
Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy


Abstract
1. Introduction
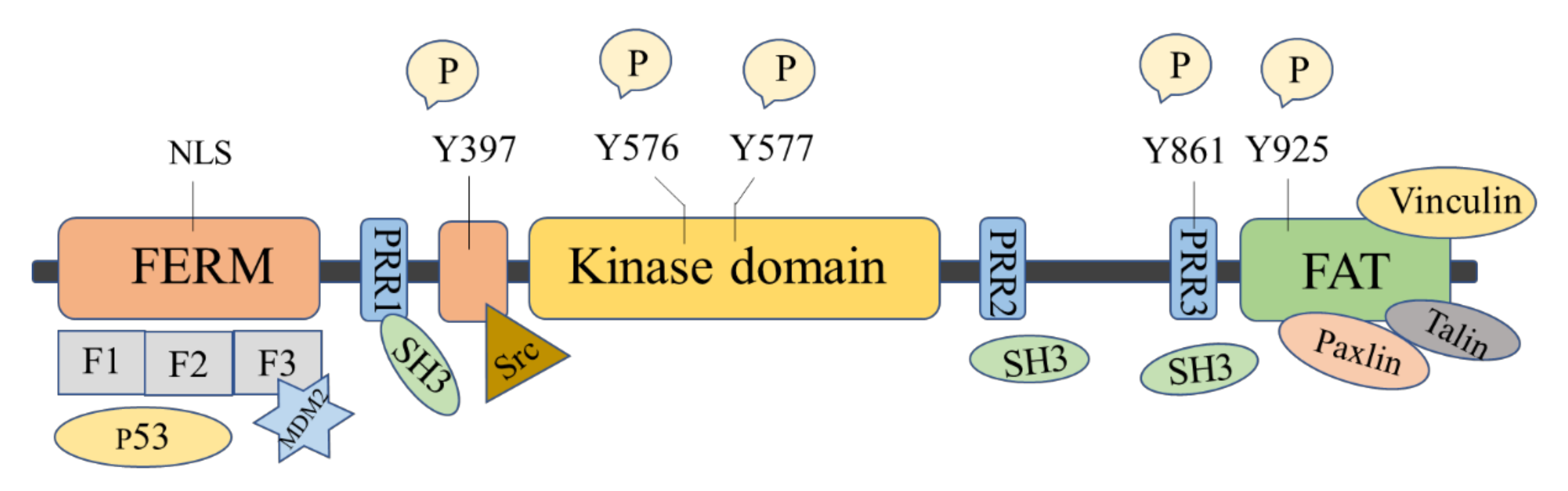
2. FAK Structure and Function
3. The Discovery of FAK Inhibitors