
Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cell Cycle
2.1. Cyclin Dependent Kinase (CDK)
2.2. Cyclin
2.3. CDK Inhibitors (CKIs)
3. Cell Cycle Dysregulation and Cancer
3.1. Cyclin
3.2. CKIs
3.3. CDK
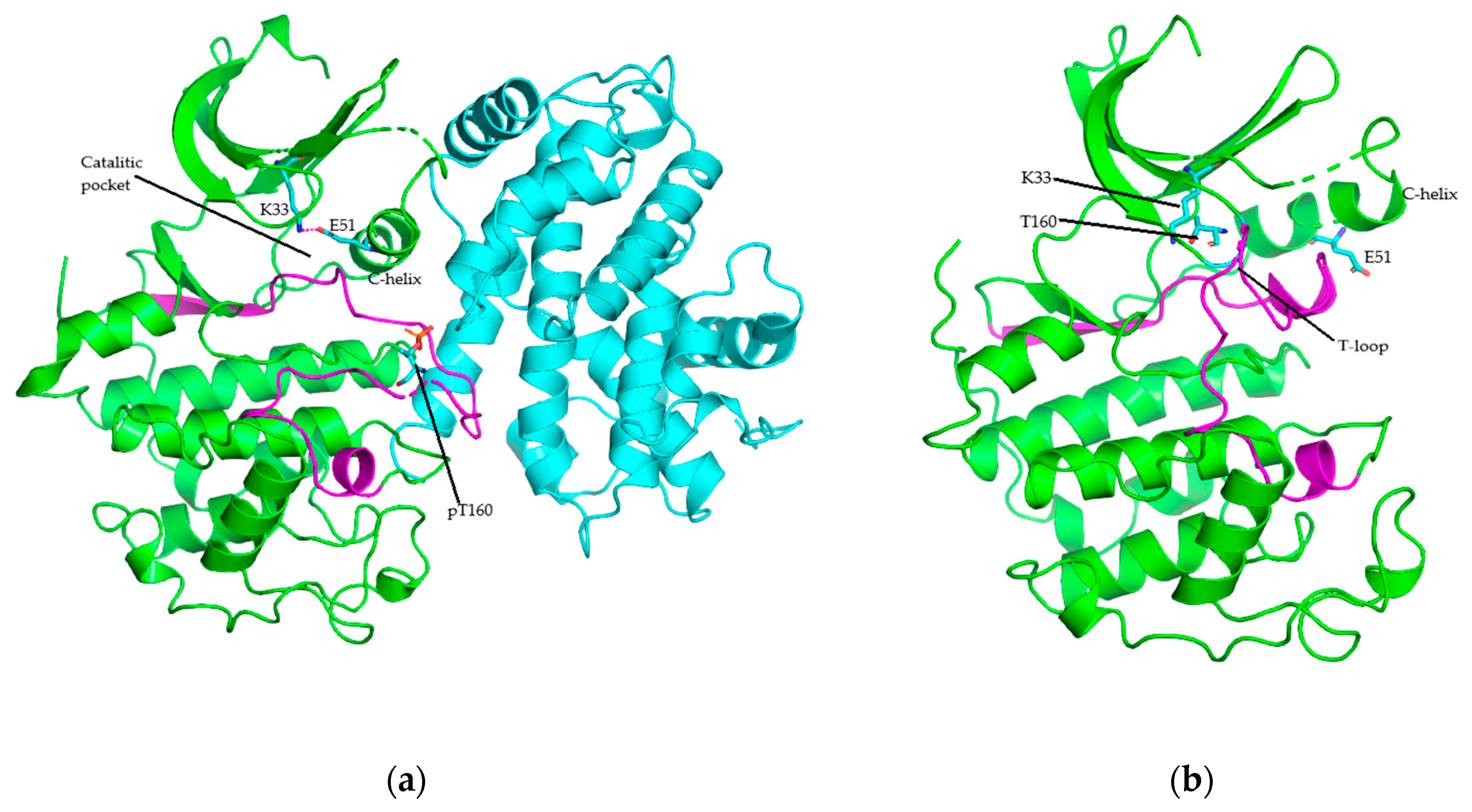
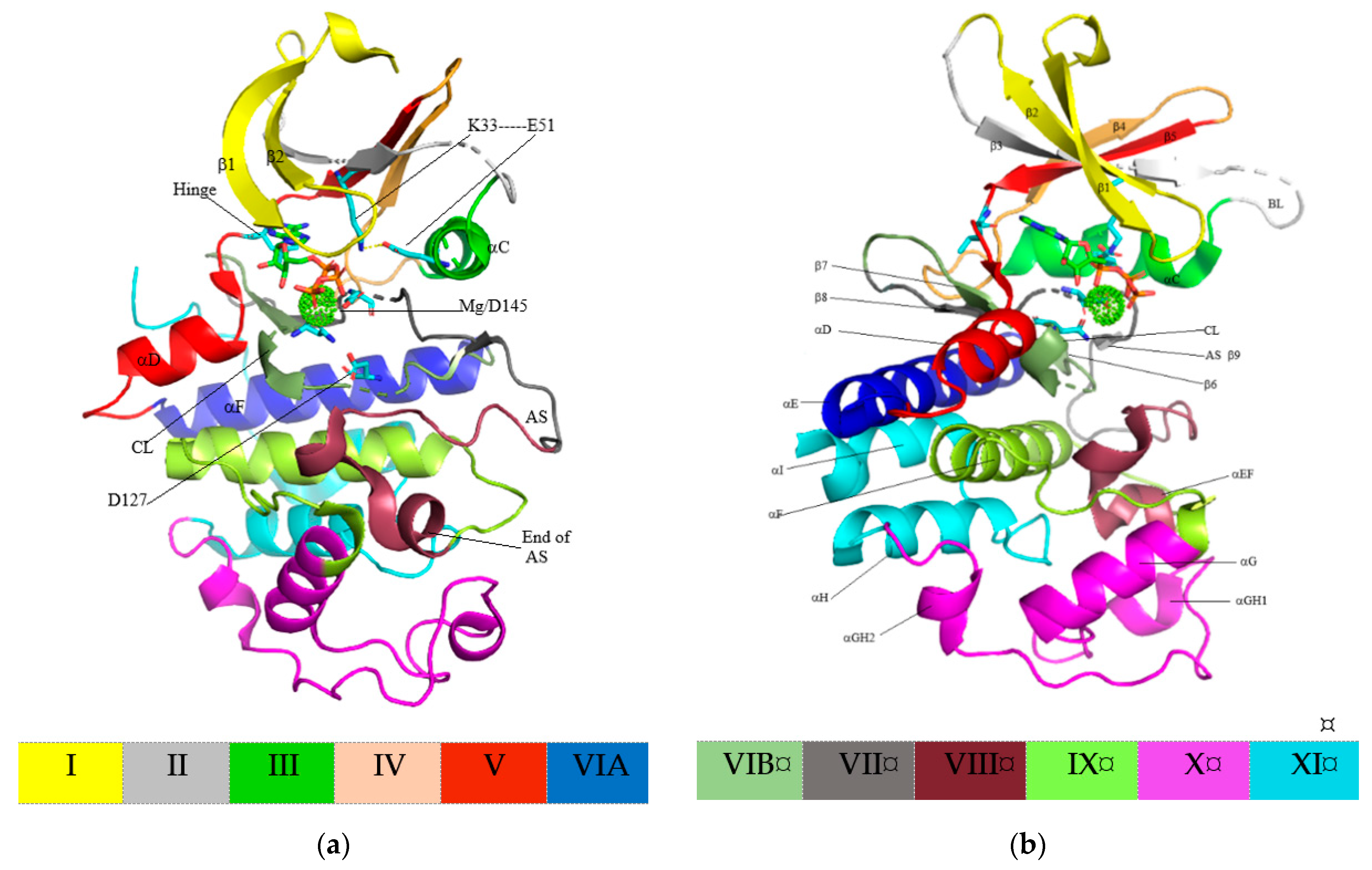
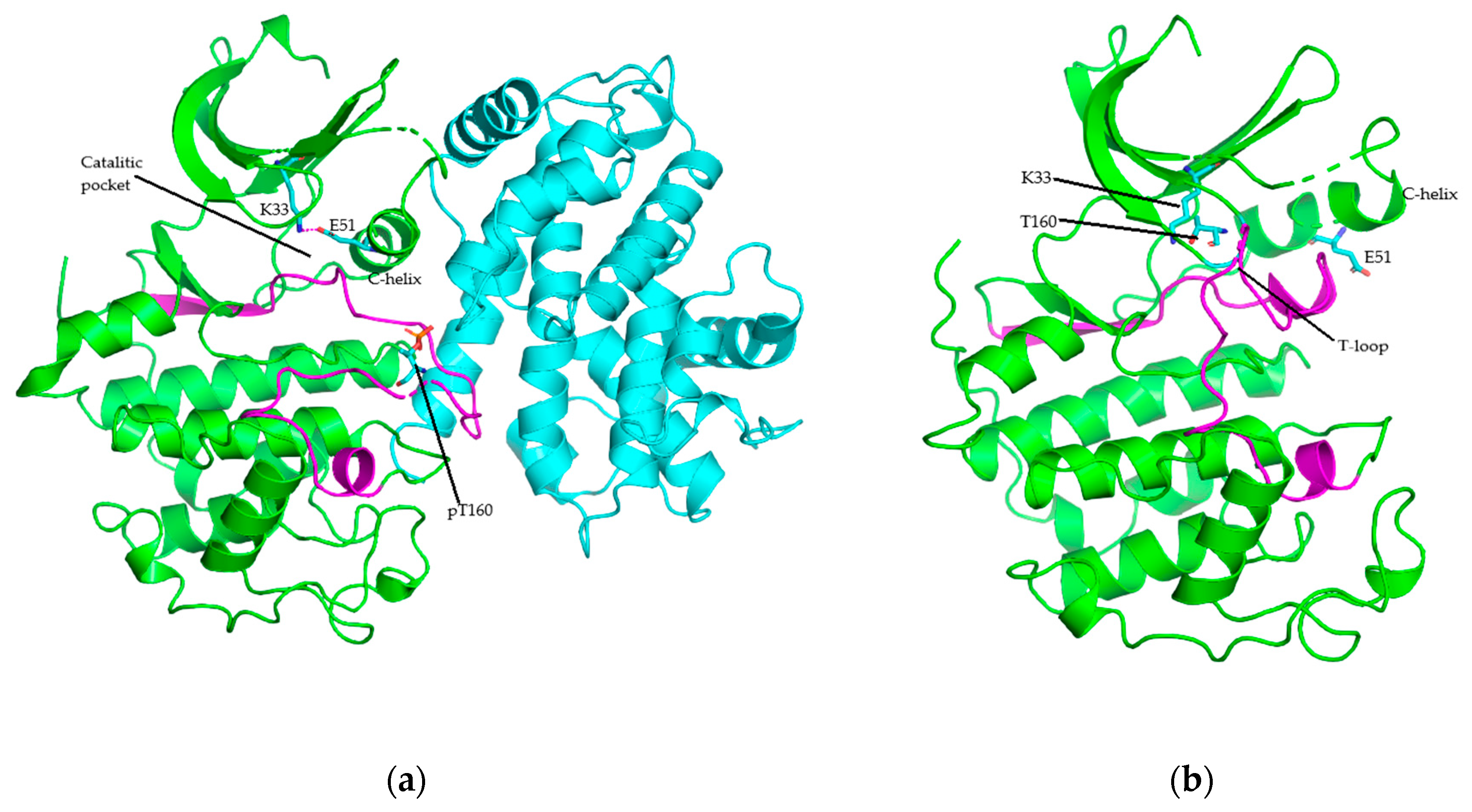
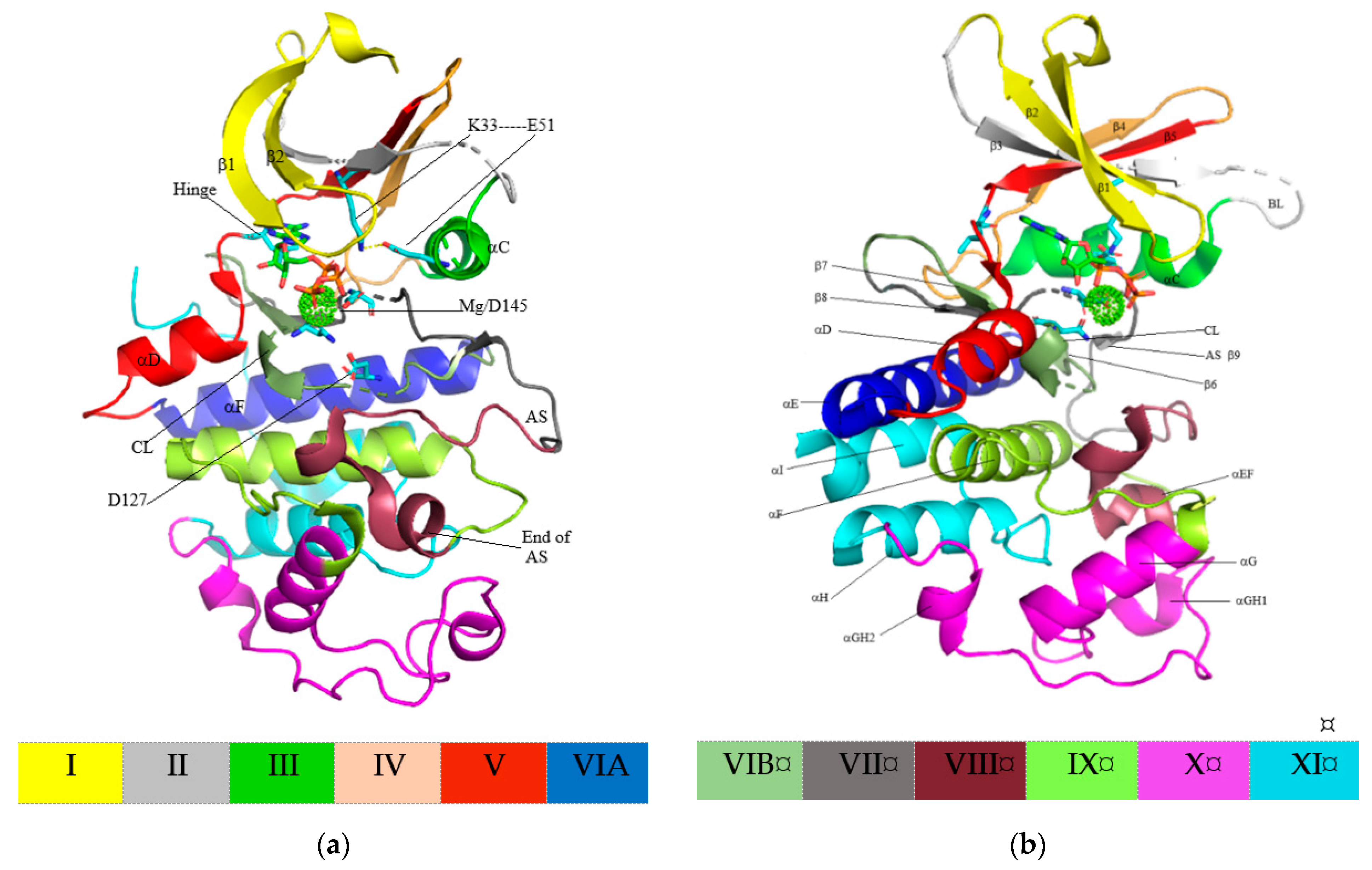
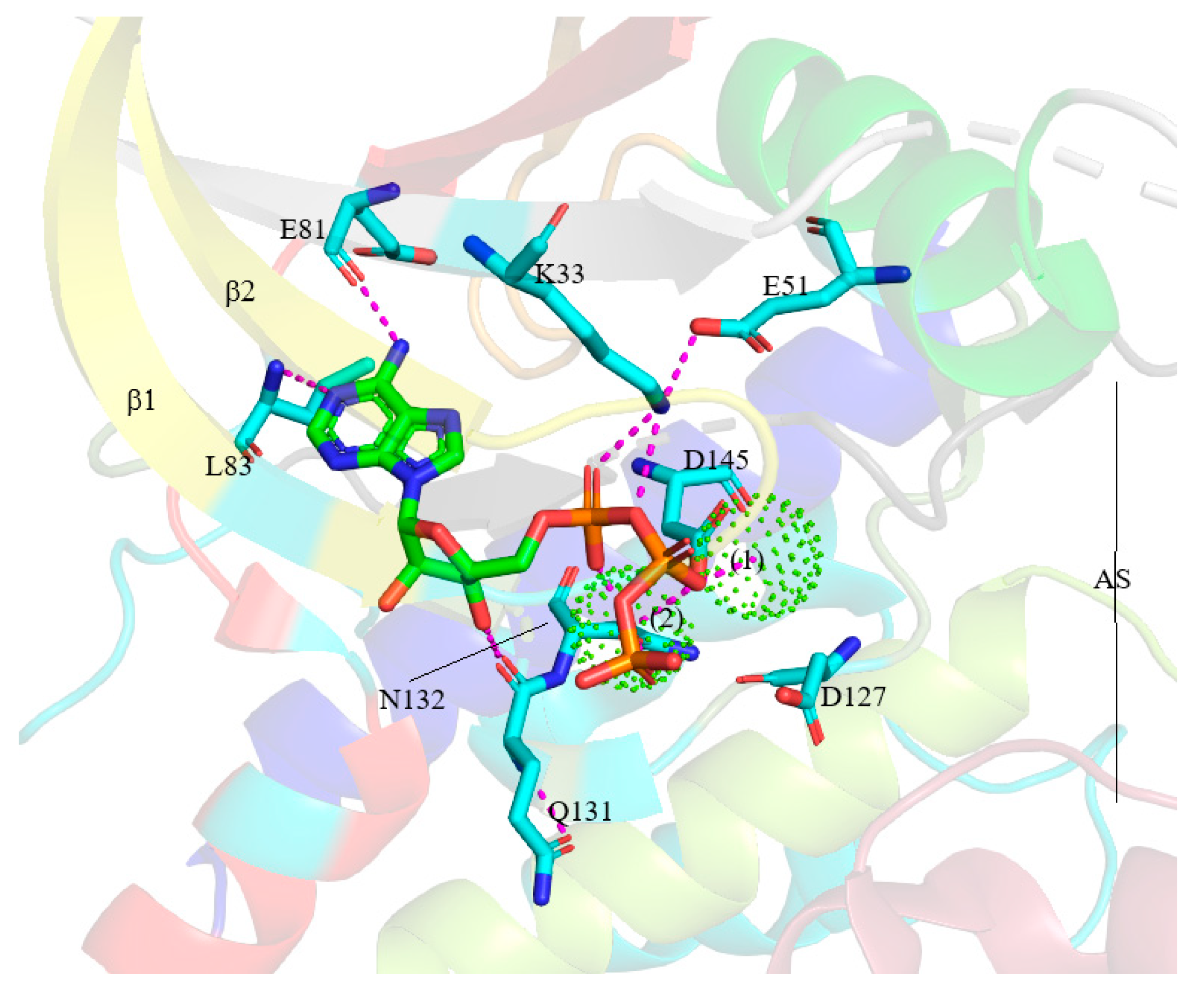
4. Structure of Cyclin-Dependent Kinase
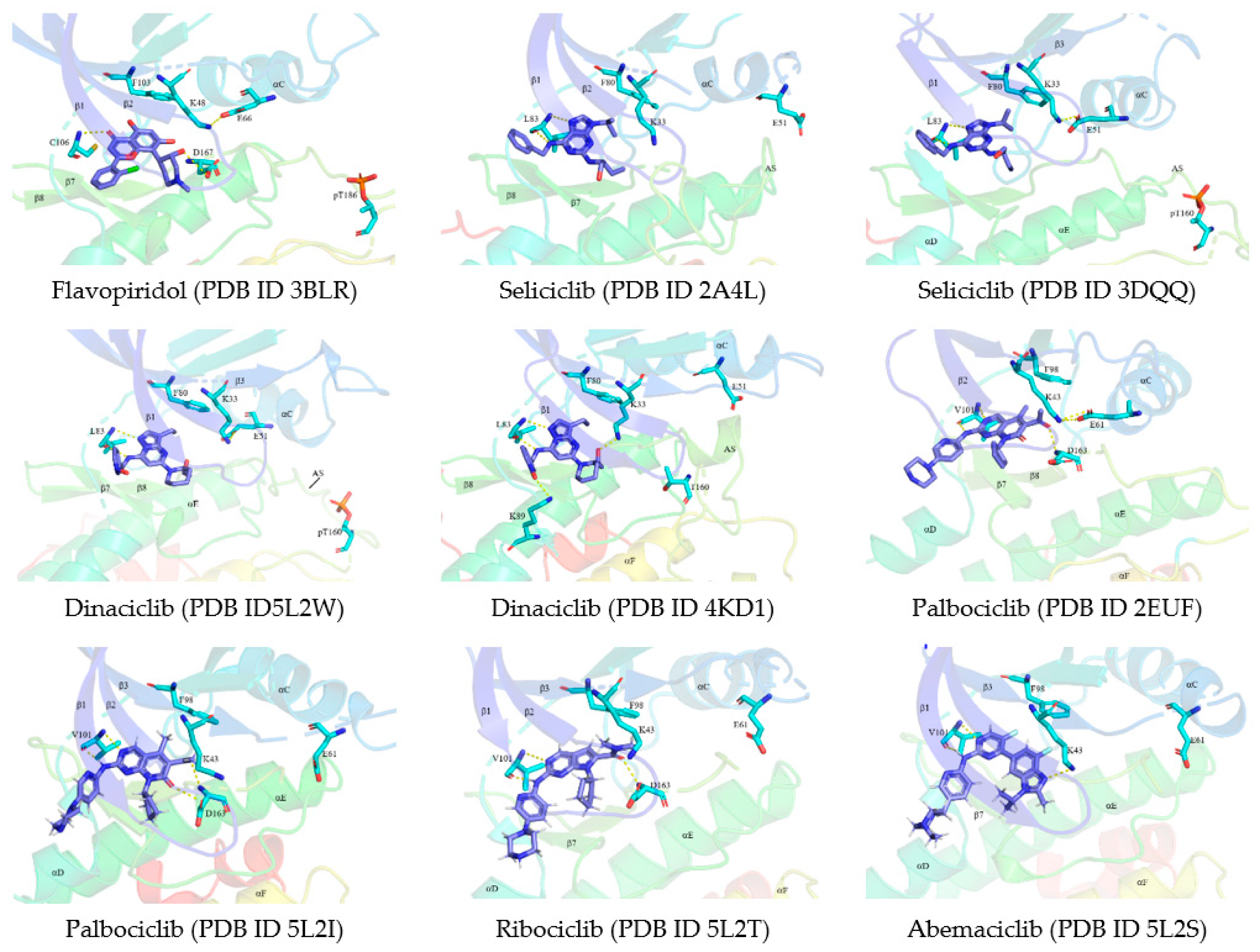
5. Protein Kinase–Drug Complex
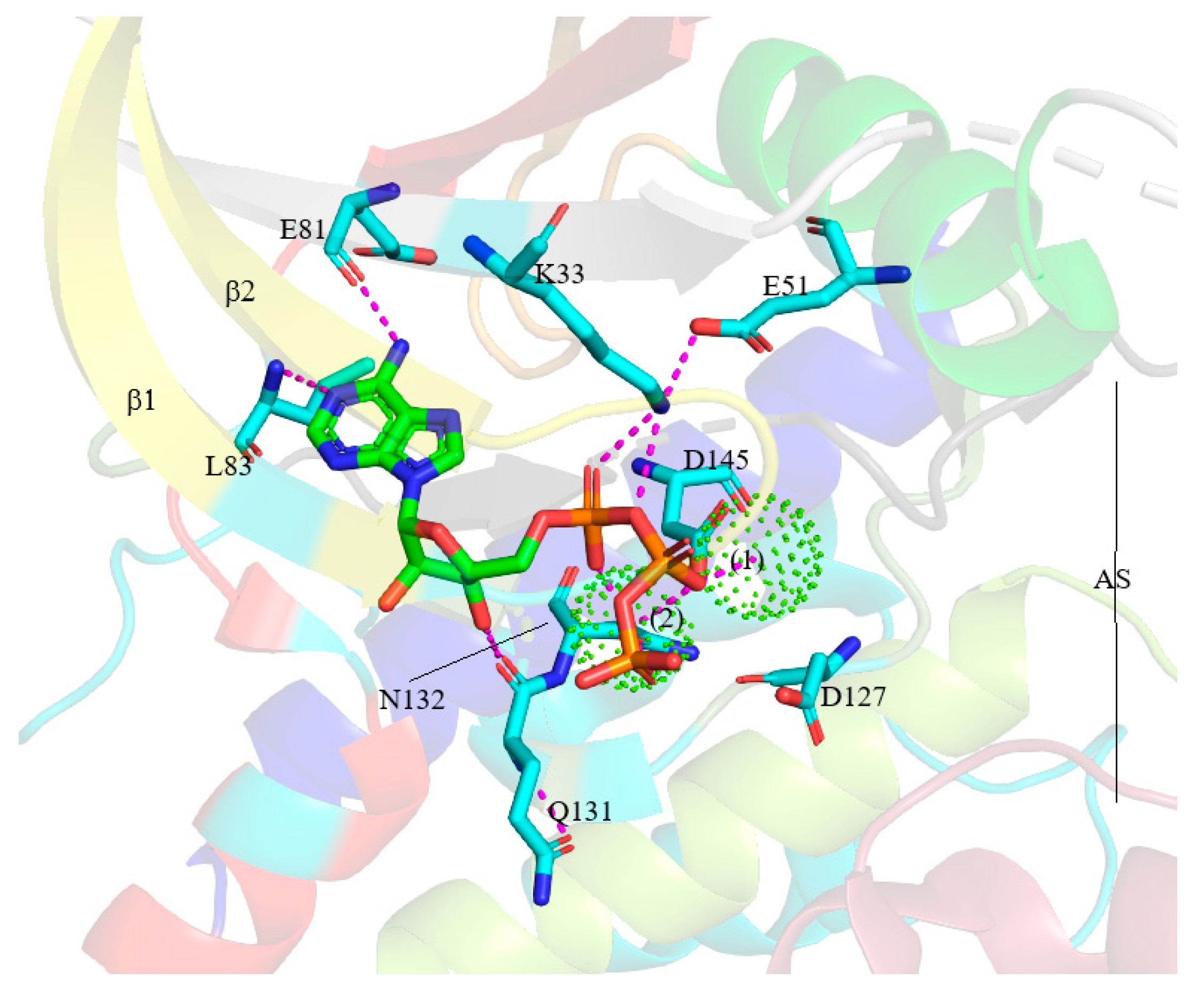
5.1. Binding Pocket within the Catalytic Cleft
5.2. Classification of Protein Kinase Inhibitors
6. Breast Cancer and Treatment
7. CDK Inhibitors
7.1. Pan CDK Inhibitors
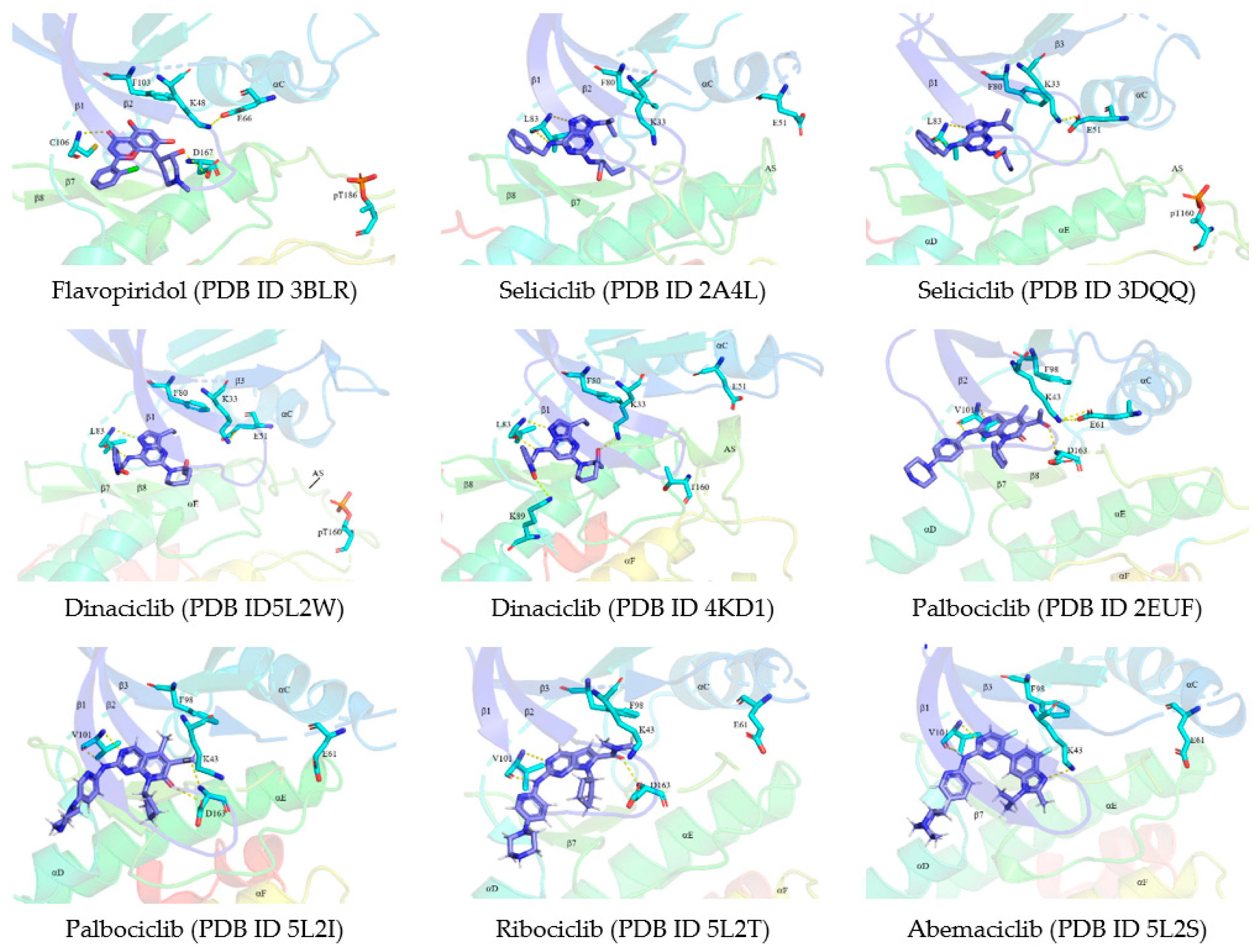
7.1.1. Flavopiridol/Alvocidib
7.1.2. Seliciclib/Roscovitine
7.1.3. Dinaciclib
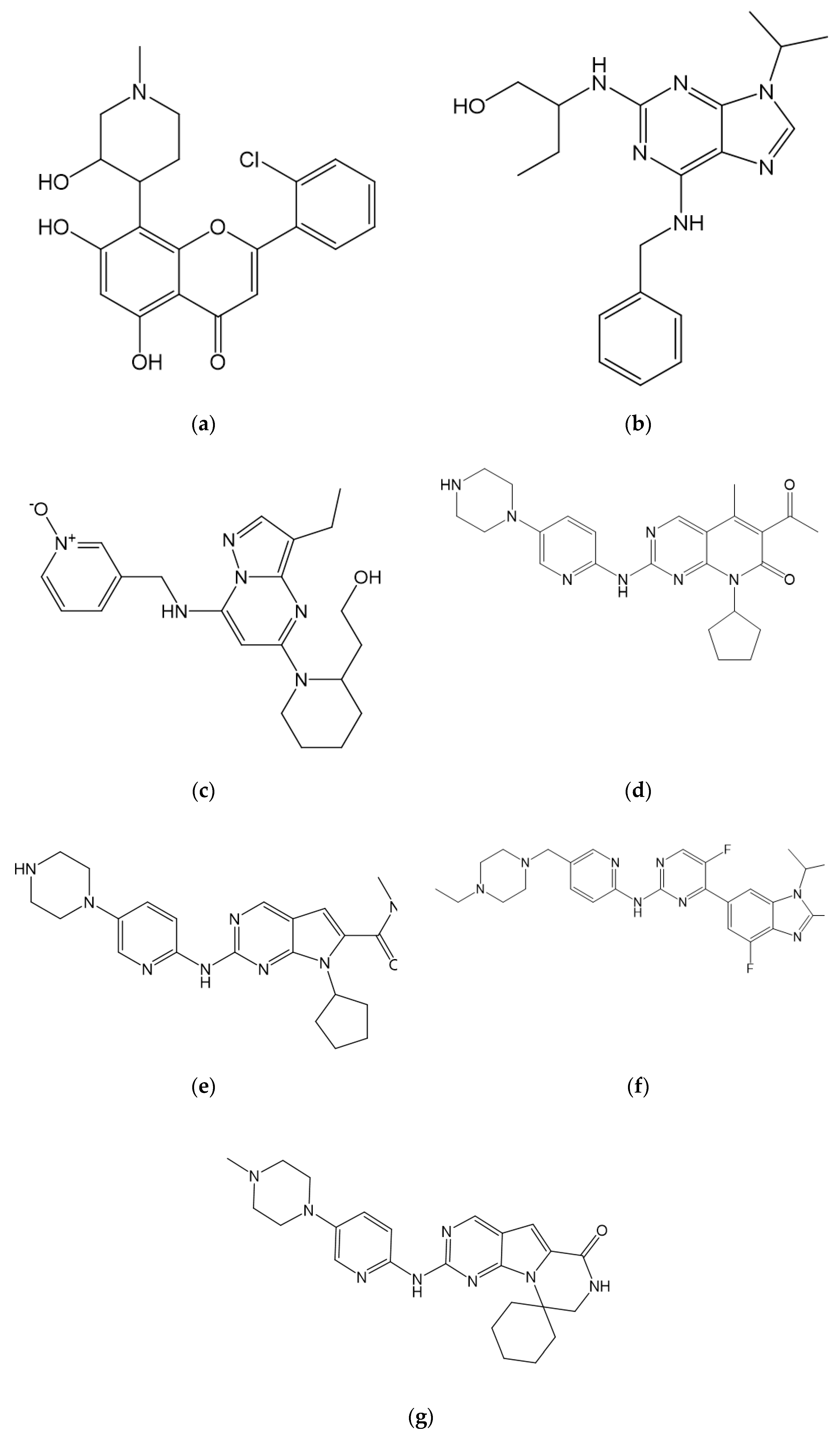
7.1.4. Other
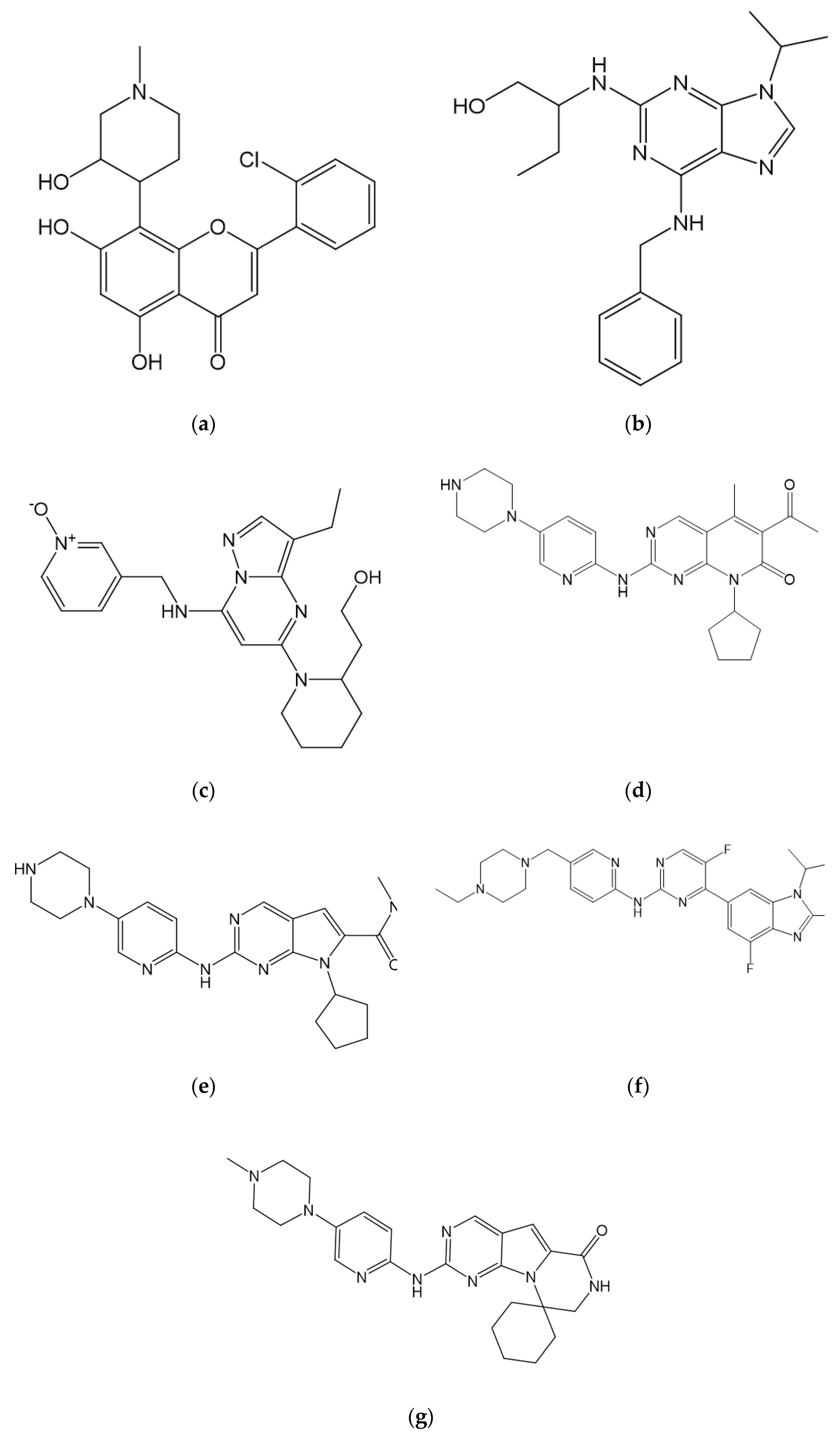
7.2. Selective CDK4/CDK6 Inhibitors
7.2.1. Palbociclib
7.2.2. Ribociclib
7.2.3. Abemaciclib
7.2.4. Trilaciclib
7.2.5. SHR6390
7.3. Other Novel CDK Inhibitors
8. CDK4/CDK6 Inhibitor Combinations
8.1. Combination with PI3K-mTOR Inhibitors
8.2. Combination with Immune Checkpoint Inhibitors
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stewart, Z.A. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181–182, 475–481. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar] [CrossRef]
- Ogino, S.; Fuchs, C.S.; Giovannucci, E. How many molecular subtypes? Implications of the unique tumor principle in personalized medicine. Expert Rev. Mol. Diagn. 2012, 12, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Galon, J.; Fuchs, C.S.; Dranoff, G. Cancer immunology—analysis of host and tumor factors for personalized medicine. Nat. Rev. Clin. Oncol. 2011, 8, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.; Chan, A.T.; Nishihara, R.; Fuchs, C.S.; Beck, A.H.; Giovannucci, E.; Ogino, S. Etiologic field effect: Reappraisal of the field effect concept in cancer predisposition and progression. Mod. Pathol. 2015, 28, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Milner, D.A., Jr.; Nishihara, R. Insights into pathogenic interactions among environment, host, and tumor at the crossroads of molecular pathology and epidemiology. Ann. Rev. Pathol. 2019, 14, 83–103. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Cyclin-dependent protein kinase inhibitors including palbociclib as anticancer drugs. Pharmacol. Res. 2016, 107, 249–275. [Google Scholar] [CrossRef]
