
MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
Example
3. Discussion
4. Materials and Methods
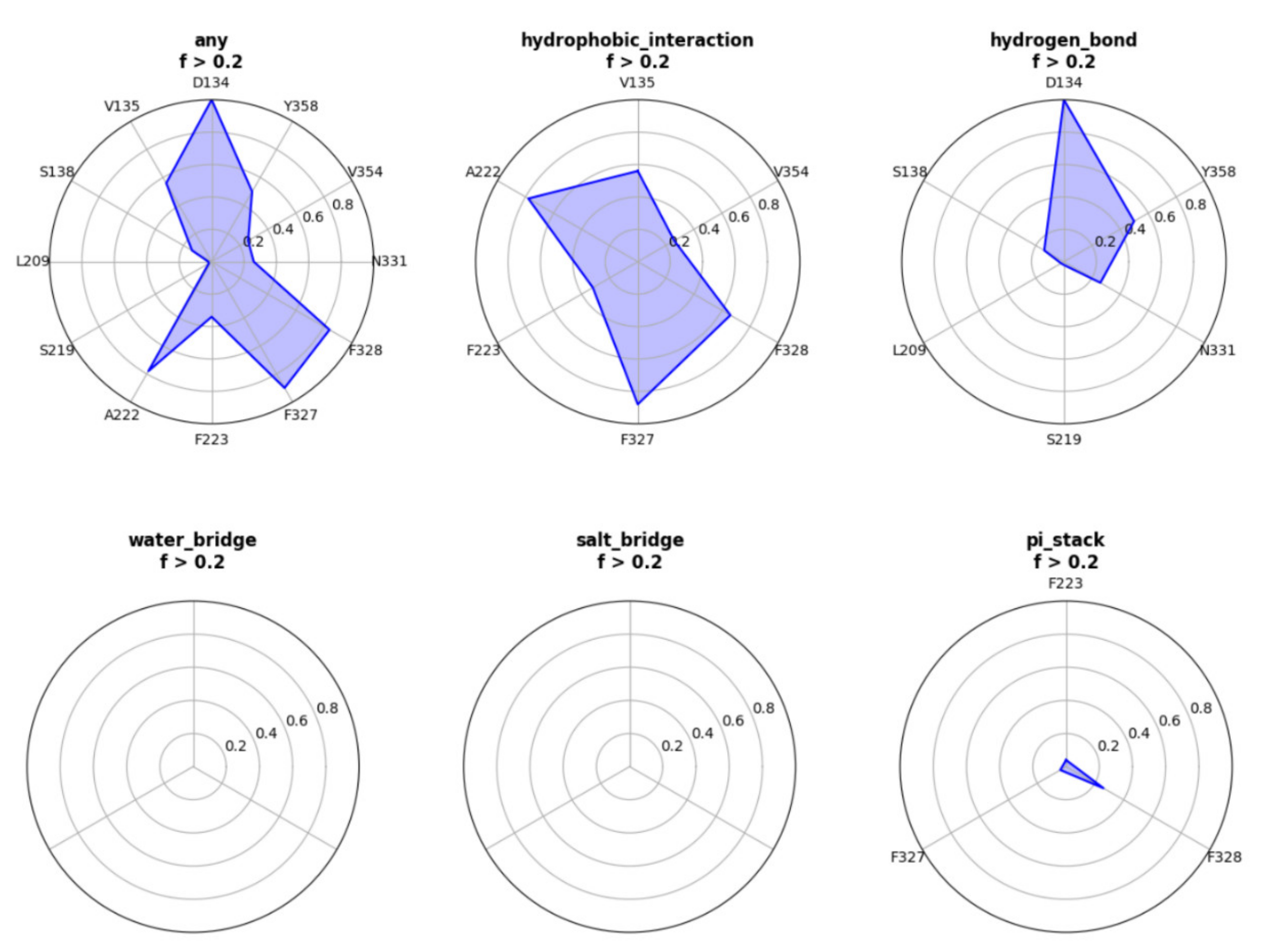