and Their Therapeutic Significance
3.1. FAK Inhibitors in Clinical Trials
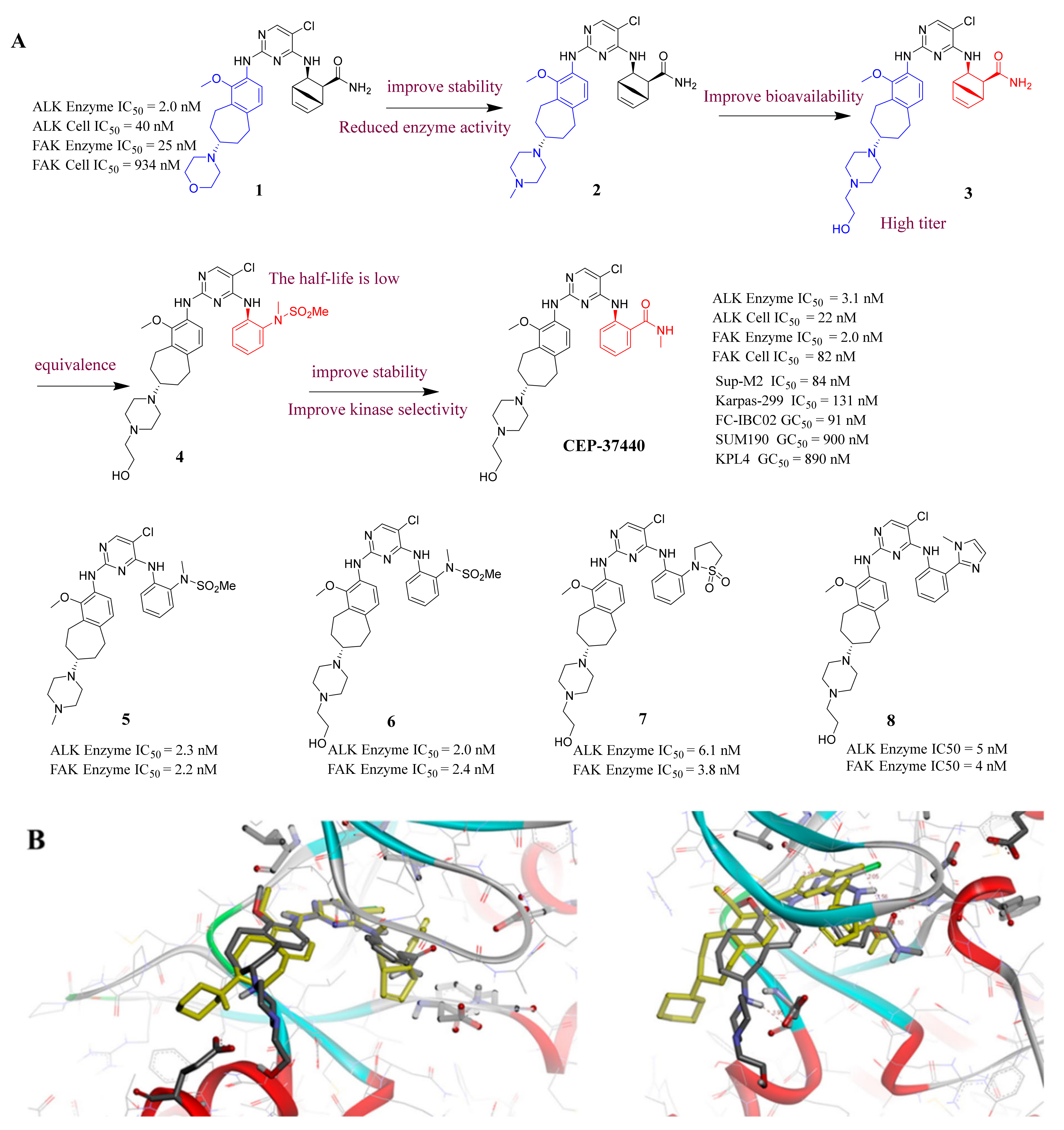
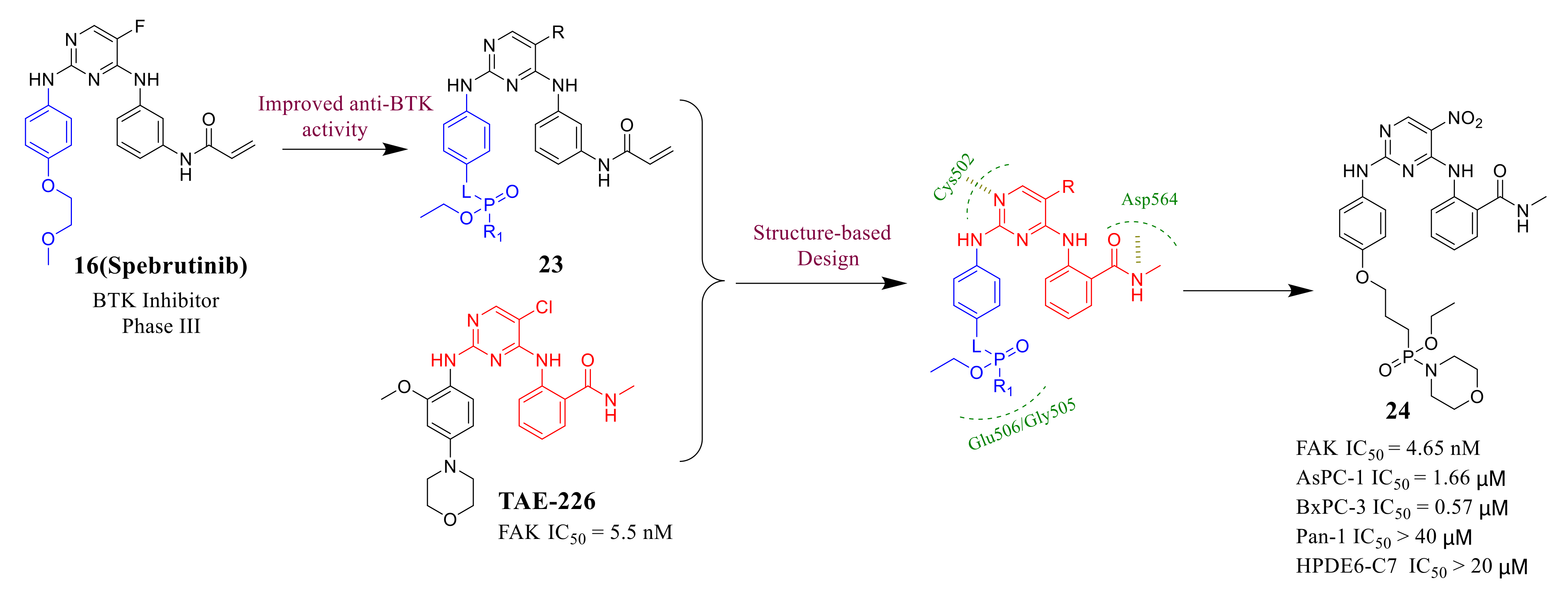
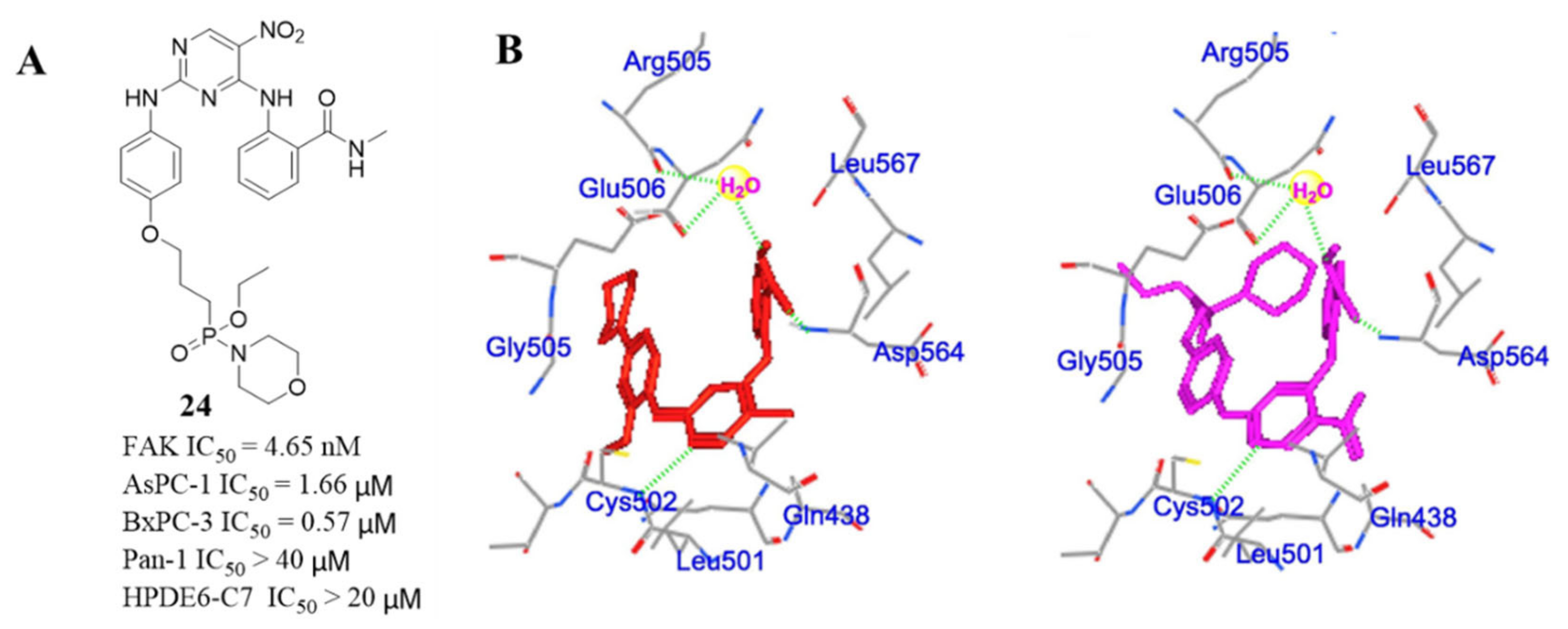