- Gogineni, K.; DeMichele, A. Current approaches to the management of Her2-negative metastatic breast cancer. Breast Cancer Res. 2012, 14, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of estrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Rocca, A.; Schirone, A.; Maltoni, R.; Bravaccini, S.; Cecconetto, L.; Farolfi, A.; Bronte, G.; Andreis, D. Progress with palbociclib in breast cancer: Latest evidence and clinical considerations. Ther. Adv. Med. Oncol. 2017, 9, 83–105. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, a phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2- metastatic breast cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [Green Version]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Weiss, J.M.; Csoszi, T.; Maglakelidze, M.; Hoyer, R.J.; Beck, J.T.; Gomez, M.D.; Lowczak, A.; Aljumaily, R.; Lima, C.M.R.; Boccia, R.V.; et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: A phase Ib/randomized phase II trial. Ann. Oncol. 2019, 30, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.R.; Wright, G.S.; Thummala, A.R.; Danso, M.A.; Popovic, L.; Pluard, T.J.; Han, H.S.; Vonjović, Ž.; Vasev, N.; Ma, L.; et al. Trilaciclib plus chemotherapy versus chemotherapy alone in patients with metastatic triple-negative breast cancer: A multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2019, 20, 1587–1601. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Cyclin-dependent protein serin/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2018, 139, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Bockstaele, L.; Bisteau, X.; Paternot, S.; Roger, P.P. Differential regulation of cyclin-dependent kinase 4 (CDK4) and CDK6, evidence that CDK4 might not be activated by CDK7, and design of a CDK6 activating mutation. Mol. Cell. Biol. 2009, 29, 4188–4200. [Google Scholar] [CrossRef] [Green Version]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef]
- Echalier, A.; Endicott, J.A.; Noble, M.E. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim. Biophys. Acta 2010, 1804, 511–519. [Google Scholar] [CrossRef]
