
Magnetism and Luminescence of a MOF with Linear Mn3 Nodes Derived from an Emissive Terthiophene-Based Imidazole Linker †

 ,
,
Abstract
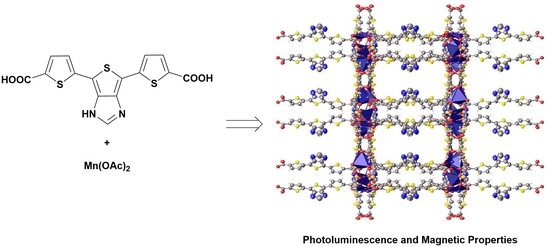
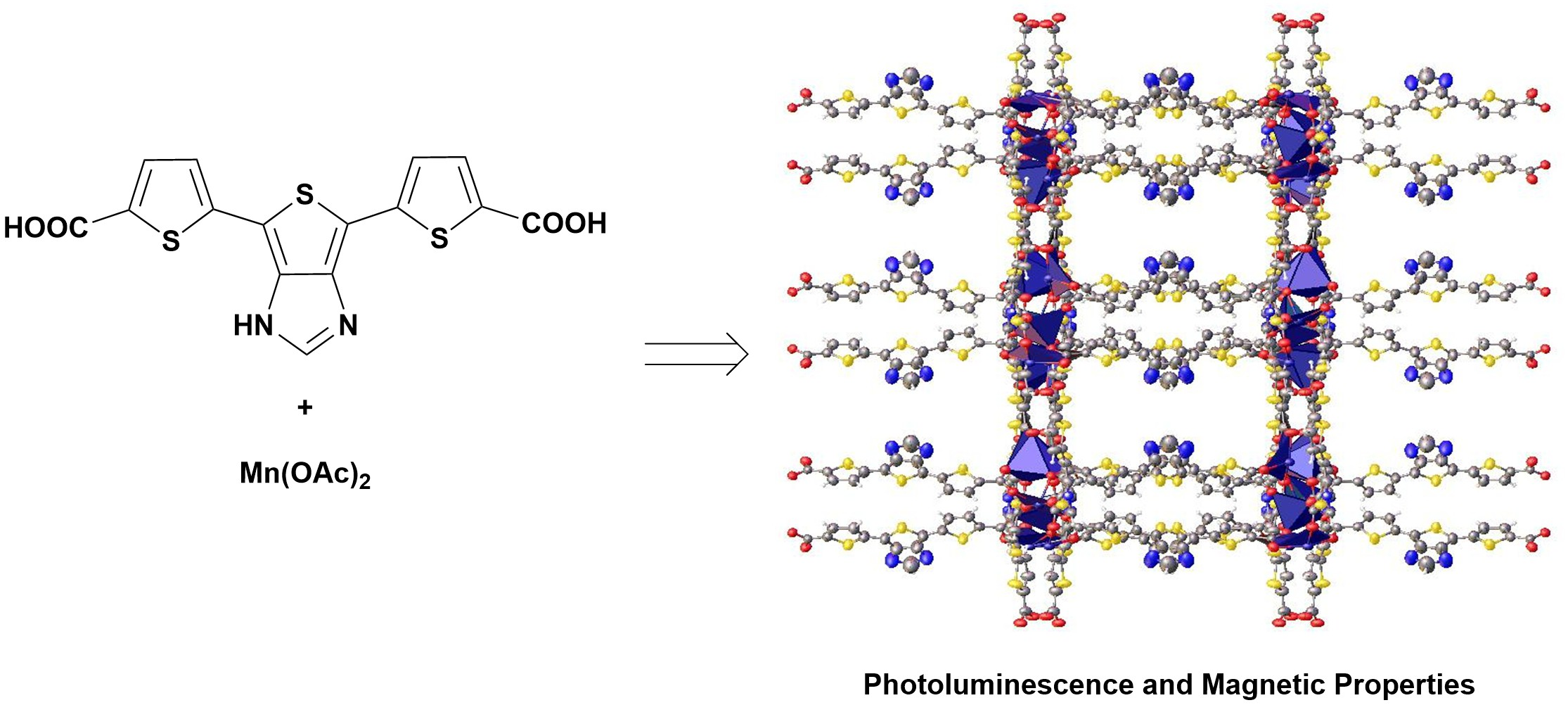
:
{kind=link}
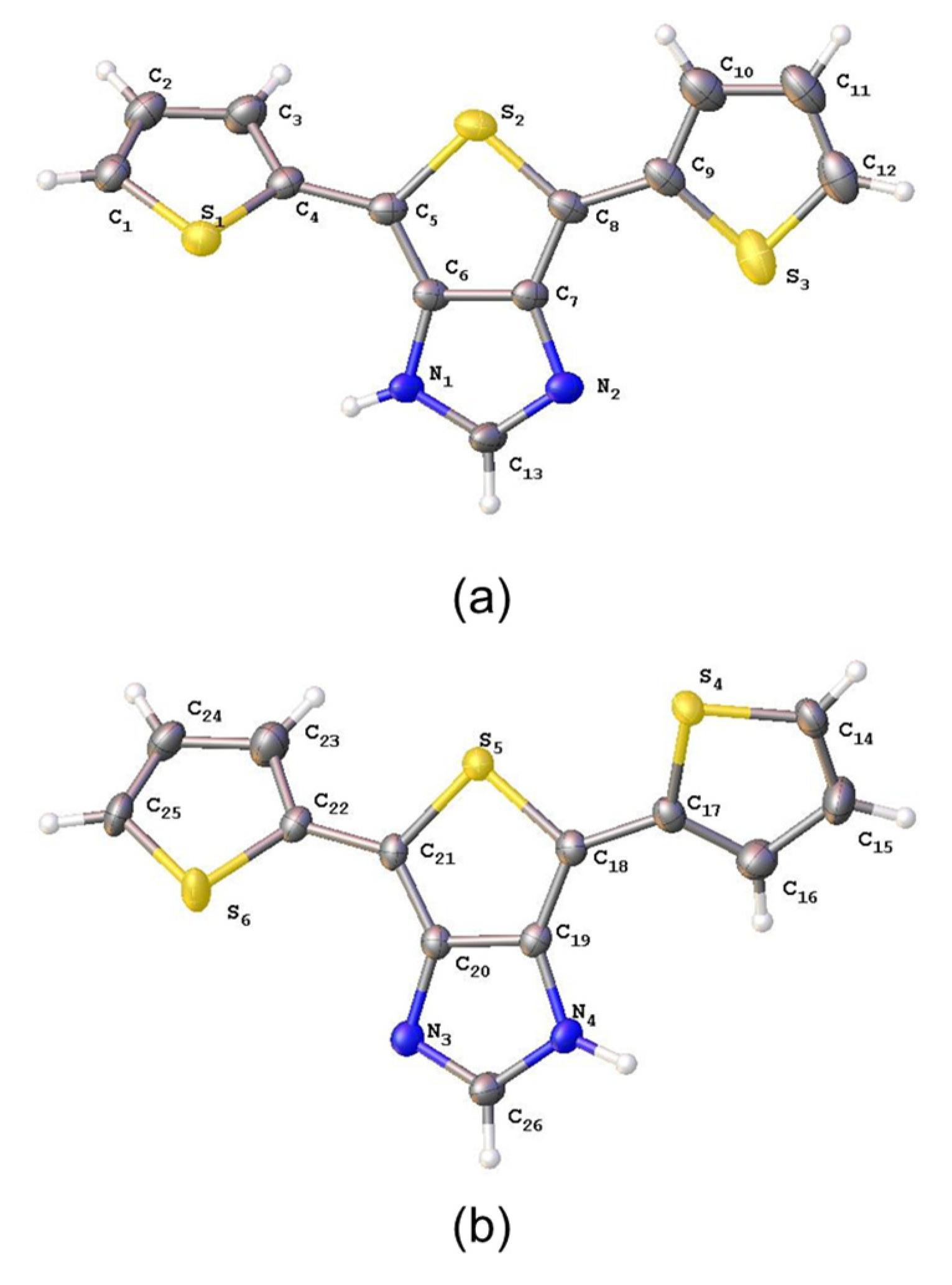{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
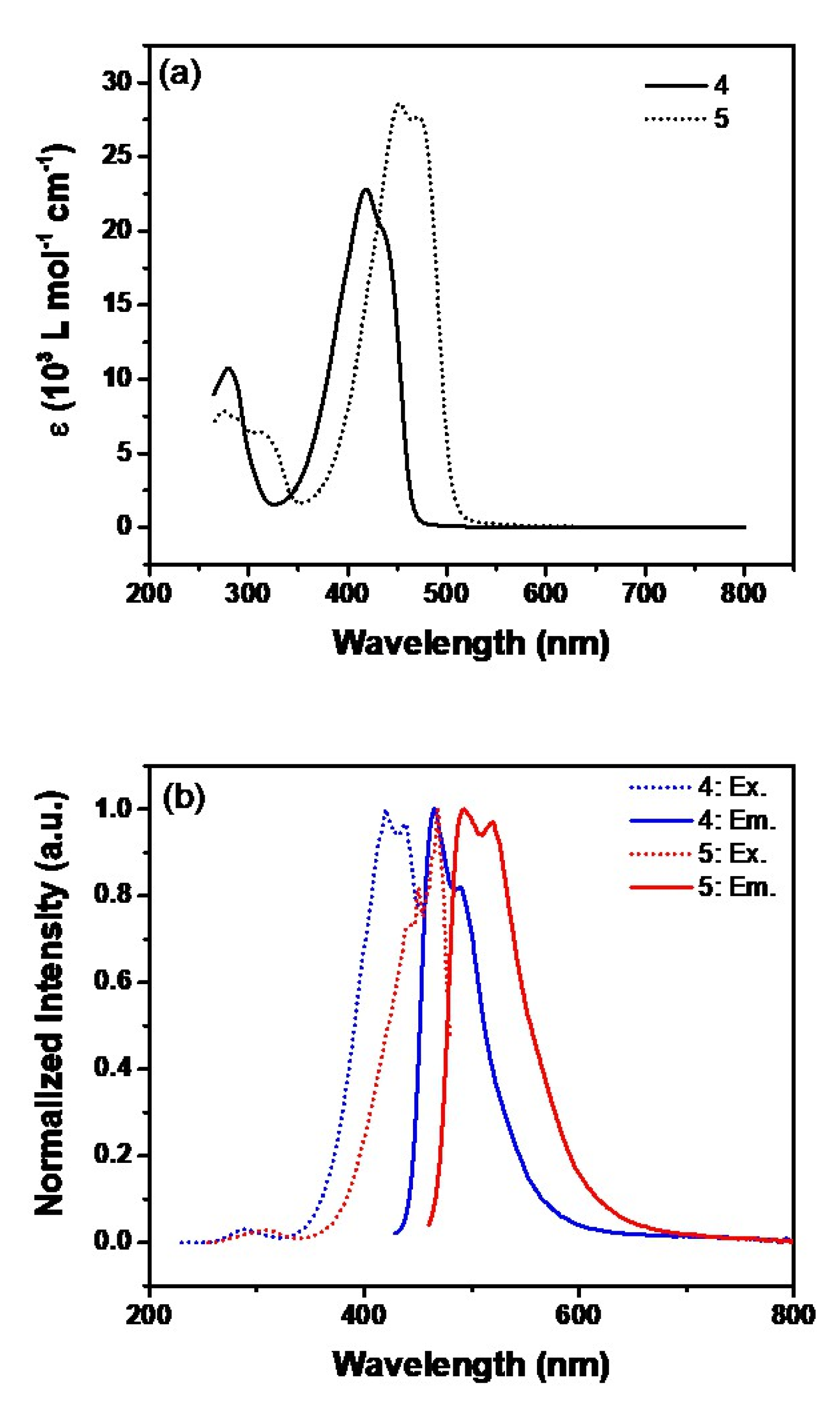
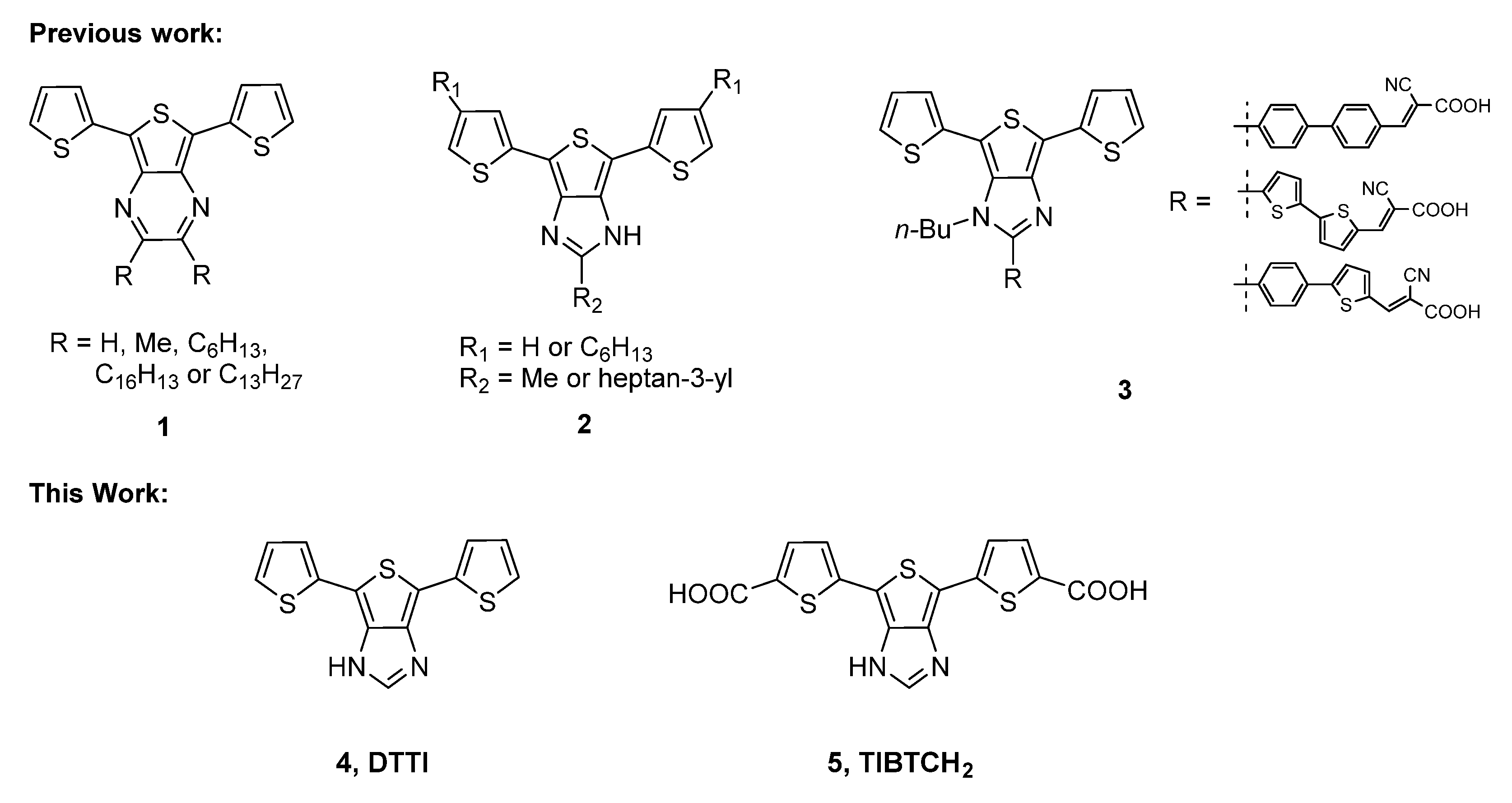
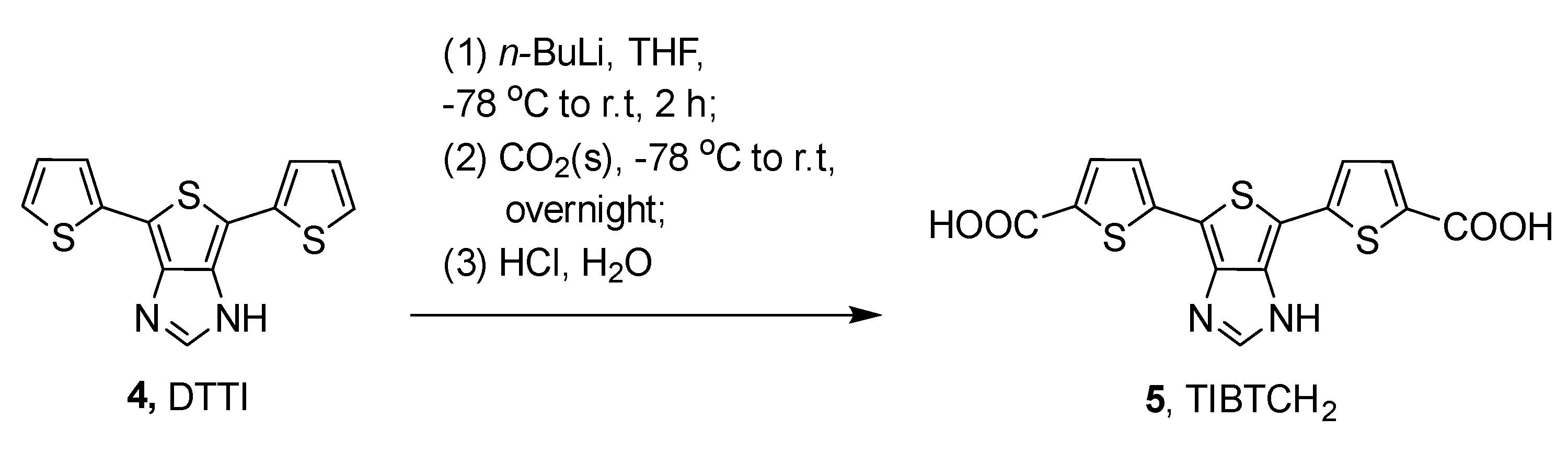
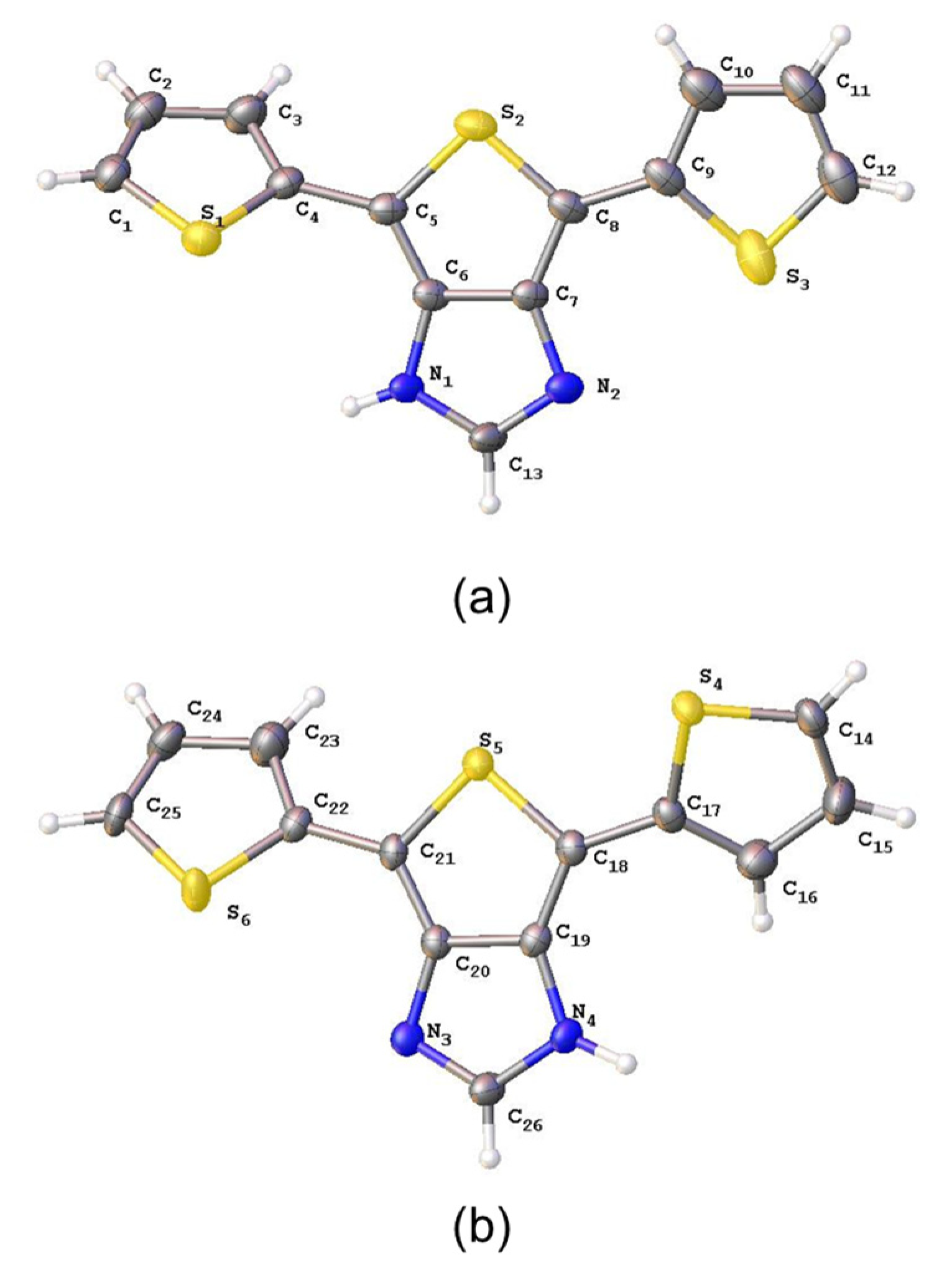
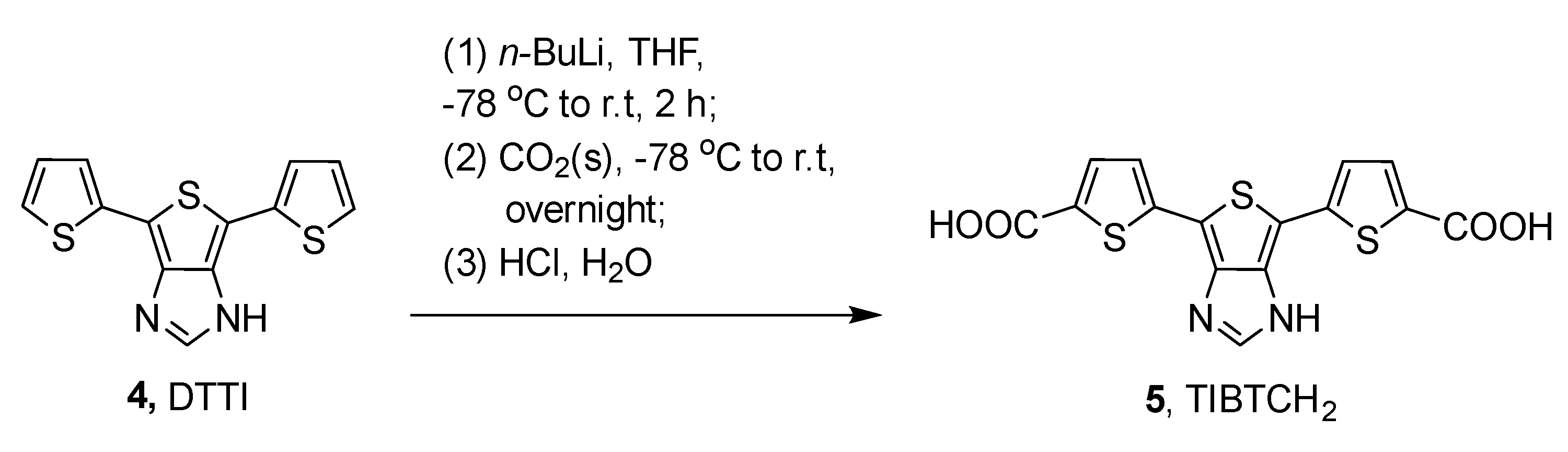
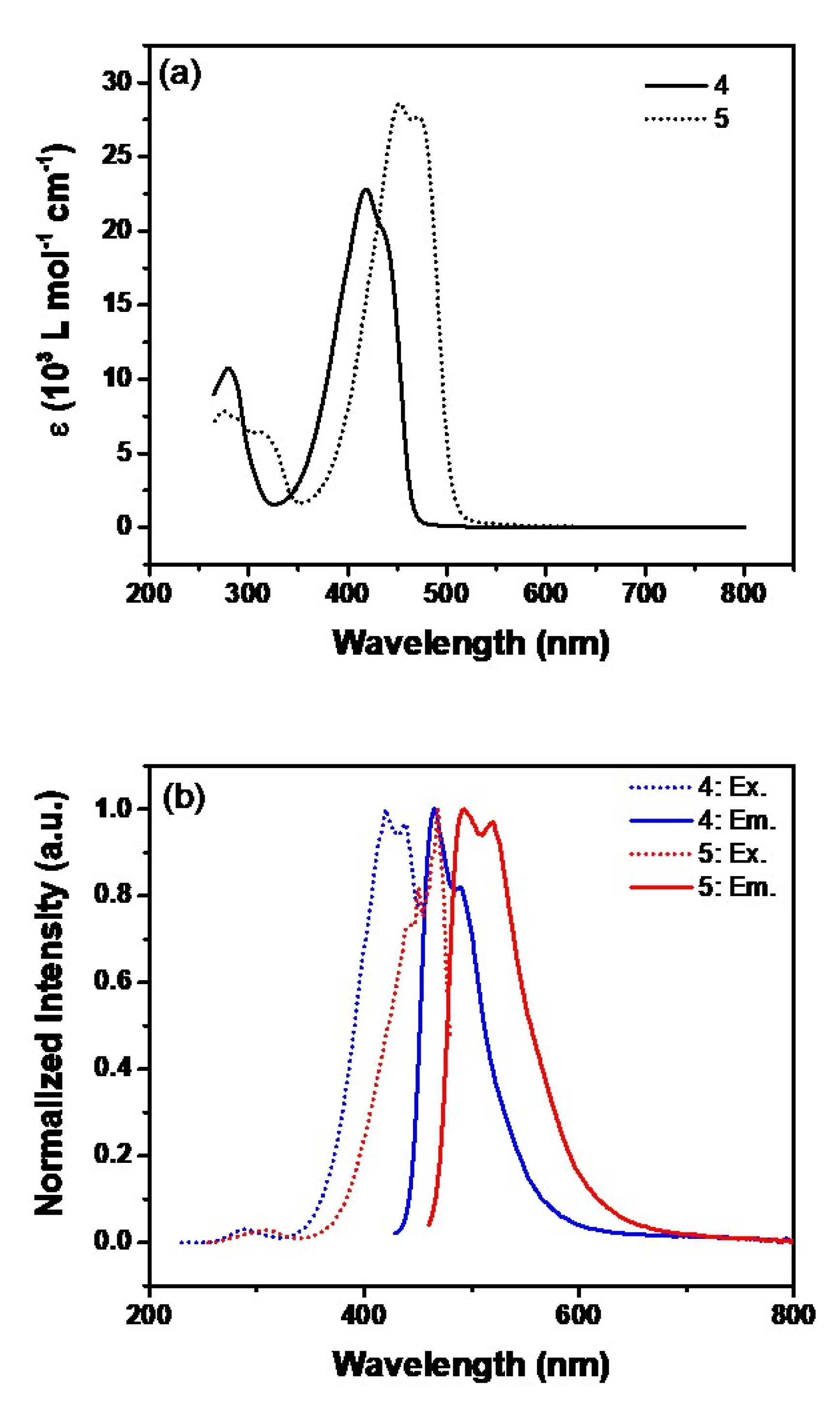
2.1. Synthesis and Photophysical Studies of New Luminophores
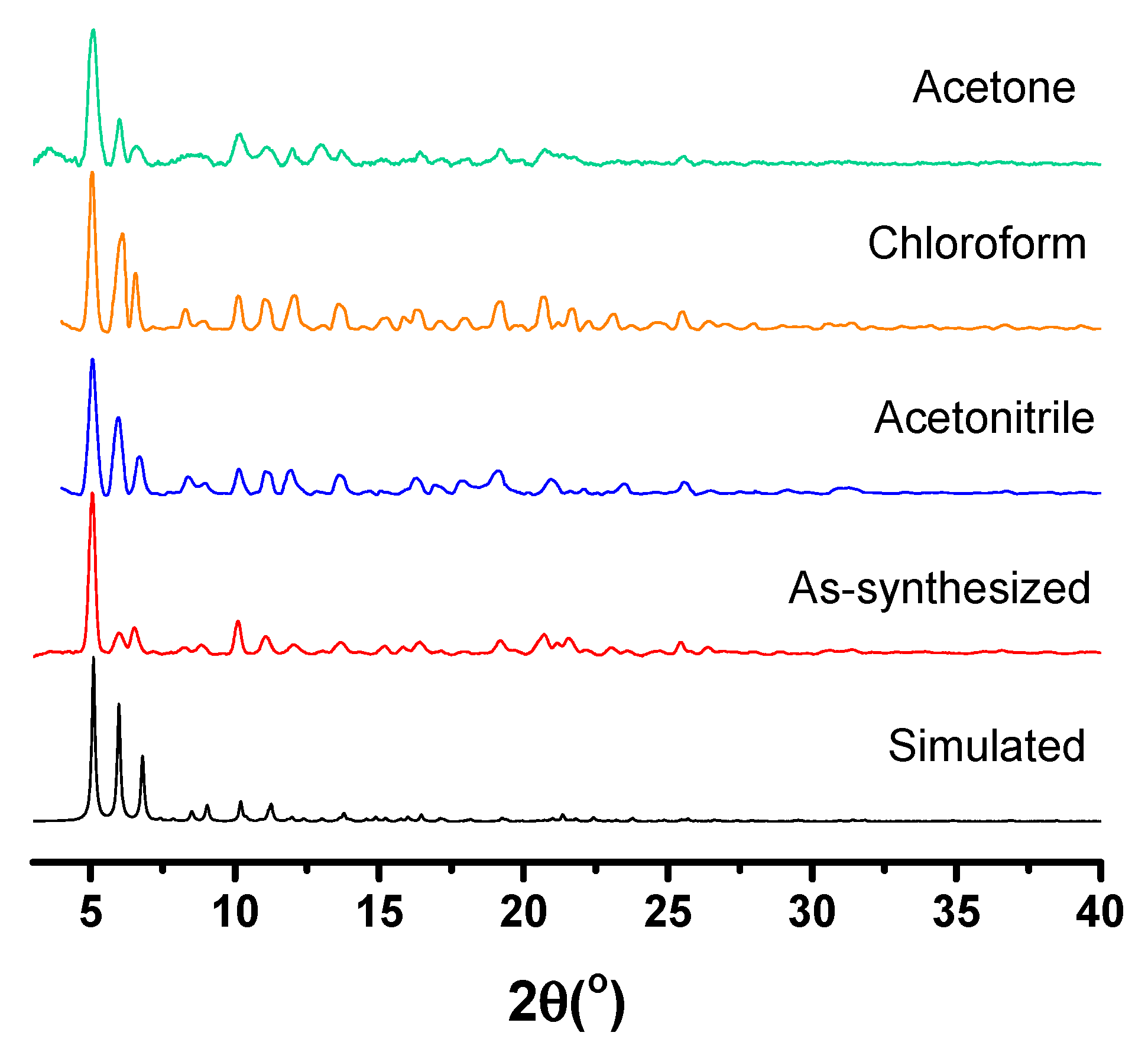
2.2. Synthesis and Characterization of 6
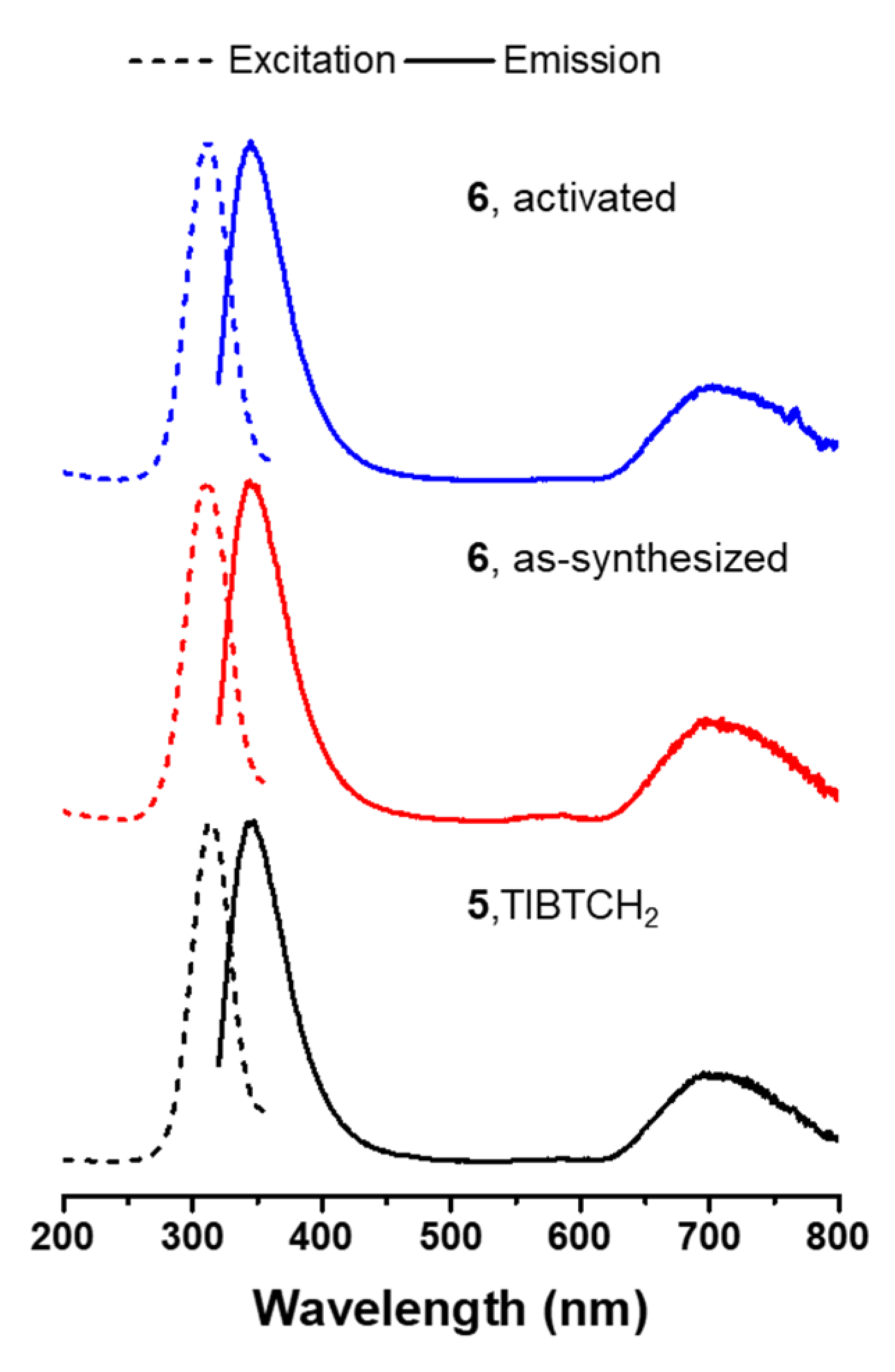
2.3. Photoluminescent Properties of 6
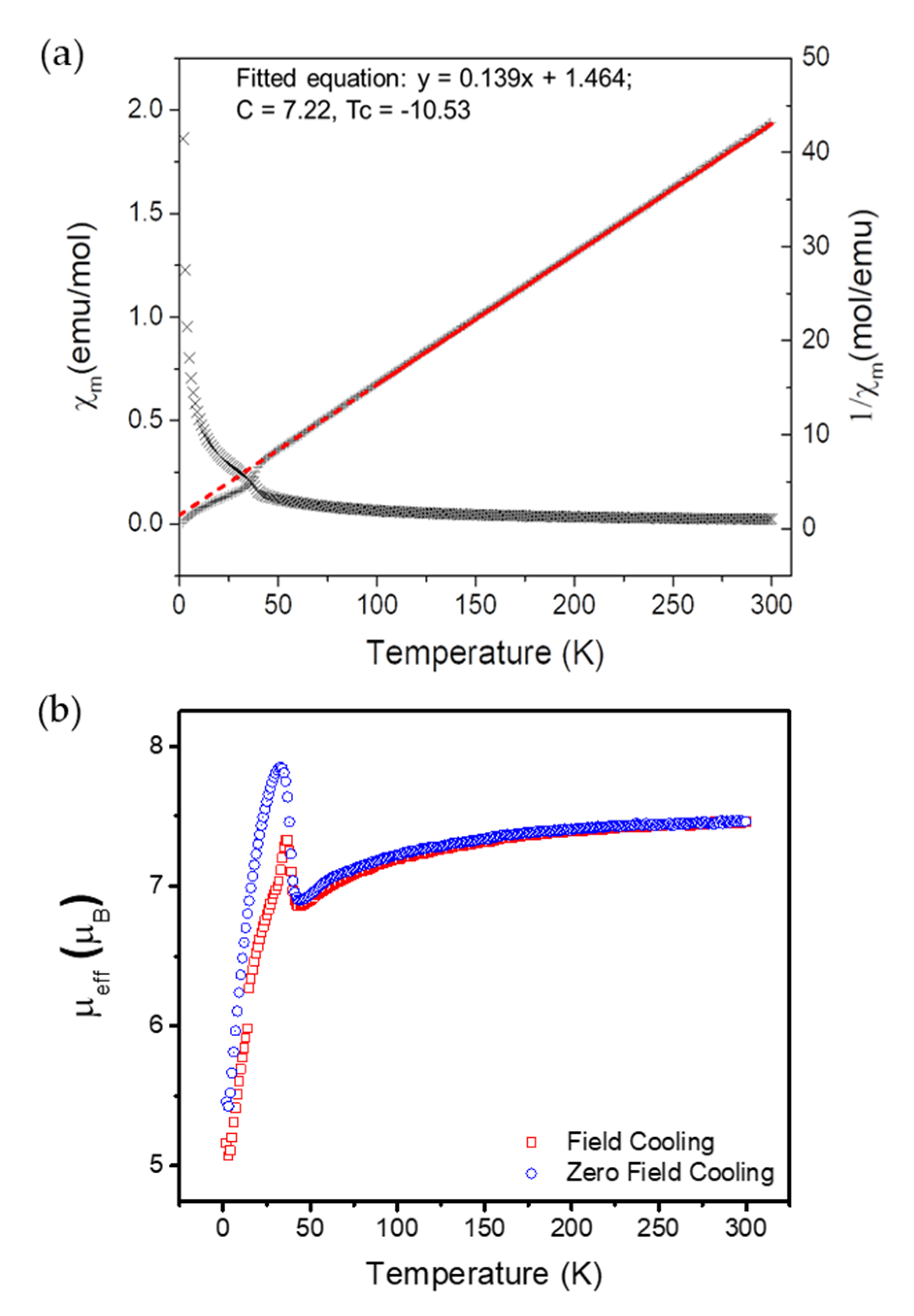
2.4. Magnetic Properties of 6
3. Conclusions
4. Experimental Section
4.1. General Methods