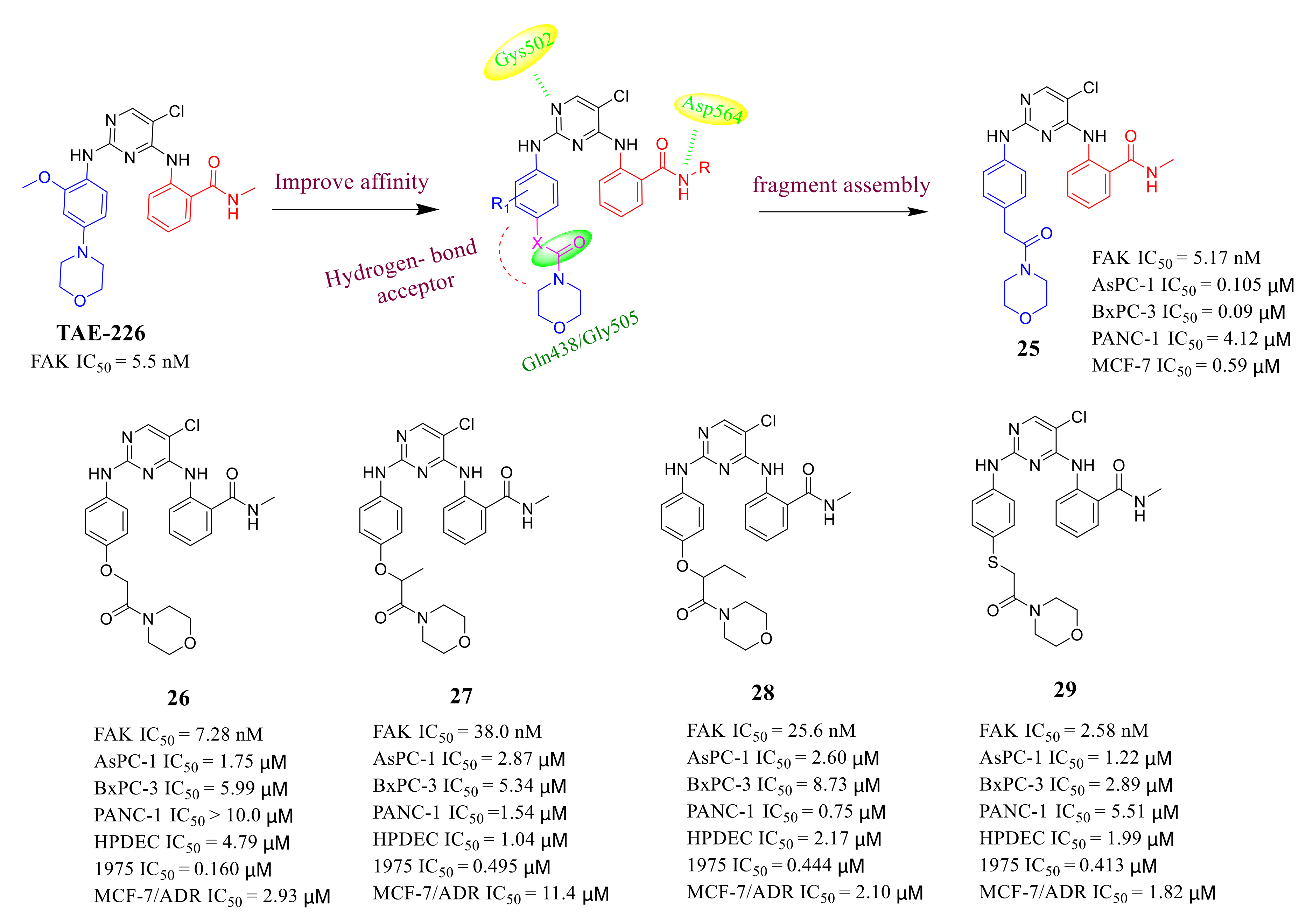
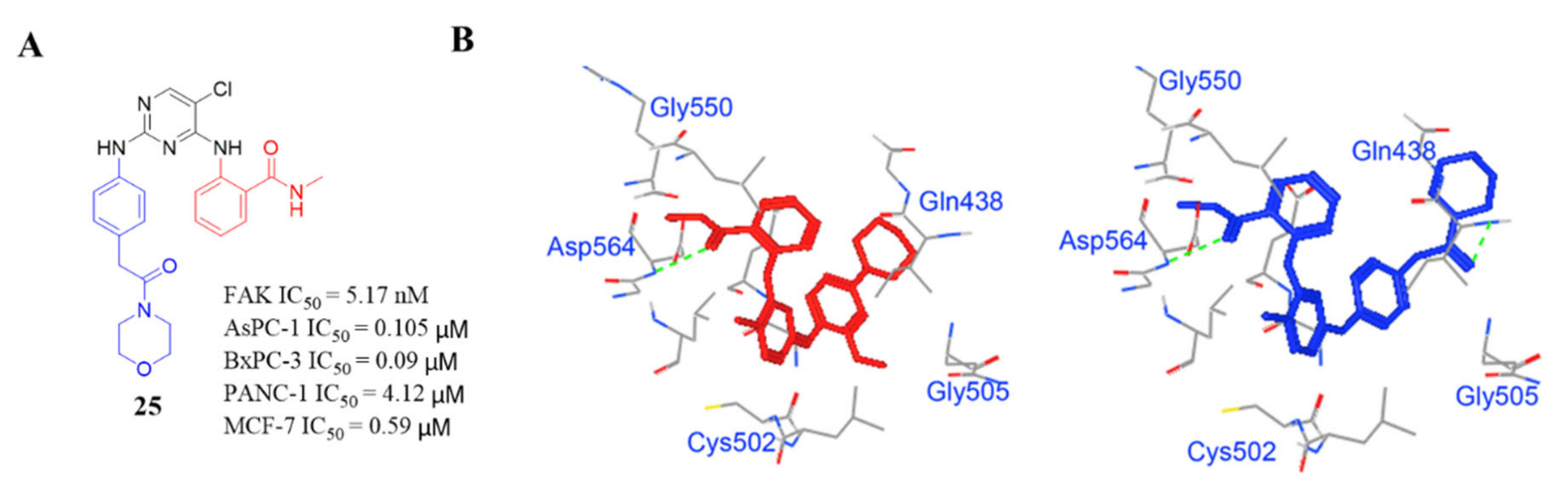
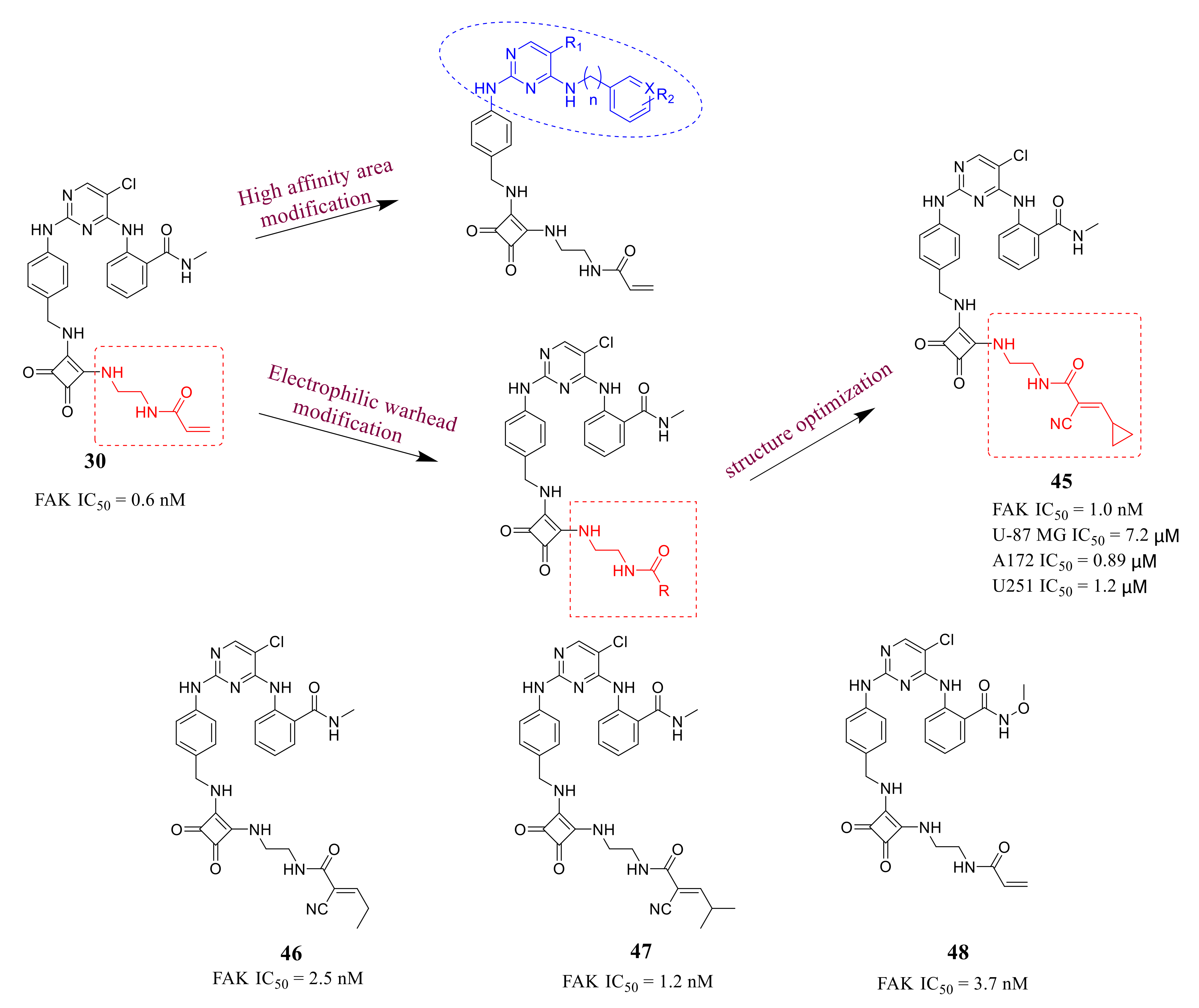
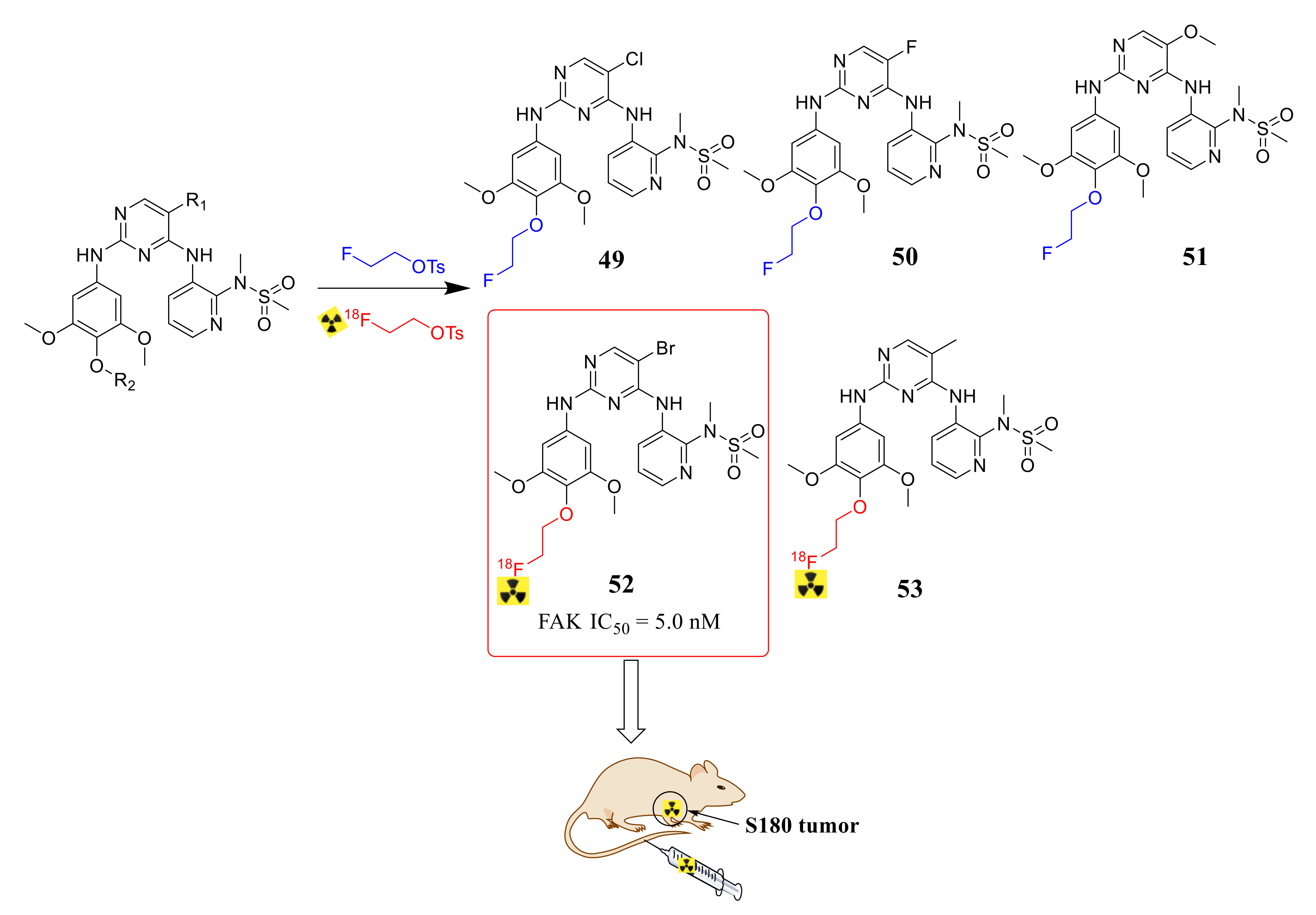