- Day, P.J.; Cleasby, A.; Tickle, I.J.; O’Reilly, M.; Coyle, J.E.; Holding, F.P.; McMenamin, R.L.; Yon, J.; Chopra, R.; Lengauer, C.; et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. USA 2009, 106, 4166–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaki, T.; Echalier, A.; Brown, N.R.; Hunt, T.; Endicott, J.A.; Noble, M.E. The structure of CDK4/cyclin D3 has implications for models of CDK activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4171–4176. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; James, M.K.; Larochelle, S.; Fisher, R.P.; Blain, S.W. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol. Cell. Biol. 2009, 29, 986–999. [Google Scholar] [CrossRef] [Green Version]
- Nolen, B.; Taylor, S.; Ghosh, G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell. 2004, 15, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Chen, F.; Yang, X.; Xu, W.; Xie, J.; Yu, L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol Biol. 2014, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Starostina, N.G.; Kipreos, E.T. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012, 22, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cánepa, E.T.; Scassa, M.E.; Ceruti, J.M.; Marazita, M.C.; Carcagno, A.L.; Sirkin, P.F.; Ogara, M.F. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life 2007, 59, 419–426. [Google Scholar] [CrossRef]
- LaPak, K.M.; Burd, C.E. The molecular balancing act of p16(INK4a) in cancer and aging. Mol. Cancer Res. 2014, 12, 167–183. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.L.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, E.; Bahadirli-Talbott, A.; Shih, I.M. Frequent CCNE1 amplification in endometrial intraepithelial carcinoma and uterine serous carcinoma. Mod. Pathol. 2014, 27, 1014–1019. [Google Scholar] [CrossRef]
- Drexler, H.G. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 1998, 12, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, C.; Wang, X.; Becker, D. Expression analysis and molecular targeting of cyclin dependent kinases in advanced melanoma. Cell Cycle 2011, 10, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.Y.; Auerbach, A.; D’Costa, A.M.; Rapoport, A.P.; Burger, A.M.; Sausville, E.A.; Stass, S.A.; Jiang, F.; Sands, A.M.; Aguilera, N.; et al. Phospho-p70S6K/p85S6K and cdc2/cdk1 are novel targets for diffuse large B-cell lymphoma combination therapy. Clin. Cancer Res. 2009, 15, 1708–1720. [Google Scholar] [CrossRef] [Green Version]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- An, H.X.; Beckmann, M.W.; Reifenberger, G.; Bender, H.G.; Niederacher, D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am. J. Pathol. 