proteomics 2015, 15, 2458–2469. [Google Scholar] [CrossRef]
- Venne, A.S.; Vögtle, F.N.; Meisinger, C.; Sickmann, A.; Zahedi, R.P. Novel Highly Sensitive, Specific, and Straightforward Strategy for Comprehensive N-Terminal Proteomics Reveals Unknown Substrates of the Mitochondrial Peptidase Icp55. J. Proteome Res. 2013, 12, 3823–3830. [Google Scholar] [CrossRef]
- Venne, A.S.; Zahedi, R.P. The potential of fractional diagonal chromatography strategies for the enrichment of post-translational modifications. EuPA Open Proteom. 2014, 4, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.H.; Wu, J.Y. Alternative splicing and programmed cell death. Proc. Soc. Exp. Biol. Med. 1999, 220, 64–72. [Google Scholar]
- Kleifeld, O.; Doucet, A.; Prudova, A.; auf dem Keller, U.; Gioia, M.; Kizhakkedathu, J.N.; Overall, C.M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011, 6, 1578–1611. [Google Scholar] [CrossRef]
- Kleifeld, O.; Doucet, A.; Auf dem Keller, U.; Prudova, A.; Schilling, O.; Kainthan, R.K.; Starr, A.E.; Foster, L.J.; Kizhakkedathu, J.N.; Overall, C.M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010, 28, 281–288. [Google Scholar] [CrossRef]
- Mommen, G.P.M.; van de Waterbeemd, B.; Meiring, H.D.; Kersten, G.; Heck, A.J.R.; de Jong, A.P.J.M. Unbiased selective isolation of protein N-terminal peptides from complex proteome samples using phospho tagging (PTAG) and TiO2-based depletion. Mol. Cell. Proteom. 2012, 11, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Schlage, P.; Kockmann, T.; Kizhakkedathu, J.N.; Auf dem Keller, U. Monitoring matrix metalloproteinase activity at the epidermal–dermal interface by SILAC-iTRAQ-TAILS. Proteomics 2015, 15, 2491–2502. [Google Scholar] [CrossRef]
- Mahrus, S.; Trinidad, J.C.; Barkan, D.T.; Sali, A.; Burlingame, A.L.; Wells, J.A. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 2008, 134, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, O.; Zhuang, M.; Wiita, A.P.; O’Donoghue, A.J.; Knudsen, G.M.; Craik, C.S.; Wells, J.A. Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc. Natl. Acad. Sci. USA 2016, 113, E2001–E2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agard, N.J.; Maltby, D.; Wells, J.A. Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell. Proteom. 2010, 9, 880–893. [Google Scholar] [CrossRef] [Green Version]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Yeretssian, G.; Doiron, K.; Hussain, S.N.; Saleh, M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J. Biol. Chem. 2007, 282, 36321–36329. [Google Scholar] [CrossRef] [Green Version]
- Dix, M.M.; Simon, G.M.; Cravatt, B.F. Global mapping of the topography and magnitude of proteolytic events in apoptosis. Cell 2008, 134, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Applications |
|---|---|
| COFRADIC | |
| Subtiligase | |
| TAILS |
|
| CHOPS |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mintoo, M.; Chakravarty, A.; Tilvawala, R. N-Terminomics Strategies for Protease Substrates Profiling. Molecules 2021, 26, 4699. https://doi.org/10.3390/molecules26154699
Mintoo M, Chakravarty A, Tilvawala R. N-Terminomics Strategies for Protease Substrates Profiling. Molecules. 2021; 26(15):4699. https://doi.org/10.3390/molecules26154699
Chicago/Turabian StyleMintoo, Mubashir, Amritangshu Chakravarty, and Ronak Tilvawala. 2021. "N-Terminomics Strategies for Protease Substrates Profiling" Molecules 26, no. 15: 4699. https://doi.org/10.3390/molecules26154699
APA StyleMintoo, M., Chakravarty, A., & Tilvawala, R. (2021). N-Terminomics Strategies for Protease Substrates Profiling. Molecules, 26(15), 4699. https://doi.org/10.3390/molecules26154699