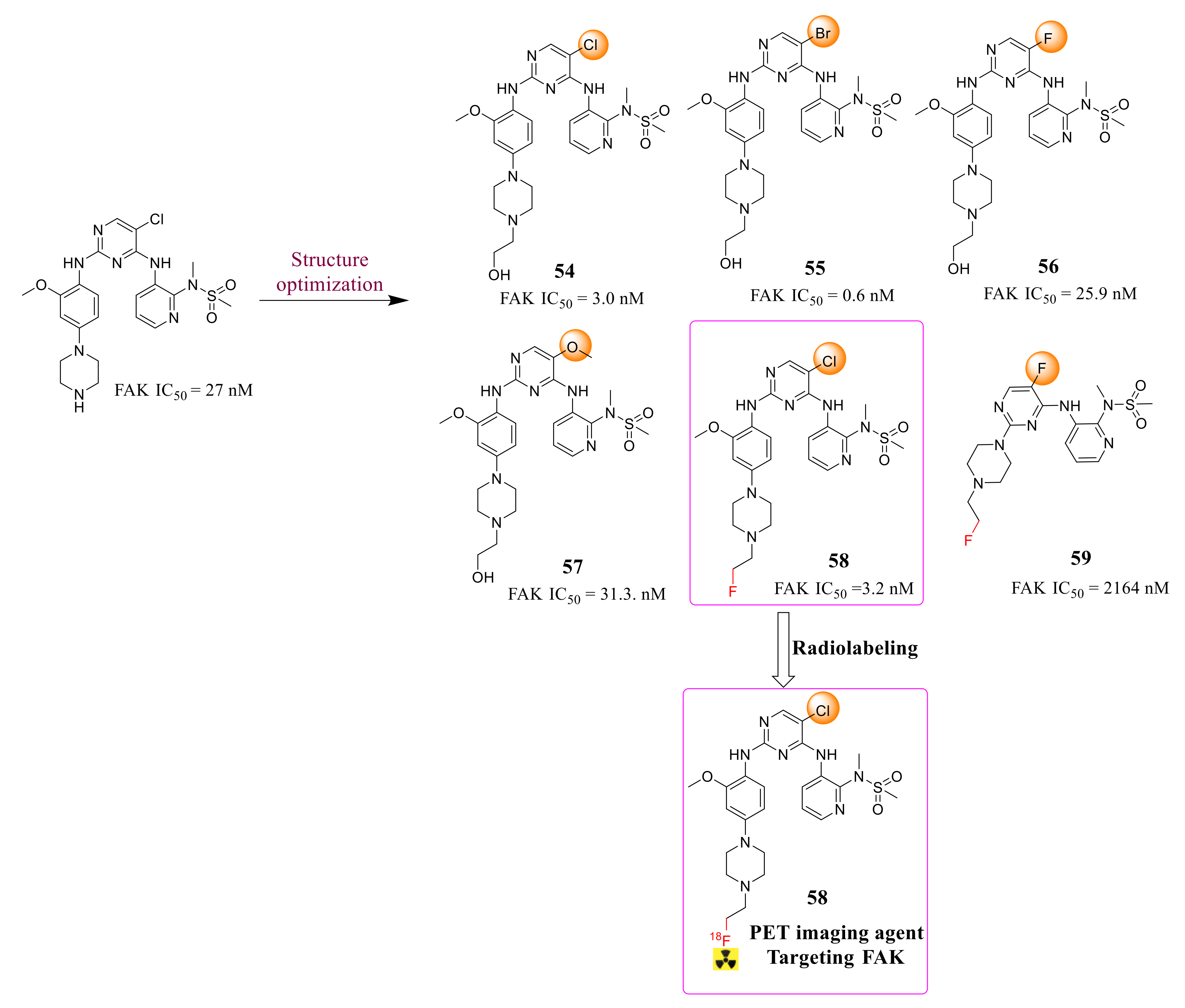
3.2. Pyrimidine FAK Inhibitors
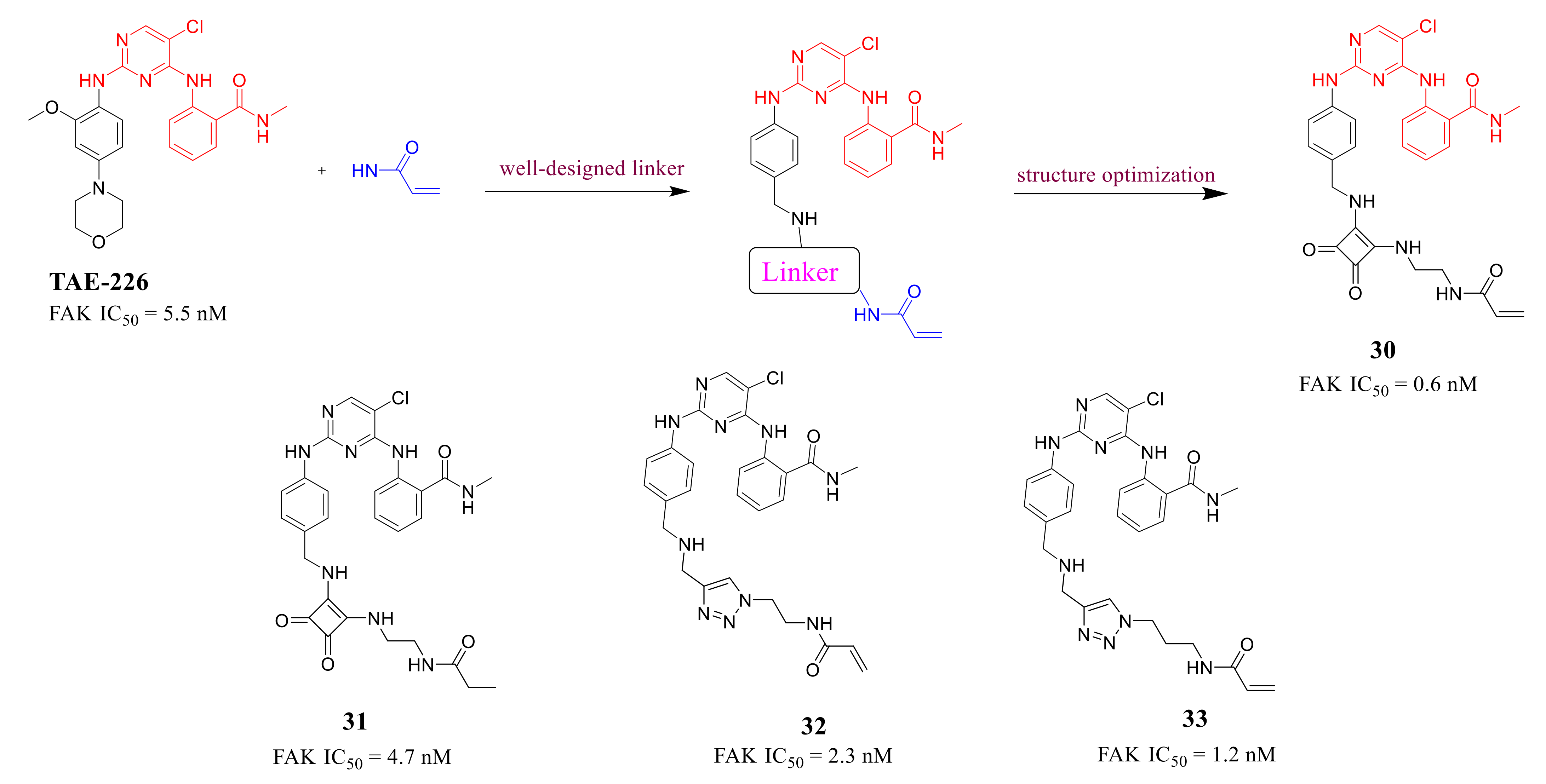
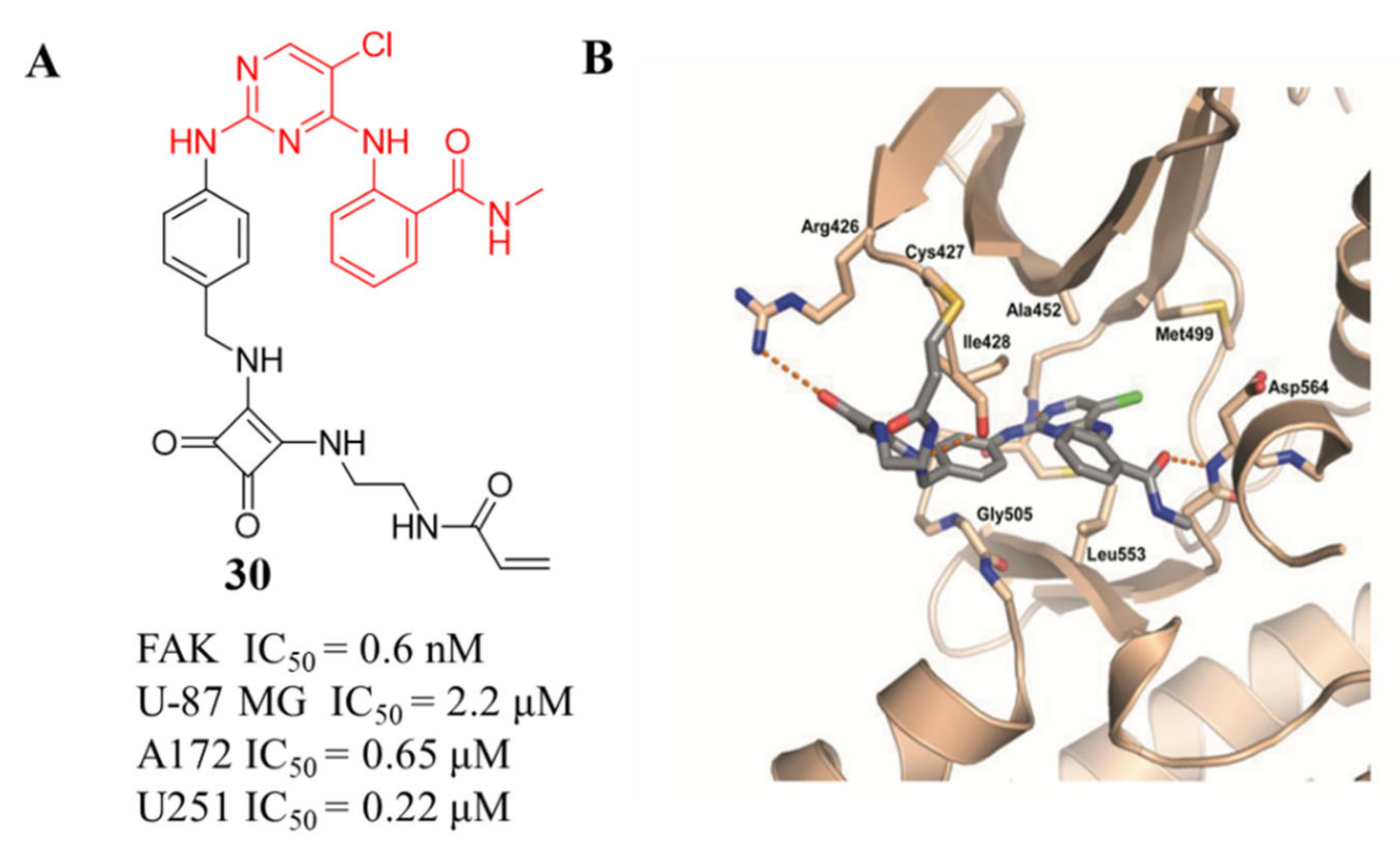
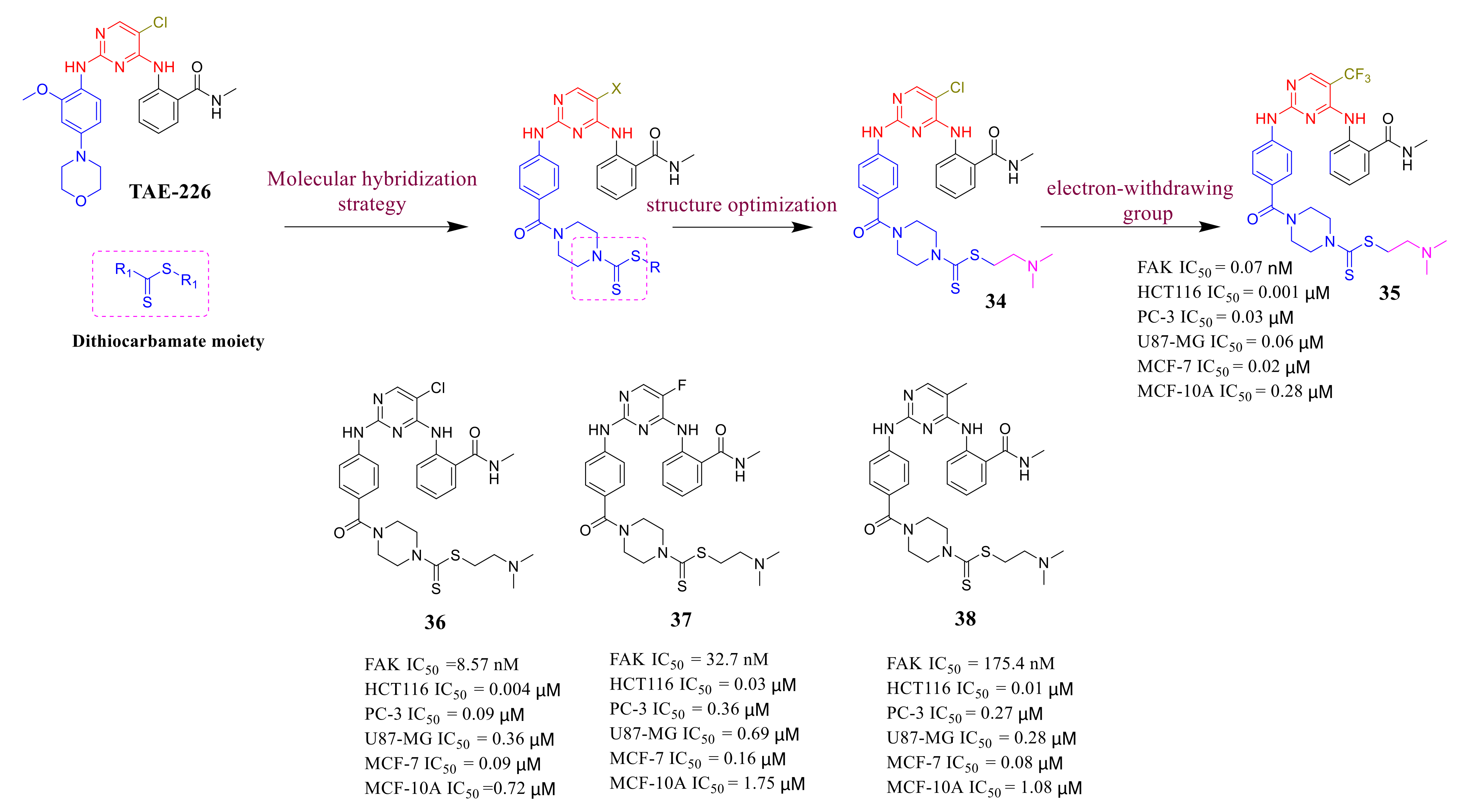