4.2. X-ray Diffraction
4.3. Compound Synthesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.K.; Houk, R.J. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Deibert, B.J.; Li, J. Luminescent metal–organic frameworks for chemical sensing and explosive detection. Chem. Soc. Rev. 2014, 43, 5815–5840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, S.G.; Nuñez, A.J.; Moore, M.D.; Steiner, A.; Lynch, V.M.; Sessler, J.L.; Holliday, B.J.; Humphrey, S.M. A Sensor for Trace H2O Detection in D2O. Chem 2017, 2, 579–589. [Google Scholar] [CrossRef]
- Lustig, W.P.; Mukherjee, S.; Rudd, N.D.; Desai, A.V.; Li, J.; Ghosh, S.K. Metal–organic frameworks: Functional luminescent and photonic materials for sensing applications. Chem. Soc. Rev. 2017, 46, 3242–3285. [Google Scholar] [CrossRef]
- Espallargas, G.M.; Coronado, E. Magnetic functionalities in MOFs: From the framework to the pore. Chem. Soc. Rev. 2018, 47, 533–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Campbell, M.G.; Dincă, M. Electrically Conductive Porous Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2016, 55, 3566–3579. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Wong, N.; Shimizu, G.K.H. MOFs as proton conductors—Challenges and opportunities. Chem. Soc. Rev. 2014, 43, 5913–5932. [Google Scholar] [CrossRef]
- Bao, S.-S.; Shimizu, G.K.; Zheng, L.-M. Proton conductive metal phosphonate frameworks. Coord. Chem. Rev. 2019, 378, 577–594. [Google Scholar] [CrossRef]
- Lee, E.Y.; Jang, A.S.Y.; Suh, M.P. Multifunctionality and Crystal Dynamics of a Highly Stable, Porous Metal−Organic Framework [Zn4O(NTB)2]. J. Am. Chem. Soc. 2005, 127, 6374–6381. [Google Scholar] [CrossRef]
- Bauer, C.A.; Timofeeva, T.V.; Settersten, T.B.; Patterson, B.D.; Liu, V.H.; Simmons, B.; Allendorf, M.D. Influence of Connectivity and Porosity on Ligand-Based Luminescence in Zinc Metal−Organic Frameworks. J. Am. Chem. Soc. 2007, 129, 7136–7144. [Google Scholar] [CrossRef]