1999, 154, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Mendrzyk, F.; Radlwimmer, B.; Joos, S.; Kokocinski, F.; Benner, A.; Stange, D.E.; Neben, K.; Fiegler, H.; Carter, N.P.; Reifenberger, G.; et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J. Clin. Oncol. 2005, 23, 8853–8862. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Jacobsen, D.M.; Bao, Z.Q.; O’Brien, P.; Brooks III, C.L.; Young, M.A. Price to be paid for two-metal catalysis: Magnesium ions that accelerate chemistry unavoidably limit product release from a protein kinase. J. Am. Chem. Soc. 2010, 134, 15357–15370. [Google Scholar] [CrossRef]
- Liao, J.J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrin, L.K.; Saiah, E. Approaches to discover non-ATP site inhibitors. Med. Chem. Commun. 2013, 4, 41–51. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef]
- Kwarcinski, F.E.; Brandvold, K.R.; Phadke, S.; Beleh, O.M.; Johnson, T.K.; Meagher, J.L.; Seeliger, M.A.; Stuckey, J.A.; Soellner, M.B. Conformation-selective analogues of dasatinib reveal insight into kinase inhibitor binding and selectivity. ACS Chem. Biol. 2016, 11, 1296–1304. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Ghosh, I. New directions in targeting protein kinases: Focusing upon true allosteric and bivalent inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, J.Y.S.; Tse, G.M. Molecular Classification of Breast Cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American society of clinical oncology/college of american pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef]
- Ethier, J.L.; Ocana, A.; Lescure, R.A.; Ruiz, A.; Alba, E.; Calvo, L.; Ruíz-Borrego, M.; Santaballa, S.; Rodríguez, C.A.; Crespo, C.; et al. Outcomes of single versus double hormone receptor-positive breast cancer. A geicam/9906 sub-study. Eur. J. Cancer. 