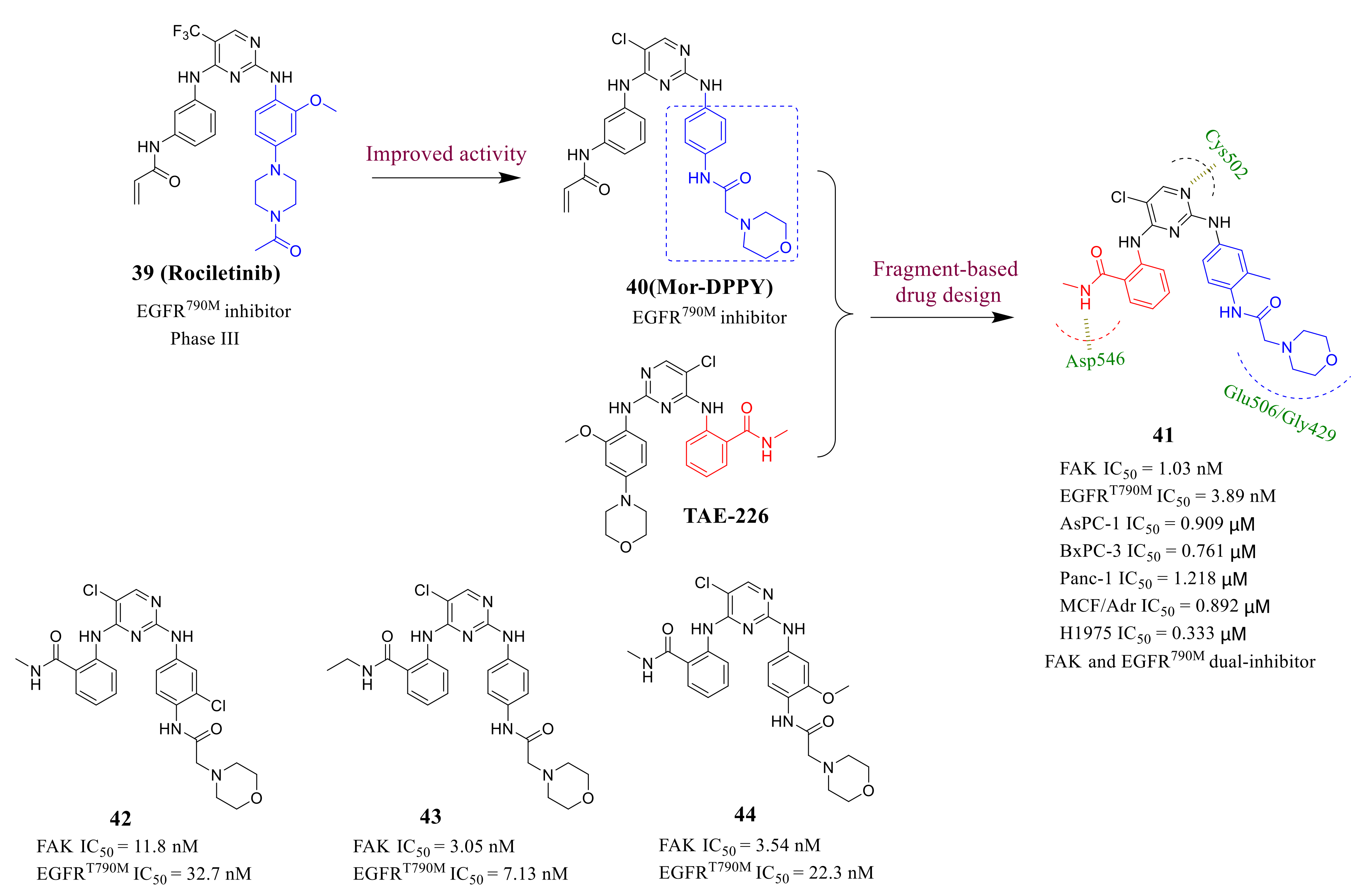
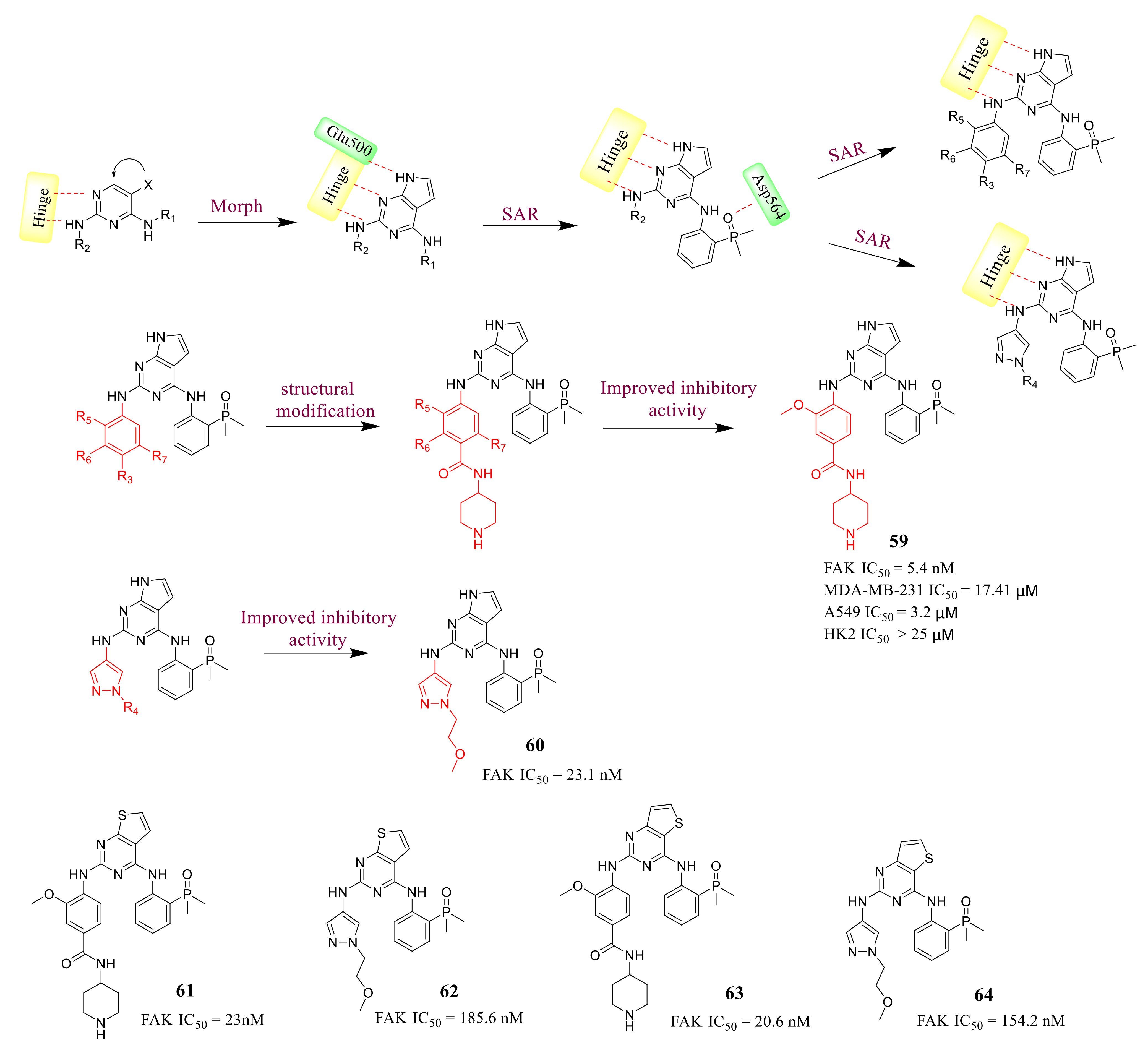
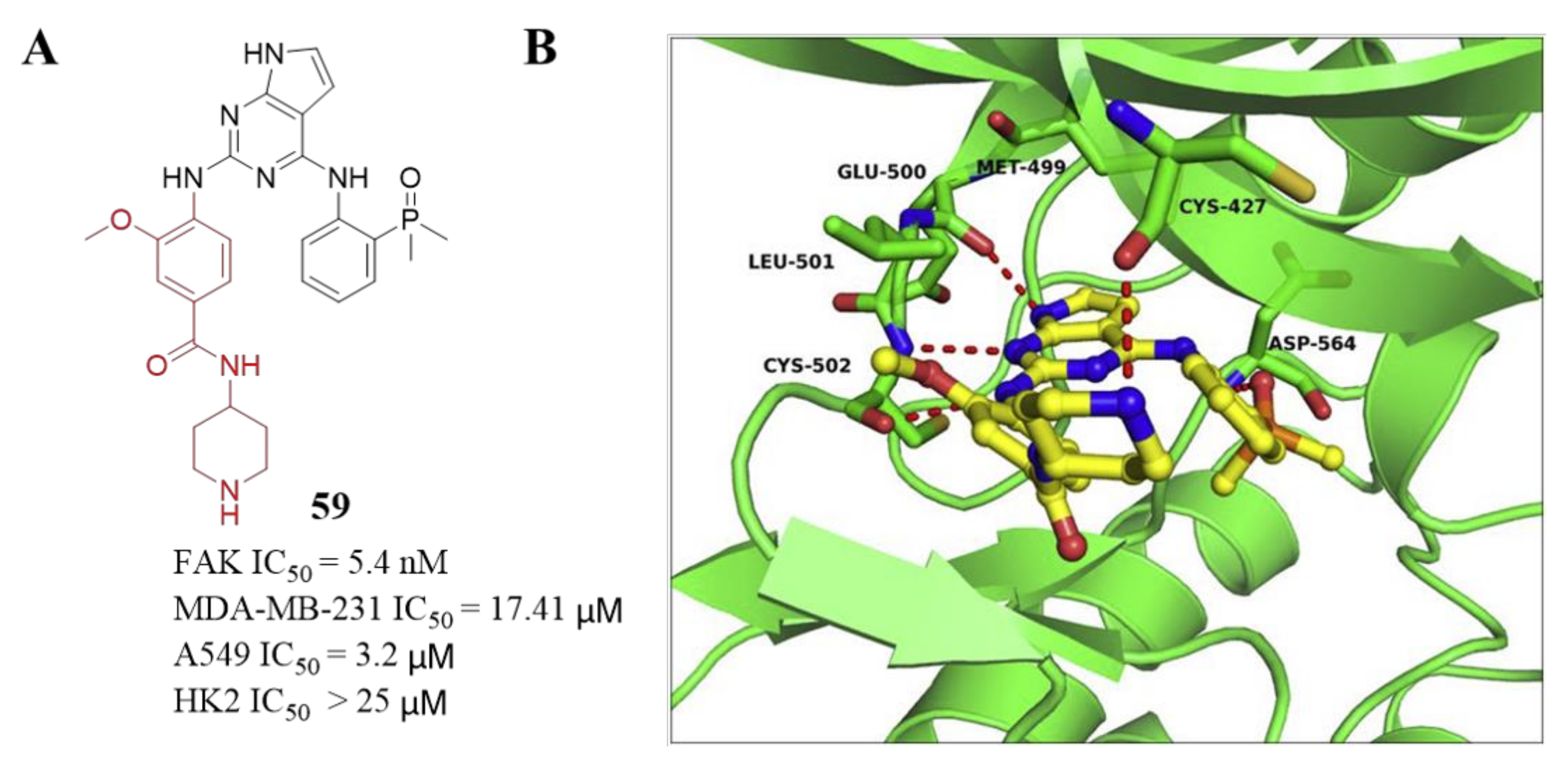
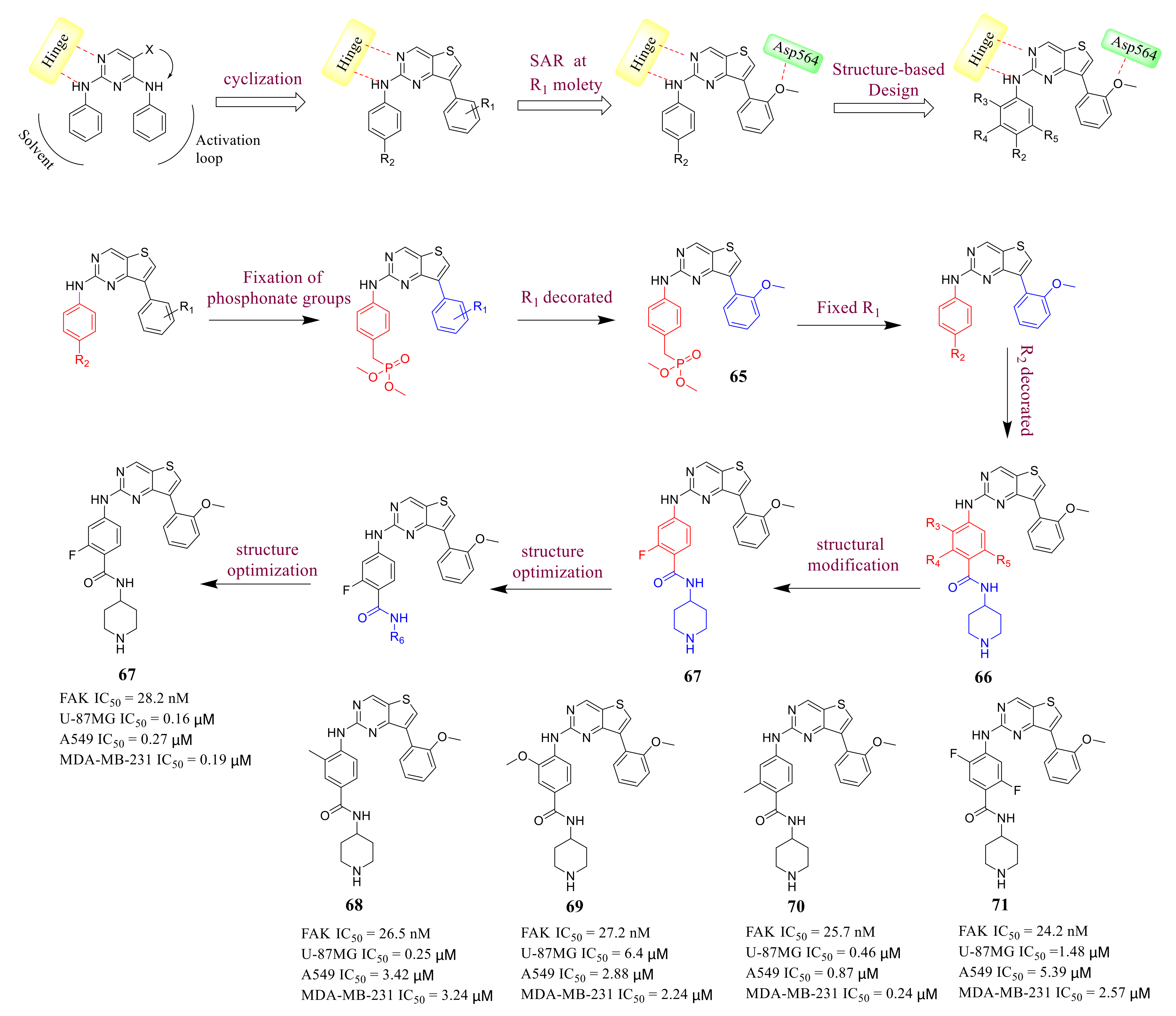
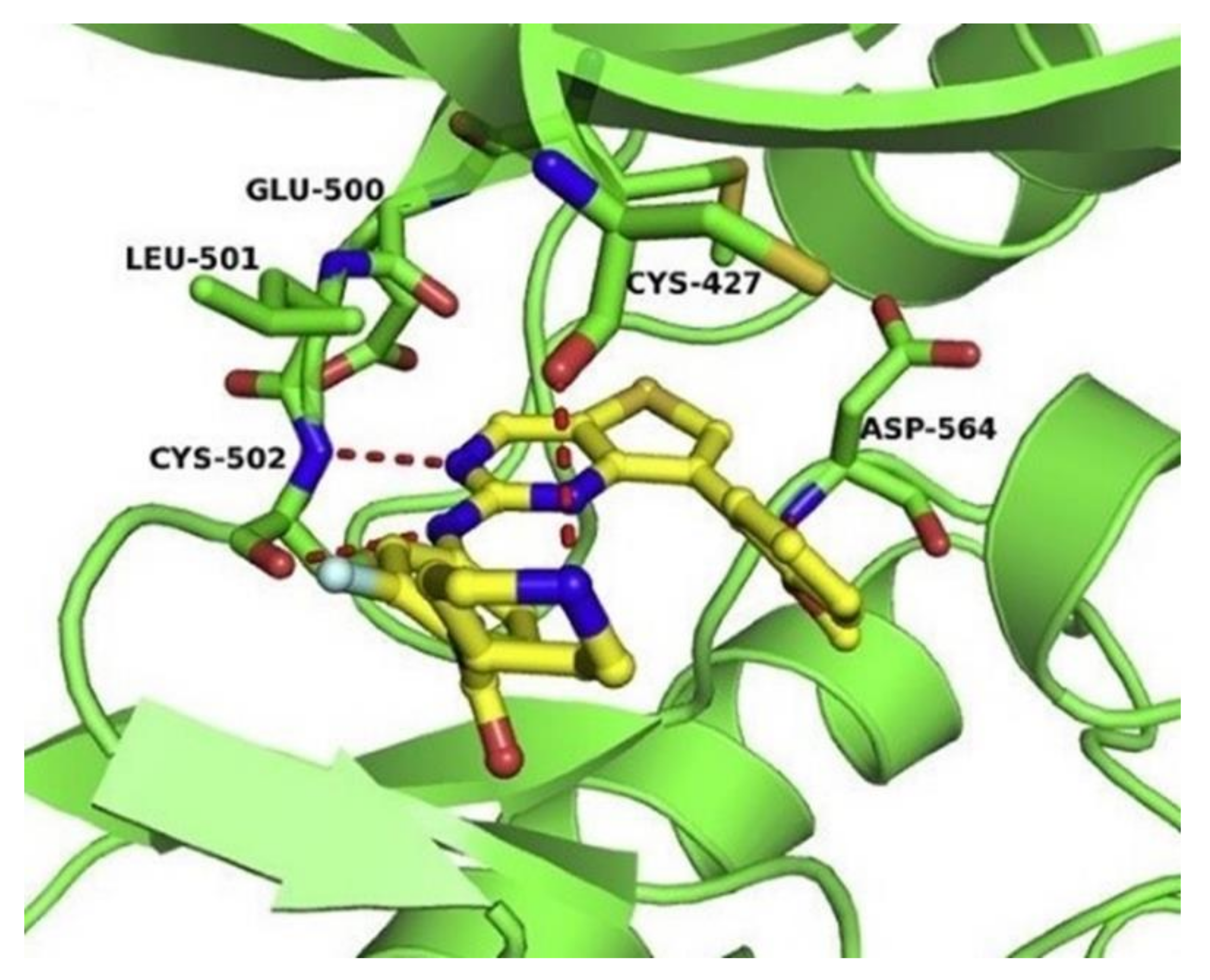