- Bauer, C.A.; Jones, S.C.; Kinnibrugh, T.L.; Tongwa, P.; Farrell, R.A.; Vakil, A.; Timofeeva, T.V.; Khrustalev, V.N.; Allendorf, M.D. Homo- and heterometallic luminescent 2-D stilbene metal–organic frameworks. Dalton Trans. 2014, 43, 2925–2935. [Google Scholar] [CrossRef]
- Pham, B.T.; Lund, L.M.; Song, D. Novel luminescent metal-organic frameworks [Eu2L3(DMSO)2(MeOH)2] x 2 DMSO x 3 H2O and [Zn2L2(DMSO)2] x 1.6 H2O (L = 4, 4’-ethyne-1, 2-diyldibenzoate). Inorg. Chem. 2008, 47, 6329–6335. [Google Scholar] [CrossRef] [PubMed]
- Shustova, N.; McCarthy, B.D.; Dinca, M. Turn-On Fluorescence in Tetraphenylethylene-Based Metal–Organic Frameworks: An Alternative to Aggregation-Induced Emission. J. Am. Chem. Soc. 2011, 133, 20126–20129. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, O. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nat. Cell Biol. 1999, 402, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Rosi, N.L.; Eckert, J.; Eddaoudi, M.; Vodak, D.T.; Kim, J.; O’Keeffe, M.; Yaghi, O.M. Hydrogen Storage in Microporous Metal-Organic Frameworks. Science 2003, 300, 1127–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaate, A.; Roy, P.; Godt, A.; Lippke, J.; Waltz, F.; Wiebcke, M.; Behrens, P. Modulated Synthesis of Zr-Based Metal-Organic Frameworks: From Nano to Single Crystals. Chemistry 2011, 17, 6643–6651. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; He, D.; Zhang, W.; Xiao, Z.; Zuo, Q.; Shi, Z.; Ding, L. Synthesis, characterization and photovoltaic properties of conjugated copolymers based on 2-alkyl-thieno[3,4-b]imidazole. Synth. Met. 2012, 162, 1694–1700. [Google Scholar] [CrossRef]
- Karthik, D.; Kumar, V.; Thomas, K.R.J.; Li, C.-T.; Ho, K.-C. Synthesis and characterization of thieno[3,4-d]imidazole-based organic sensitizers for photoelectrochemical cells. Dye. Pigment. 2016, 129, 60–70. [Google Scholar] [CrossRef]