2018, 94, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Cardoso, F.; Piccart-Gebhart, M.J. Stemming resistance to her-2 targeted therapy. J. Mammary Gland. Biol. Neoplasia 2009, 14, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Yam, C.; Mani, S.A.; Moulder, S.L. Targeting the molecular subtypes of triple negative breast cancer: Understanding the diversity to progress the field. Oncologist 2017, 22, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Pernas, S.; Tolaney, S.M.; Winer, E.P.; Goel, S. CDK4/6 inhibition in breast cancer: Current practice and future directions. Ther. Adv. Med. Oncol. 2018, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Chohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.; Sonke, A.G.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as first-line therapy for HR positive, advanced breast cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.; Manso, L.; et al. MONARCH 3: Abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Bose, P.; Simmons, G.L.; Grant, S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin. Investig. Drugs 2013, 22, 723–738. [Google Scholar] [CrossRef]
- Tourneau, C.L.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclindependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef]
- Benson, C.; White, J.; Bono, D.J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J.J.; Small, K.A.; Kirschmeier, P.; Zhang, D.; Zhu, Y.; Jou, Y.; Statkevich, P.; Yao, S.; Bannerji, R. A first-in-human, Phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J. Transl. Med. 2013, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Blachly, J.S.; Byrd, J.C. Emerging drug profile: Cyclin-dependent kinase inhibitors. Leuk. Lymphoma 2013, 54, 2133–2143. [Google Scholar] [CrossRef]
- Mita, M.M.; Joy, A.A.; Mita, A.; Sankhala, K.; Jou, Y.; Zhang, D.; Statkevich, P.; Zhu, Y.; Yao, S.; Small, K.; et al. Randomized Phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin. Breast Cancer 2014, 14, 169–176. [Google Scholar] [CrossRef]
- Tong, W.G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef]
- Heath, E.I.; Bevara, G.B.; Kumar, A.N.D.; Koteshwaramma, K.L.; Badana, A.; Kumari, S.; Malla, R.R. C-glycosyl flavone from Urginea indica inhibits proliferation & angiogenesis & induces apoptosis via cyclin-dependent kinase 6 in human breast, hepatic & colon cancer cell lines. Indian J. Med. Res. 2018, 147, 158–168. [Google Scholar] [CrossRef]
- Liu, D.; You, P.; Luo, Y.; Yang, M.; Liu, Y. Galangin induces apoptosis in MCF-7 human breast cancer cells through mitochondrial pathway and phosphatidylinositol 3-kinase/Akt inhibition. Pharmacology 2018, 102, 58–66. [Google Scholar] [CrossRef]