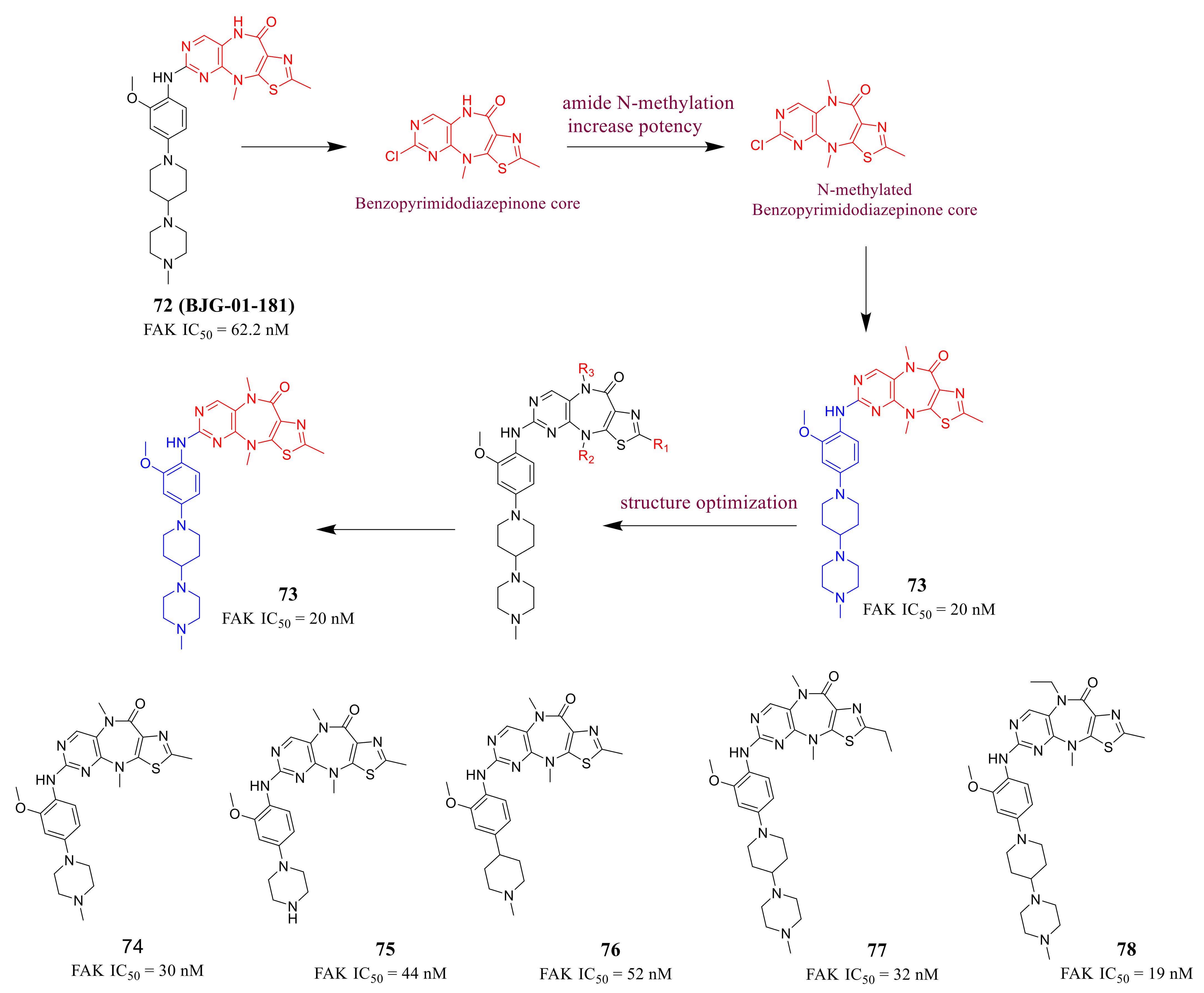
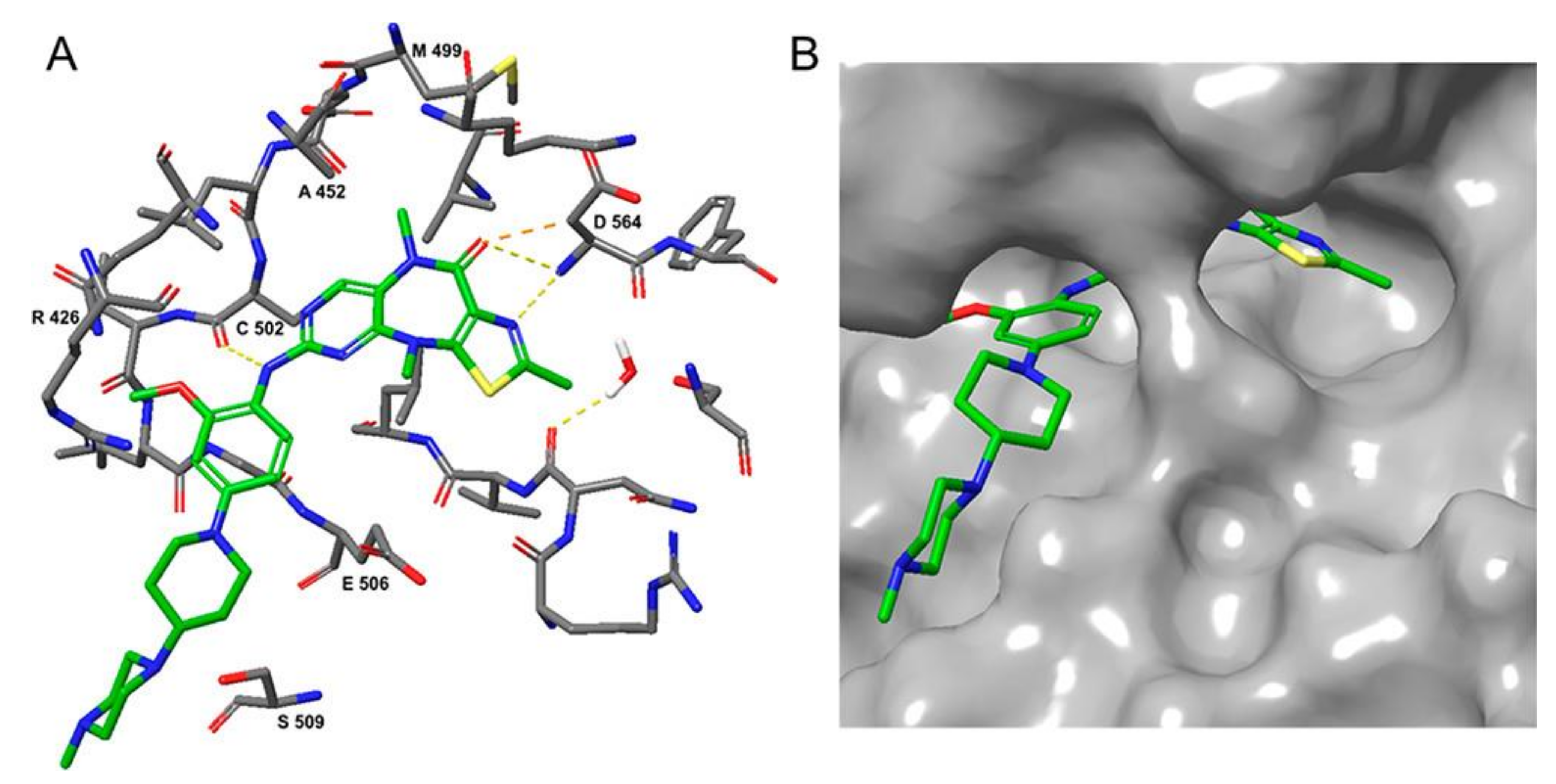
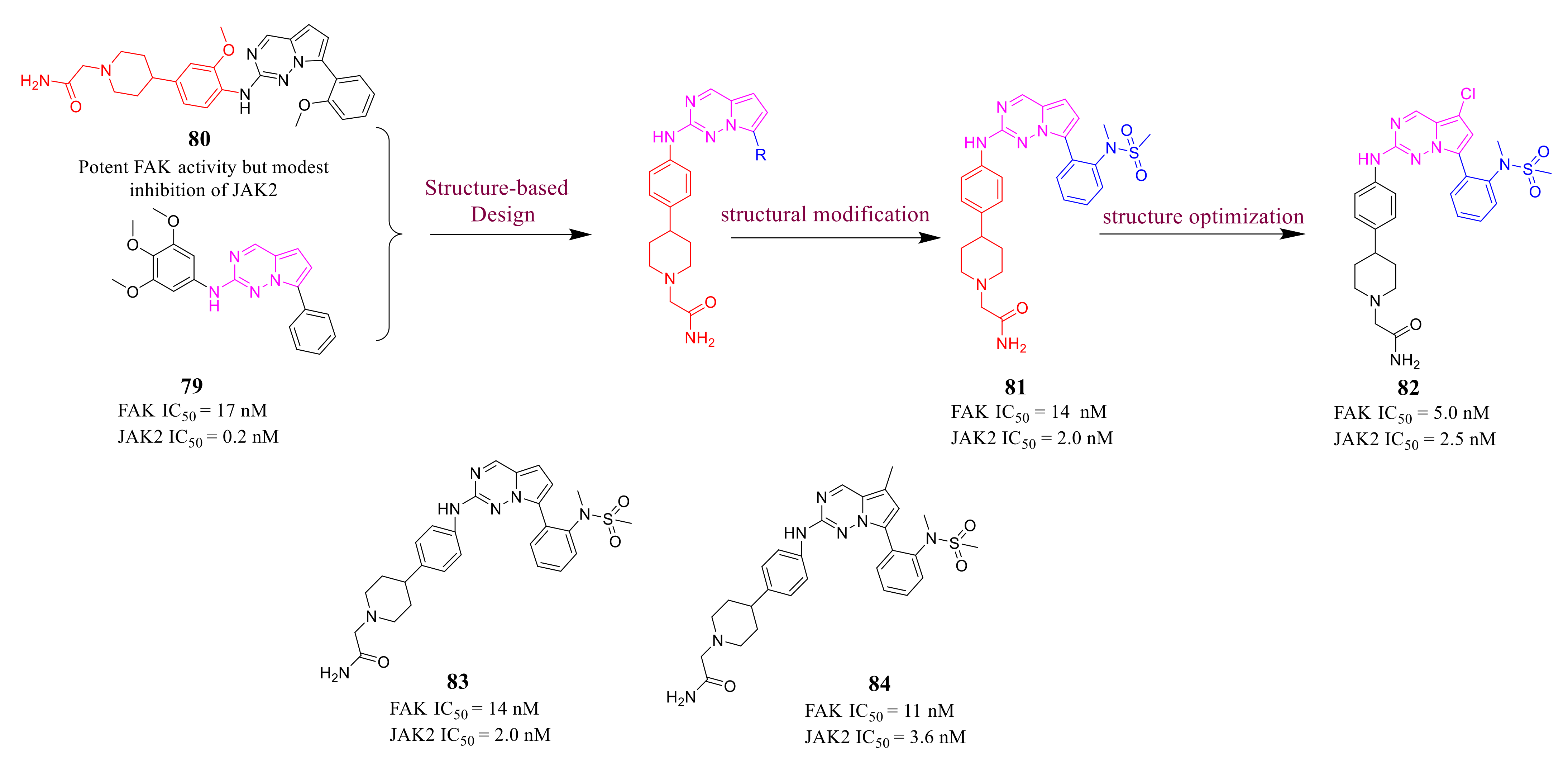
3.3. FAK Inhibitors of Pyrimidine Heterocyclic Compounds