- Wang, W.; Guo, H.; Jones, R.A. Synthesis and electropolymerization of N-heterocyclic carbene complexes of Pd (ii) and Pt (ii) from an emissive imidazolium salt with a terthiophene backbone. Dalton Trans. 2019, 48, 14440–14449. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, C.; Tanaka, S.; Yamashita, Y. Design of Narrow-Bandgap Polymers. Syntheses and Properties of Monomers and Polymers Containing Aromatic-Donor ando-Quinoid-Acceptor Units. Chem. Mater. 1996, 8, 570–578. [Google Scholar] [CrossRef]
- Gagné, O.C.; Hawthorne, F. Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2015, 71, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junta, J.L.; Hochella, M.F. Manganese (II) oxidation at mineral surfaces: A microscopic and spectroscopic study. Geochim. Cosmochim. Acta 1994, 58, 4985–4999. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Ladrak, T.; Smulders, S.; Roubeau, O.; Teat, S.J.; Gamez, P.; Reedijk, J. Manganese-Based Metal-Organic Frameworks as Heterogeneous Catalysts for the Cyanosilylation of Acetaldehyde. Eur. J. Inorg. Chem. 2010, 2010, 3804–3812. [Google Scholar] [CrossRef]
- Xu, G.; Lv, J. Two Mn3 cluster-based frameworks with porosity tuned by solvent coordination/non-coordination: Structural correlation and sorption properties. Inorg. Chem. Commun. 2013, 37, 214–218. [Google Scholar] [CrossRef]
- Waggoner, N.W.; Saccoccia, B.; Ibarra, I.; Lynch, V.M.; Wood, P.; Humphrey, S.M. Magnetism of Linear [Ln3]9+ Oxo-Bridged Clusters (Ln = Pr, Nd) Supported inside a [R3PR′] + Phosphonium Coordination Material. Inorg. Chem. 2014, 53, 12674–12676. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; He, J.; Guo, H.; Dunning, S.G.; Humphrey, S.M.; Jones, R.A. Magnetism and Luminescence of a MOF with Linear Mn3 Nodes Derived from an Emissive Terthiophene-Based Imidazole Linker. Molecules 2021, 26, 4286. https://doi.org/10.3390/molecules26144286
Wang W, He J, Guo H, Dunning SG, Humphrey SM, Jones RA. Magnetism and Luminescence of a MOF with Linear Mn3 Nodes Derived from an Emissive Terthiophene-Based Imidazole Linker. Molecules. 2021; 26(14):4286. https://doi.org/10.3390/molecules26144286
Chicago/Turabian StyleWang, Weiran, Junpeng He, Hongyu Guo, Samuel G. Dunning, Simon M. Humphrey, and Richard A. Jones. 2021. "Magnetism and Luminescence of a MOF with Linear Mn3 Nodes Derived from an Emissive Terthiophene-Based Imidazole Linker" Molecules 26, no. 14: 4286. https://doi.org/10.3390/molecules26144286
APA StyleWang, W., He, J., Guo, H., Dunning, S. G., Humphrey, S. M., & Jones, R. A. (2021). Magnetism and Luminescence of a MOF with Linear Mn3 Nodes Derived from an Emissive Terthiophene-Based Imidazole Linker. Molecules, 26(14), 4286. https://doi.org/10.3390/molecules26144286