- Jeong, C.H.; Ryu, H.; Kim, D.H.; Cheng, W.N.; Yoon, J.E.; Kang, S.; Han, S.G. Piperlongumine induces cell cycle arrest via reactive oxygen species accumulation and IKKbeta suppression in human breast cancer cells. Antioxidants 2019, 8, 553. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Tan, S.; Duan, F.; Yuan, Q.; Li, Q.; Deng, G. Icariin induces apoptosis by suppressing autophagy in tamoxifen-resistant breast cancer cell line MCF-7/TAM. Breast Cancer 2019, 26, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Jiang, H.; Chen, Y.; Ren, F. Resveratrol chemosensitizes adriamycin resistant breast cancer cells by modulating miR-122-5p. J. Cell Biochem. 2019, 120, 16283–16292. [Google Scholar] [CrossRef]
- Chen, J.; Ko, J.; Kim, J.T.; Cho, J.S.; Qiu, S.; Kim, G.D. β-Thujaplicin inhibits basal-like mammary tumor growth by regulating glycogen synthase kinase-3β/β-catenin signaling. Food Funct. 2019, 10, 2691–2700. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming therapeutic resistance in HER2 positive breast cancers with CDK4/6 inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-cell dynamics determines response to CDK4/6 inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, Z.L.; Versaci, S.; Dushyanthen, S.; Caramia, F.; Savas, P.; Mintoff, C.P.; Zethoven, M.; Virassamy, B.; Luen, S.J.; McArthur, G.A.; et al. Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017, 77, 6340–6352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabre, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 inhibition augments antitumor immunity by enhancing T-cell activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugo, H.S.; Kabos, P.; Dickler, M.N.; John, W.J.; Smith, I.; Lu, Y.; Young, S.; Tolaney, S.M. A phase 1b study of abemaciclib plus pembrolizumab for patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2 negative (HER2) metastatic breast cancer (MBC). Cancer Res. 2018, 78, P1-09-01. [Google Scholar] [CrossRef]
- Rugo, H.S.; Kabos, P.; Beck, J.T.; Chisamore, M.J.; Hossain, A.; Chen, Y.; Tolaney, S.M. A phase Ib study of abemaciclib in combination with pembrolizumab for patients with hormone receptor positive (HR+), human epidermal growth factor receptor 2 negative (HER2-) locally advanced or metastatic breast cancer (MBC) (NCT02779751): Interim results. J. Clin. Oncol. 2020, 38, 1051. [Google Scholar] [CrossRef]