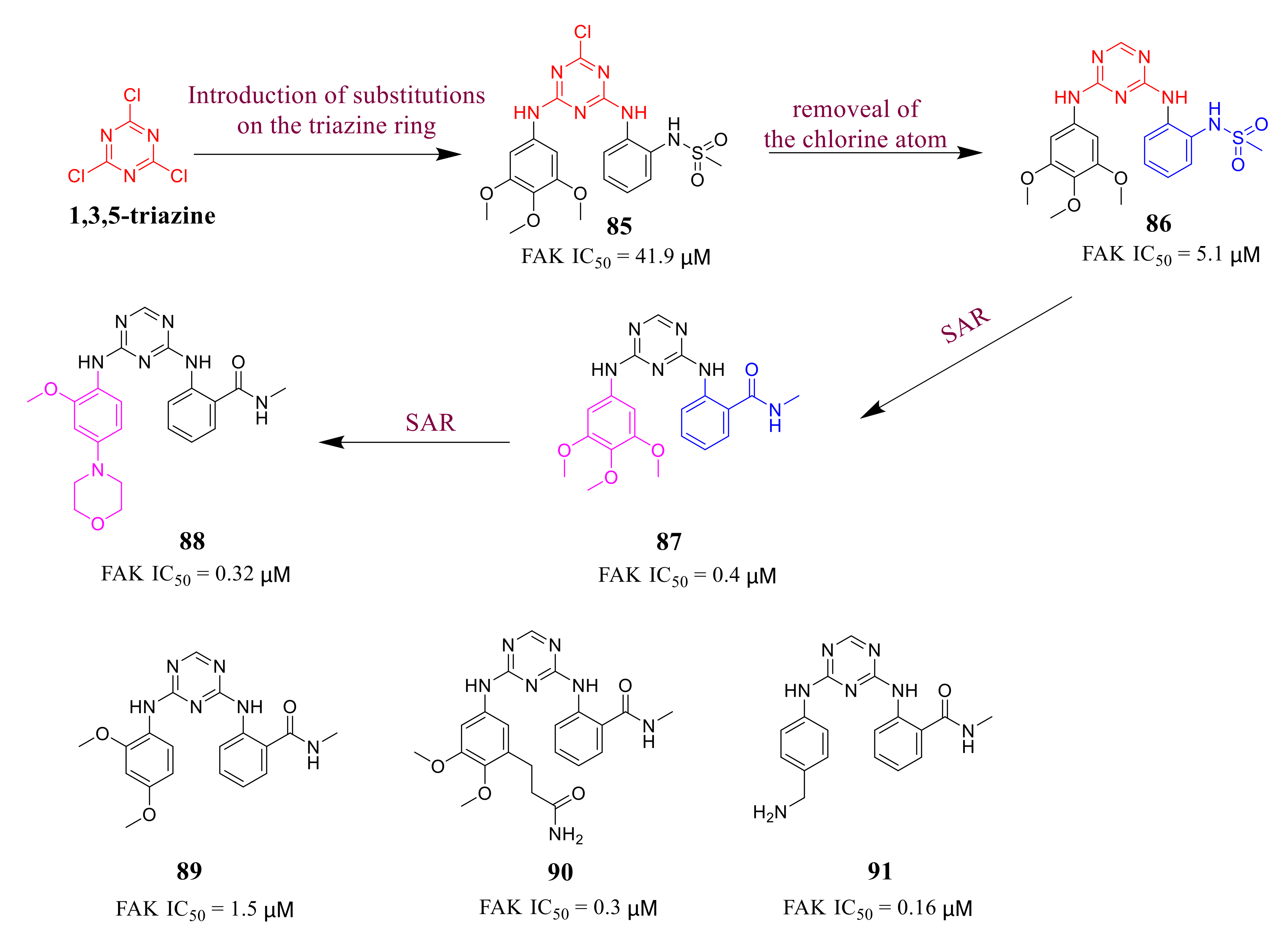
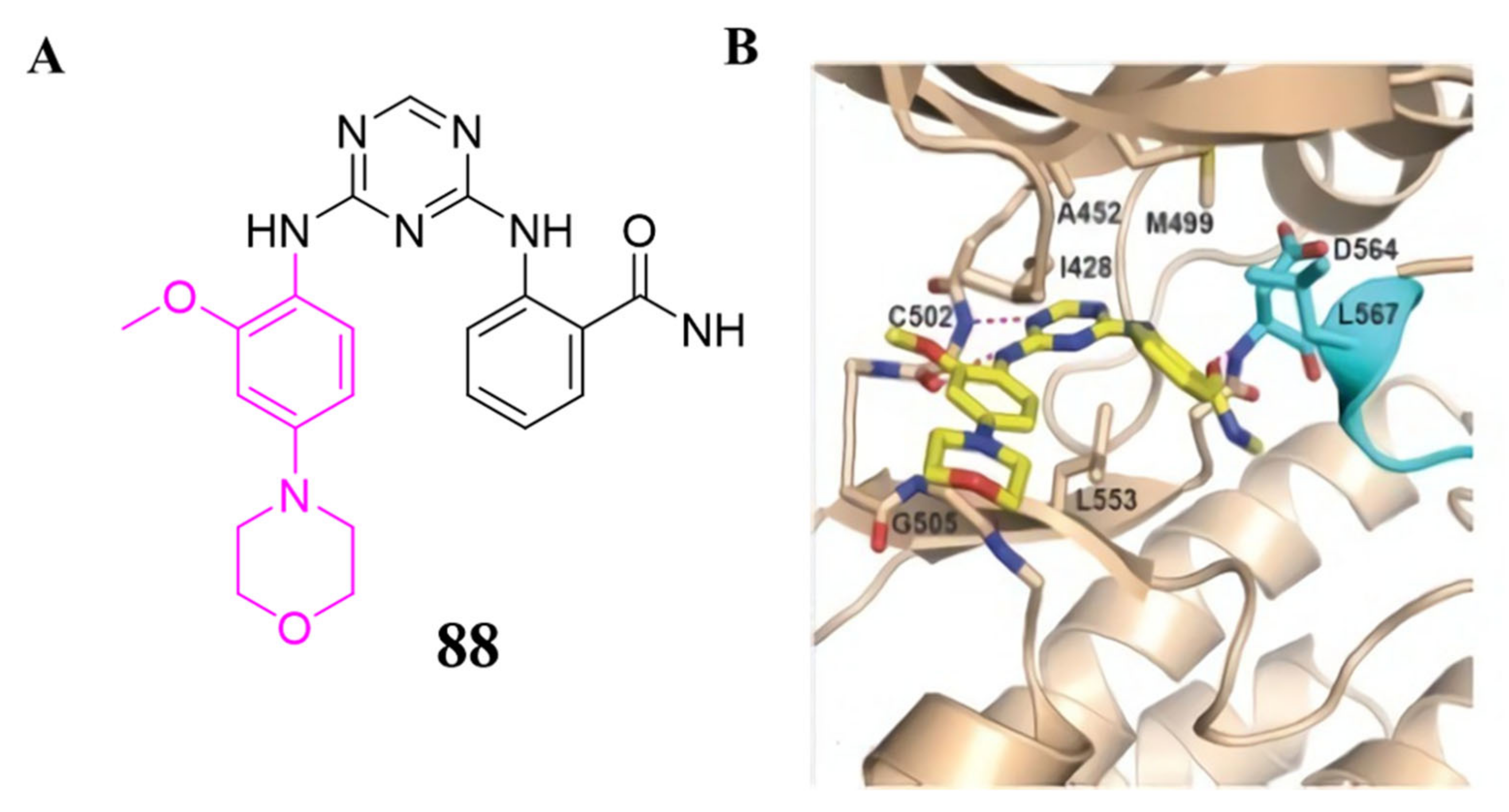
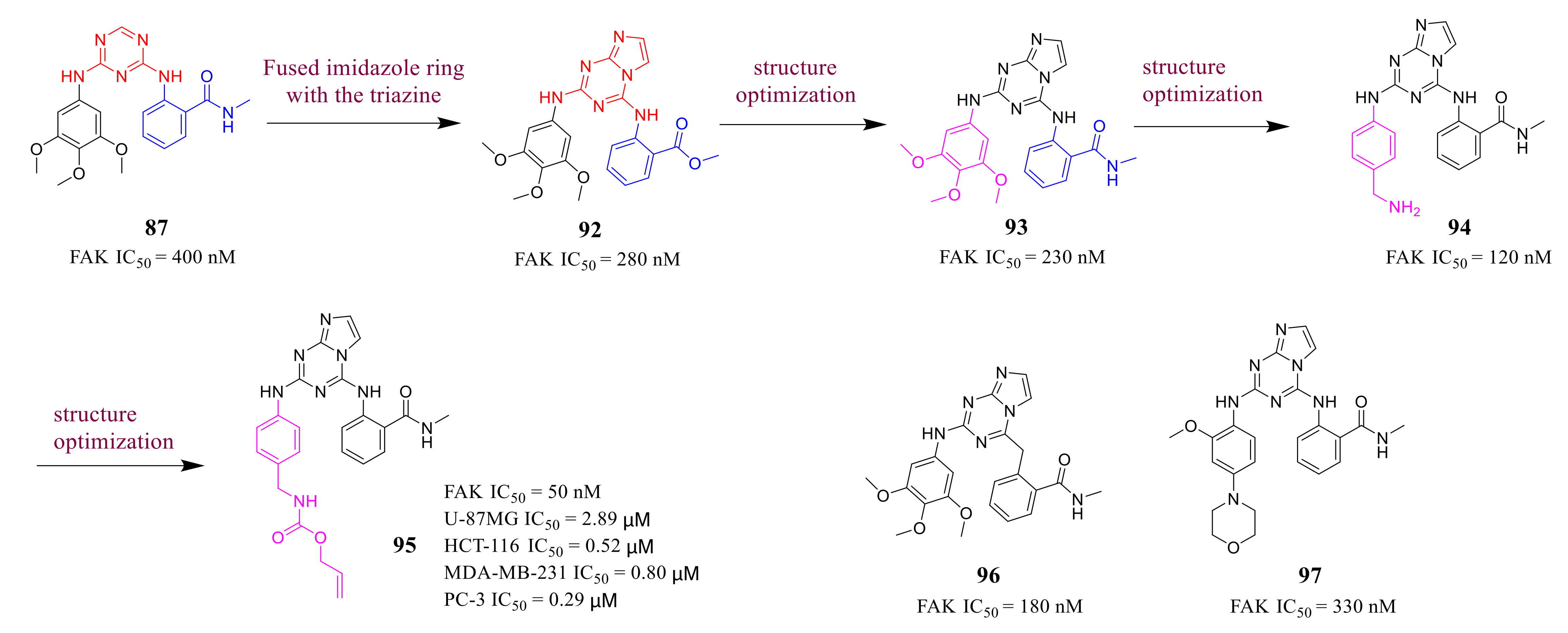
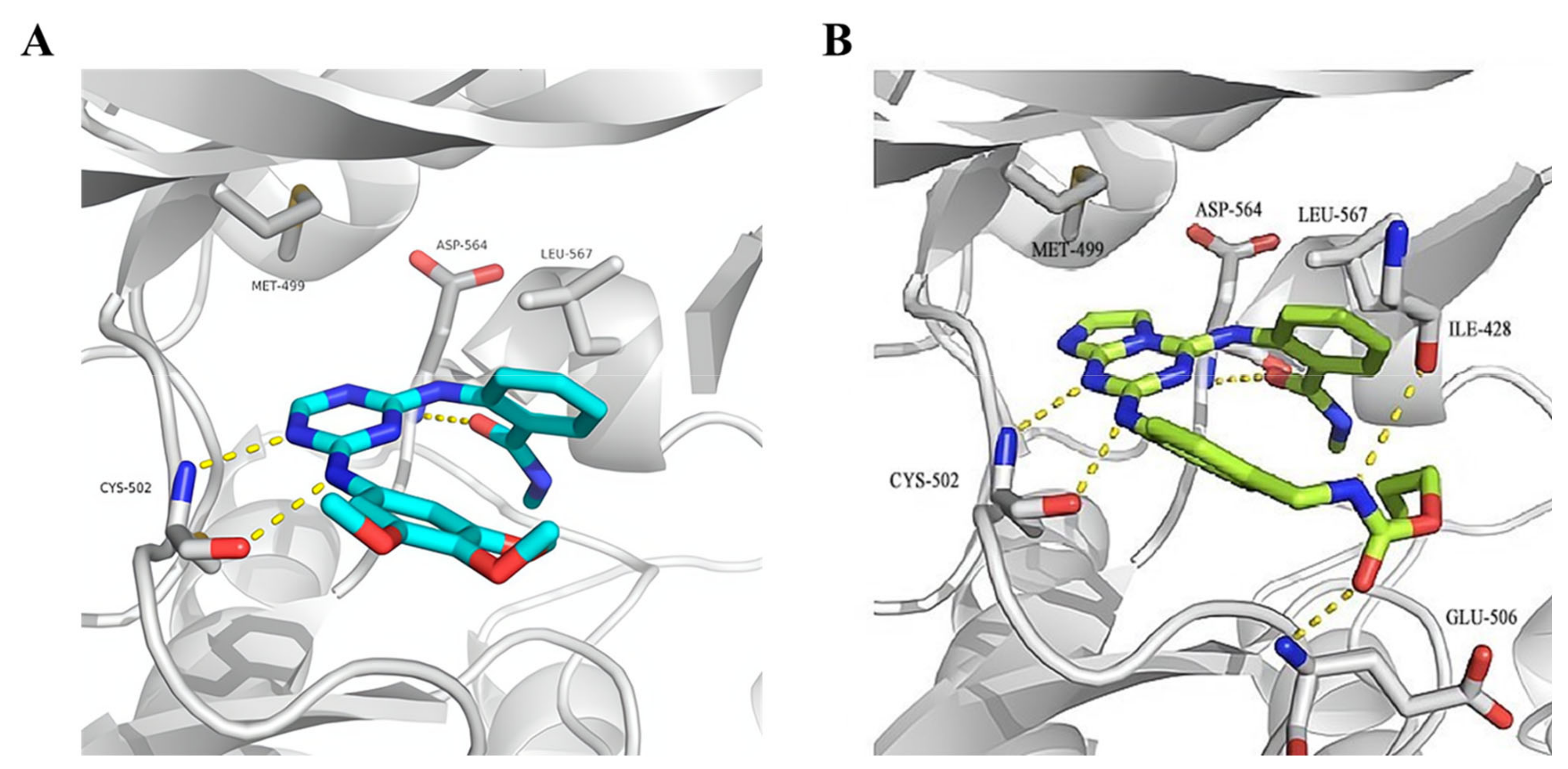
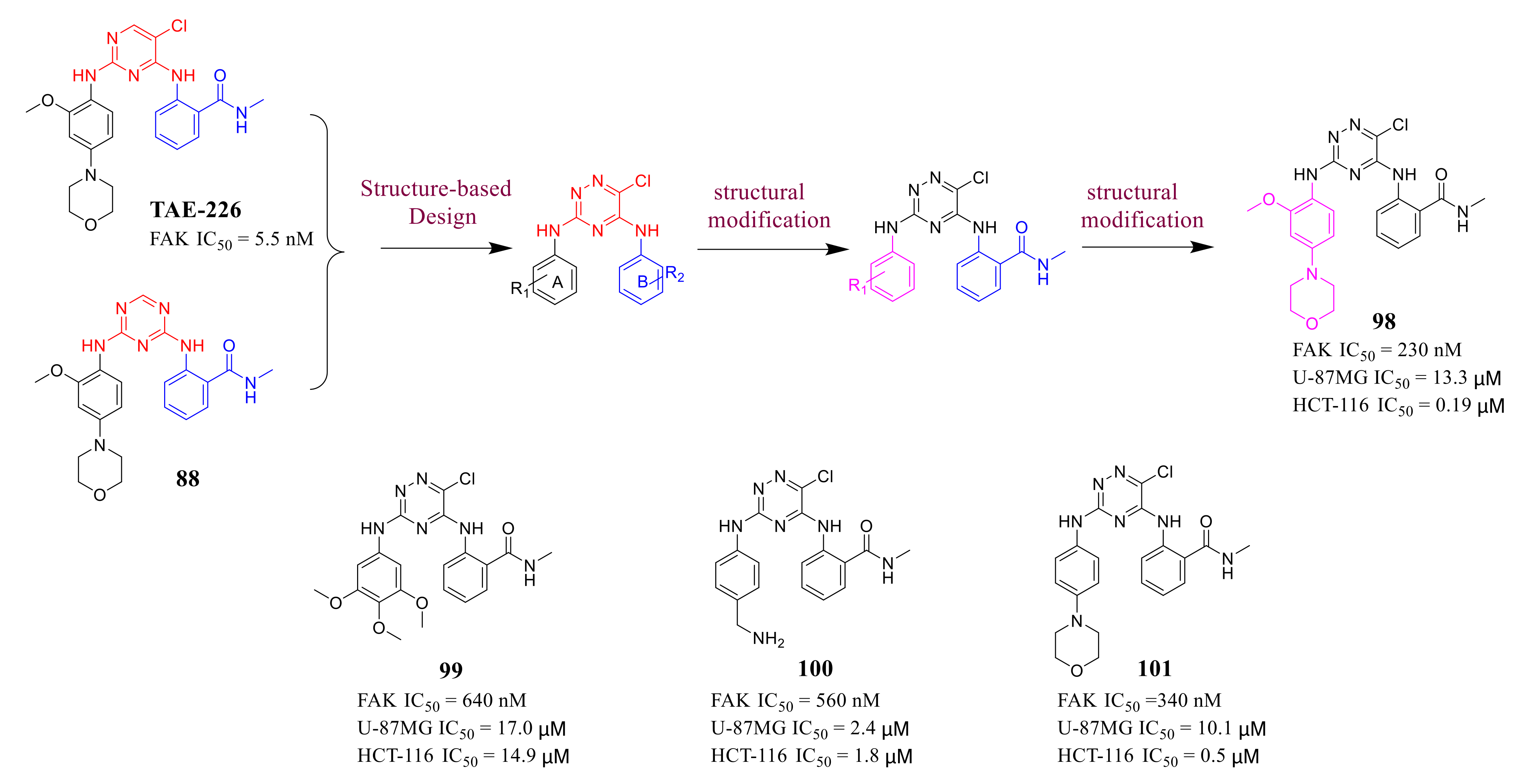