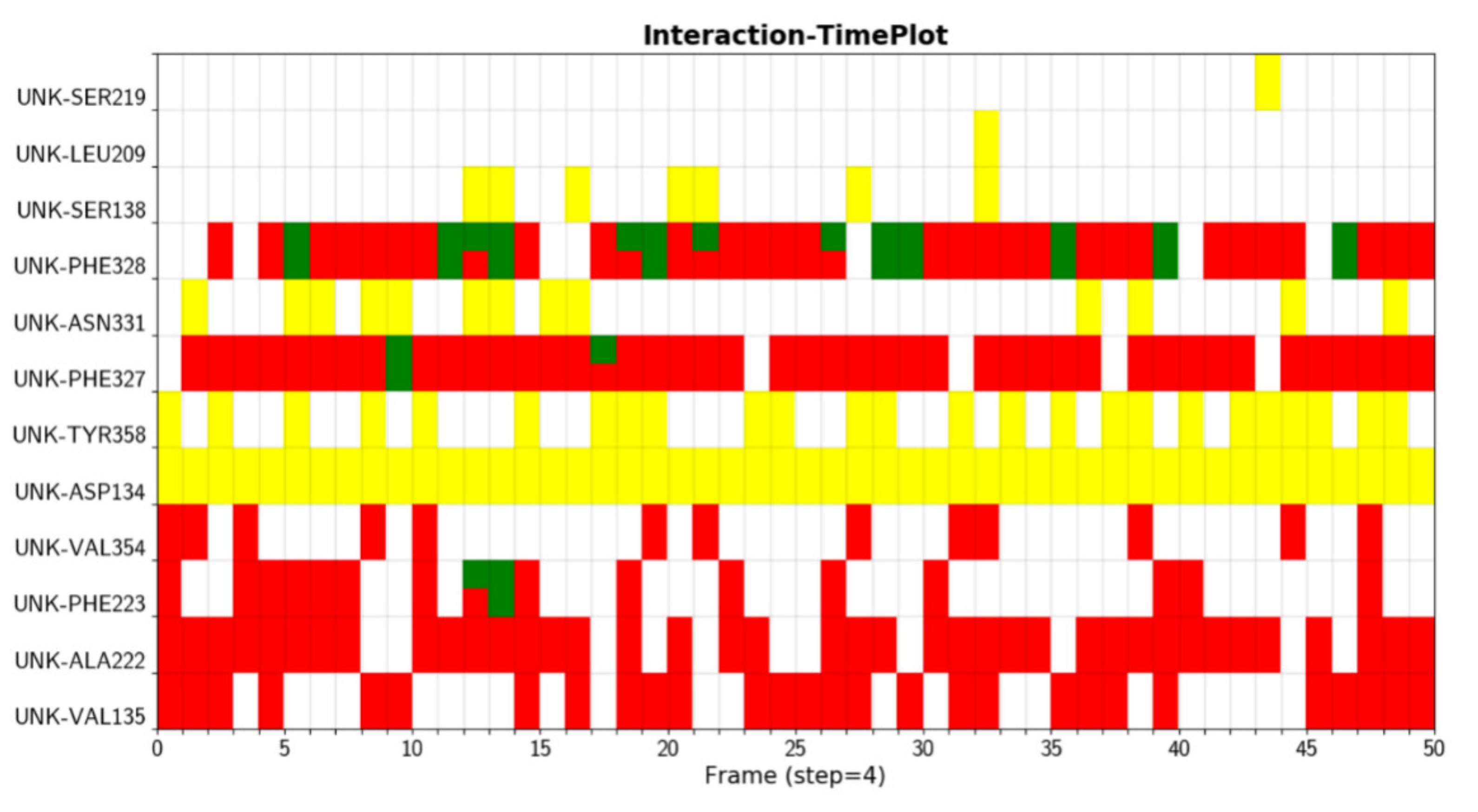
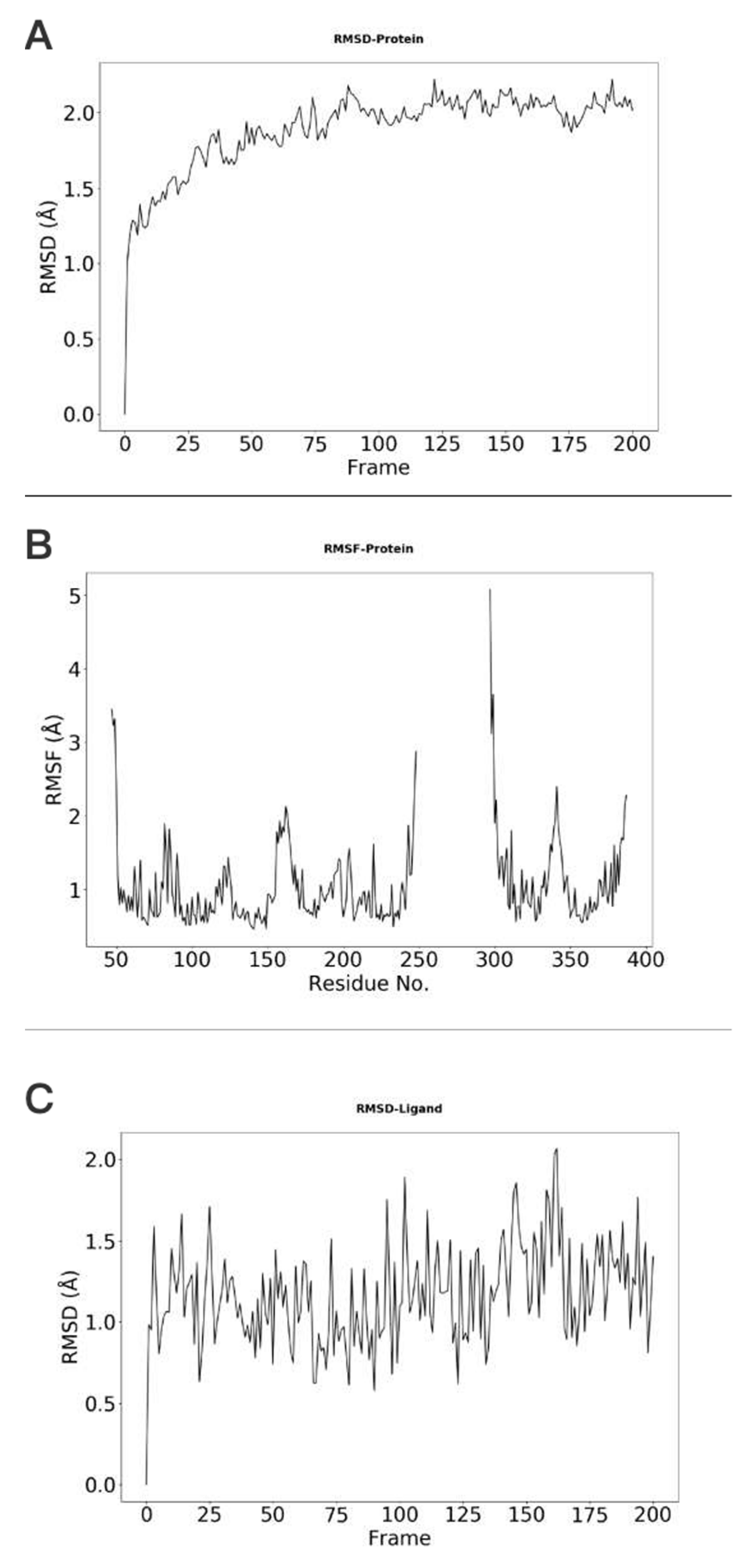
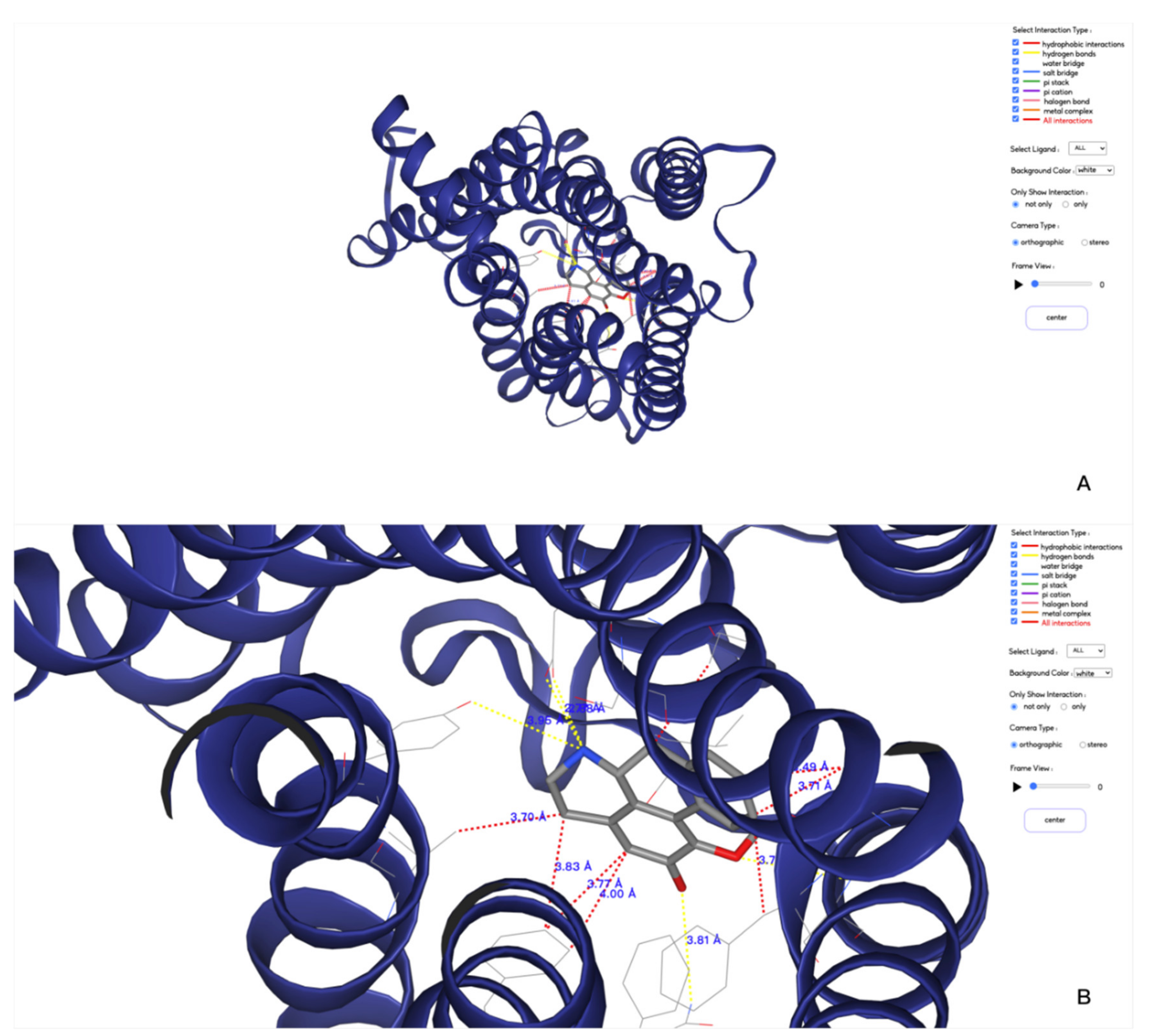
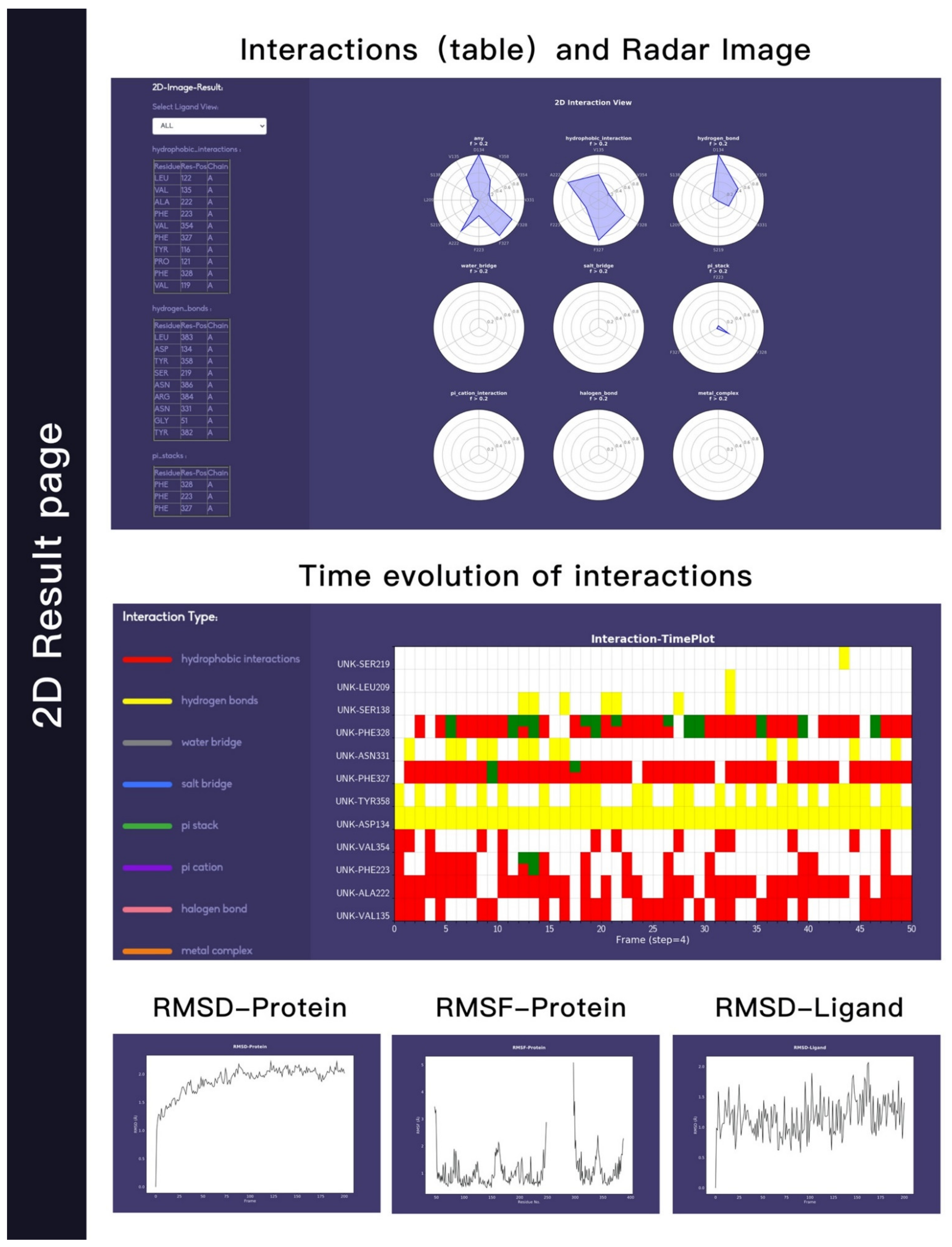
- Customizing the display information about the types of interactions of interest. There are nine selection boxes in this functional area in total, including the eight specific interaction types, as well as an option to display all of the interactions or not. Users can click on the selection box of the corresponding types to decide whether to display them. The color corresponding to the interaction type is also marked in this area, so that users can understand the results better.