- Heath, E.I.; Bible, K.; Martell, R.E.; Adelman, D.C.; LoRusso, P.M. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Investig. New Drugs 2008, 26, 59–65. [Google Scholar] [CrossRef]
- Byth, K.F.; Thomas, A.; Hughes, G.; Folder, C.; McGregor, A.; Geh, C.; Oakes, S.; Green, C.; Walker, M.; Newcombe, N.; et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 2009, 8, 1856–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boss, D.S.; Schwartz, G.K.; Middleton, M.R.; Amakye, D.D.; Swaisland, H.; Midgley, R.S.; Ranson, M.; Danson, S.; Calvert, H.; Plummer, R.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of the oral cyclindependent kinase inhibitor AZD5438 when administered at intermittent and continuous dosing schedules in patients with advanced solid tumours. Ann. Oncol. 2010, 21, 884–894. [Google Scholar] [CrossRef]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar]
- VanderWel, S.N.; Harvey, P.J.; McNamara, D.J.; Repine, T.J.; Keller, P.R.; Quin, J., 3rd; Booth, R.J.; Elliott, W.L.; Dobrusin, E.M.; Fry, D.W.; et al. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J. Med. Chem. 2005, 48, 2371–2387. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-dependent kinases 4/6 inhibitors in breast cancer: Current status, resistance, and combination strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; Laurentiis, M.D.; Im, S.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: MONALEESA-3. J. Clin. Oncol. 2018, 36, 2465–2472. [Google Scholar] [CrossRef]
- Tripathy, D.; Im, S.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Prado, M.D.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle dependent/independent antitumor activities alone/in combination with gemcitabine. Investg. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; Dios, A.D. Brain exposure of two selective dual CDK4 and CDK6 inhibitors and the antitumor activity of CDK4 and CDK6 inhibition in combination with temozolomide in an intracranial glioblastoma xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [Green Version]