- Customizing small molecules for selections. This function is used to select which interaction information of small molecule will be displayed. All interactions are displayed by default. All the small molecules are listed here. By selecting the interaction of a specific small molecule of interest to display, the contents of two regions (2D region and 3D region) will correspondingly be changed to show the information of the selected small molecule.
- Option to display the interaction information only. This function is for users to easily observe the interaction information. By default, the structure diagram will be displayed. However, if the interaction information is too complicated, the structure information may mask the interaction information. This function can be used to display only the interaction information to better observe the corresponding interaction information.
- Center. This function is used to locate the 3D result to the center of the page to facilitate the observation of the result.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Berman, H.M. The protein data bank: A historical perspective. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Janežič, D. ProBiS-ligands: A web server for prediction of ligands by examination of protein binding sites. Nucleic Acids Res. 2014, 42, W215–W220. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein–ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein–small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Shin, W.-H.; Lee, M.S.; Seok, C. GalaxySite: Ligand-binding-site prediction by using molecular docking. Nucleic Acids Res. 2014, 42, W210–W214. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Cesnik, T.; Konc, J.T.; Penca, M.; Janezic, D. ProBiS-database: Precalculated binding site similarities and local pairwise alignments of PDB structures. J. Chem. Inf. Model. 2012, 52, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Hildebrand, P.W. NGL Viewer: A web application for molecular visualization. Nucleic Acids Res. 2015, 43, W576–W579. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Kinoshita, K. GIANT: Pattern analysis of molecular interactions in 3D structures of protein–small ligand complexes. BMC Bioinform. 2014, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, V.; Sorokine, A.; Prilusky, J.; Abola, E.E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, A.; Blundell, T. CREDO: A protein–ligand interaction database for drug discovery. Chem. Biol. Drug Des. 2009, 73, 157–167. [Google Scholar] [CrossRef]
- Gallina, A.M.; Bisignano, P.; Bergamino, M.; Bordo, D. PLI: A web-based tool for the comparison of protein–ligand interactions observed on PDB structures. Bioinformatics 2013, 29, 395–397. [Google Scholar] [CrossRef][Green Version]
- Hendlich, M.; Bergner, A.; Günther, J.; Klebe, G. Relibase: Design and development of a database for comprehensive analysis of protein–ligand interactions. J. Mol. Biol. 2003, 326, 607–620. [Google Scholar] [CrossRef]
- De Beer, T.A.; Berka, K.; Thornton, J.M.; Laskowski, R.A. PDBsum additions. Nucleic Acids Res. 2014, 42, D292–D296. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Weisel, M.; Bitter, H.-M.; Diederich, F.; So, W.V.; Kondru, R. Prolix: Rapid mining of protein–ligand interactions in large crystal structure databases. J. Chem. Inf. Model. 2012, 52, 1450–1461. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P. 2D depiction of protein−Ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 1–7. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, B.; Zou, R.; Chan, H.C.S.; Li, H.; Yuan, S. MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions. Molecules 2021, 26, 4625. https://doi.org/10.3390/molecules26154625
Bai B, Zou R, Chan HCS, Li H, Yuan S. MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions. Molecules. 2021; 26(15):4625. https://doi.org/10.3390/molecules26154625
Chicago/Turabian StyleBai, Bing, Rongfeng Zou, H. C. Stephen Chan, Hongchun Li, and Shuguang Yuan. 2021. "MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions" Molecules 26, no. 15: 4625. https://doi.org/10.3390/molecules26154625
APA StyleBai, B., Zou, R., Chan, H. C. S., Li, H., & Yuan, S. (2021). MolADI: A Web Server for Automatic Analysis of Protein–Small Molecule Dynamic Interactions. Molecules, 26(15), 4625. https://doi.org/10.3390/molecules26154625