- Tolaney, S.M.; Lin, N.U.; Thornton, D.; Klise, S.; Costigan, T.M.; Turner, P.K.; Anders, C.K. Abemaciclib for the treatment of brain metastases (BM) secondary to hormone receptor positive (HR+), HER2 negative breast cancer. J. Clin. Oncol. 2017, 35, 1019. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Carezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases CDK4 and CDK6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, M.R.; Krishnamurthy, J.; Pei, X.H.; Torrice, C.; Lin, W.; Carrasco, D.R.; Ligon, K.L.; Xiong, Y.; Sharpless, N.E. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6-dependent tumors and tissues. Cancer Res. 2007, 67, 4732–4741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Roberts, P.J.; Sorrentino, J.A.; Bisi, J.E.; Storrie-Whiteet, H.; Tiessen, R.G.; Makhuli, K.M.; Wargin, W.A.; Tadema, H.; Hoogdalem, E.V.; et al. Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci. Transl. Med. 2017, 9, eaal3986. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; He, Y.; Fu, H.; Li, Y.; Bao, X.; Wang, Q.; Wang, Y.; Xie, C.; Lou, L. Preclinical characterization of SHR6390, a novel CDK 4/6 inhibitor, in vitro and in human tumor xenograft models. Cancer Sci. 2019, 110, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xu, B.; Gui, L.; Wang, W.; Xiu, M.; Zhang, X.; Sun, G.; Zhu, X.; Zou, J. A phase I study of SHR6390, a cyclin-dependent kinase 4/6 inhibitor in patients with advance breast cancer (ABC). J. Clin. Oncol. 2020, 38, 1095. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Cao, Y.; Zhou, P.; Gui, S.; Wu, X.; Xia, Y.; Tu, J. Panduratin A inhibits cell proliferation by inducing G0/G1 phase cell cycle arrest and induces apoptosis in breast cancer cells. Biomol. Ther. 2018, 26, 328–334. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Susanti, N.M.P.; Tjahjono, D.H. Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment. Molecules 2021, 26, 4462. https://doi.org/10.3390/molecules26154462
Susanti NMP, Tjahjono DH. Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment. Molecules. 2021; 26(15):4462. https://doi.org/10.3390/molecules26154462
Chicago/Turabian StyleSusanti, Ni Made Pitri, and Daryono Hadi Tjahjono. 2021. "Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment" Molecules 26, no. 15: 4462. https://doi.org/10.3390/molecules26154462
APA StyleSusanti, N. M. P., & Tjahjono, D. H. (2021). Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment. Molecules, 26(15), 4462. https://doi.org/10.3390/molecules26154462