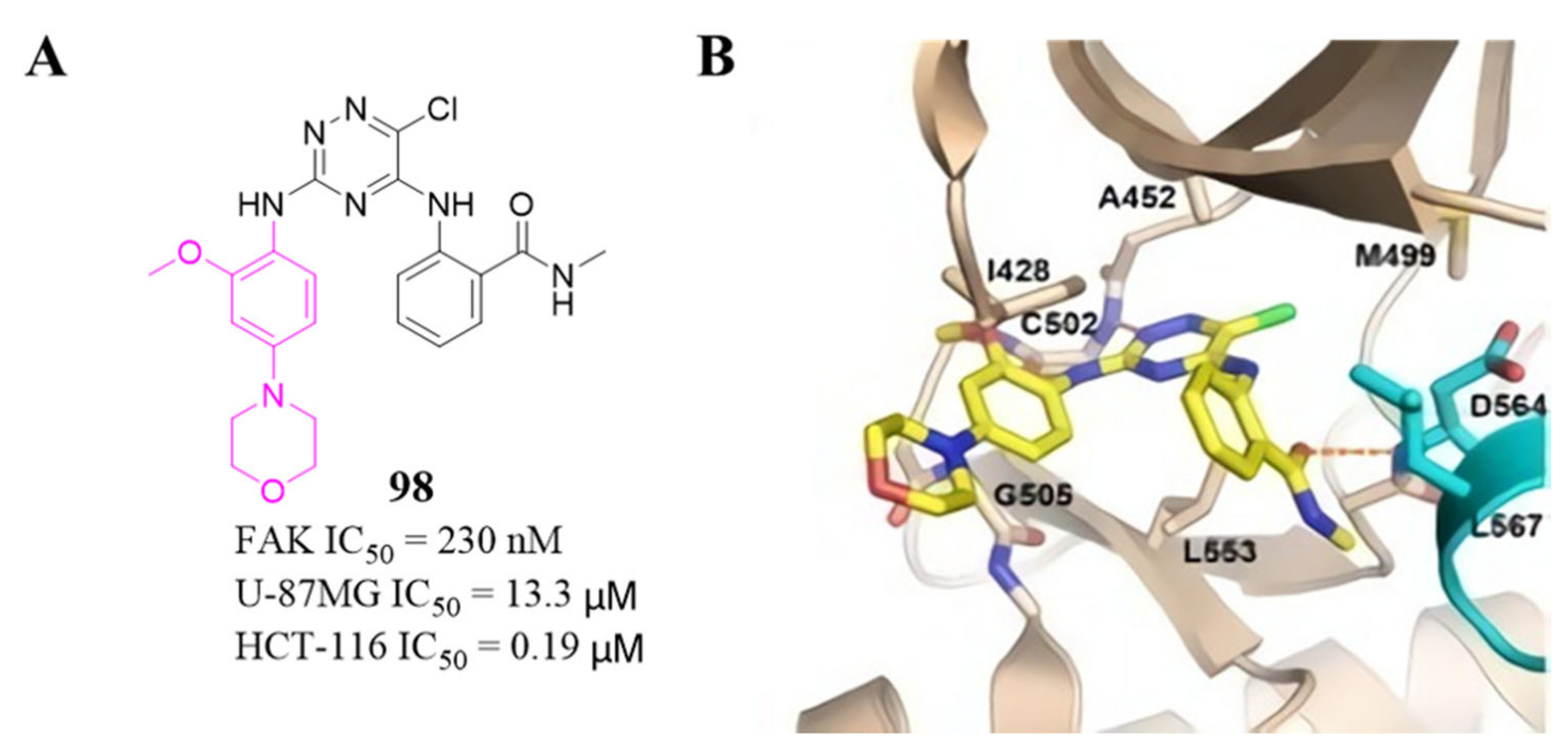
3.4. FAK Inhibitors with a Triazine Scaffold
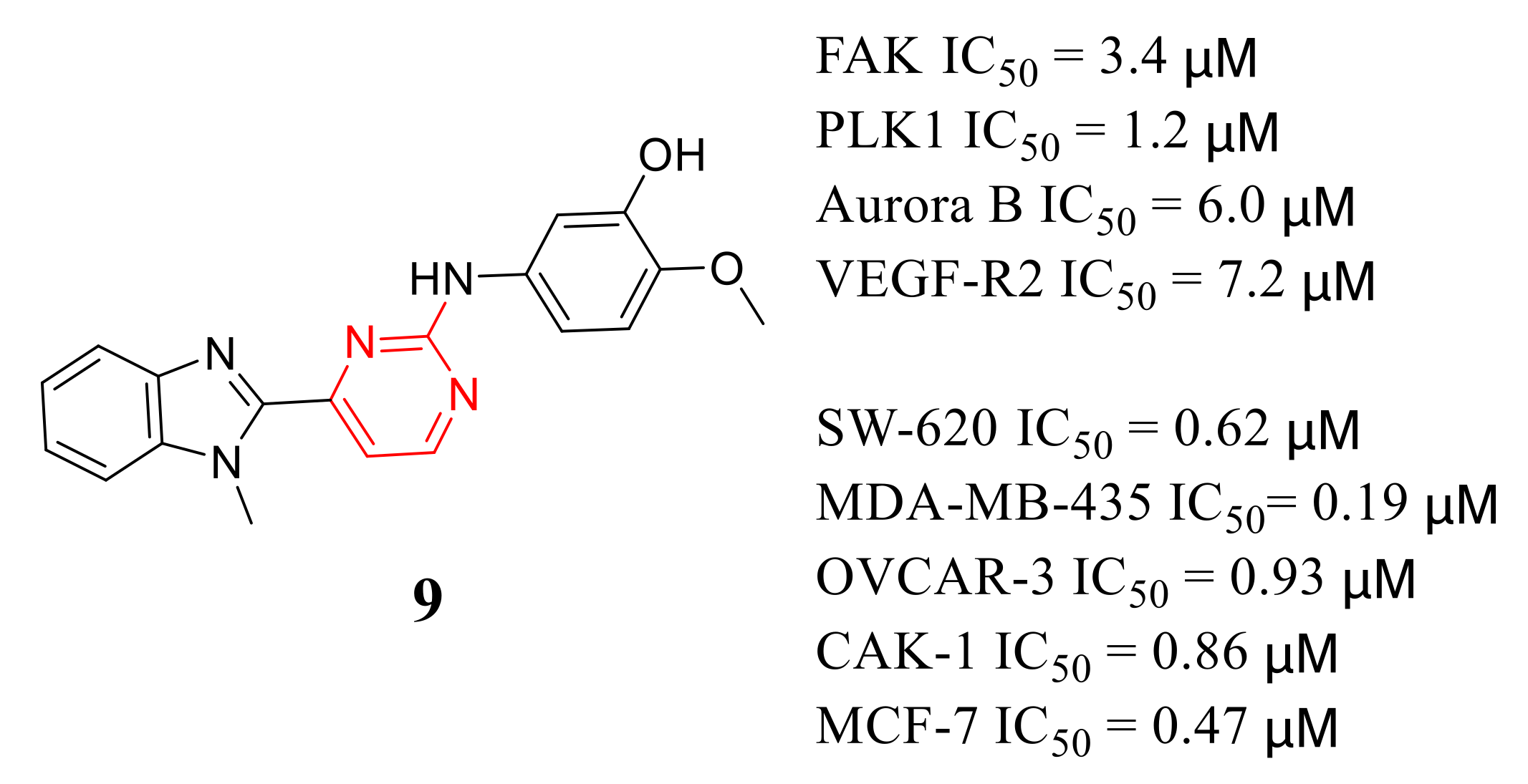
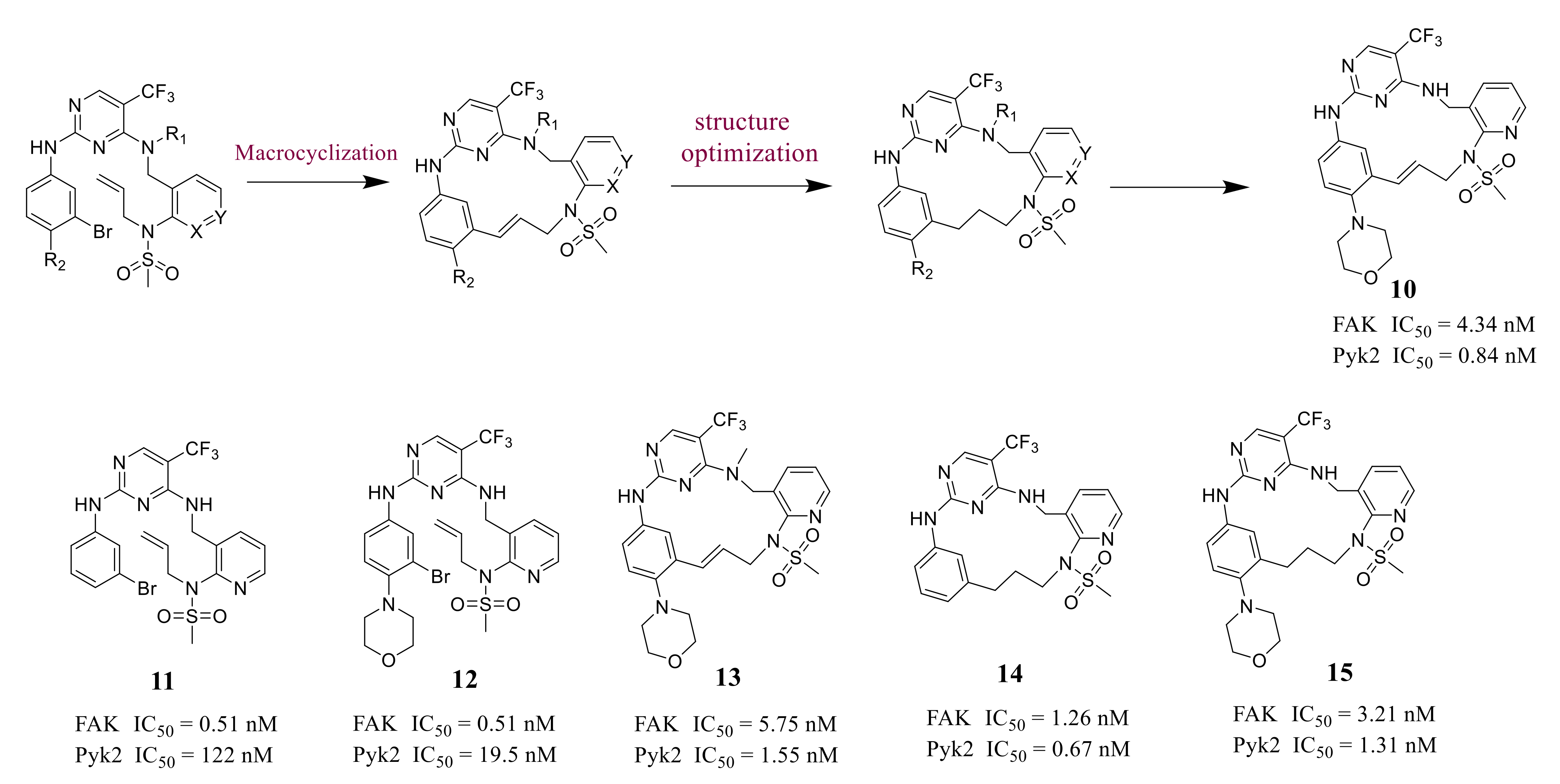
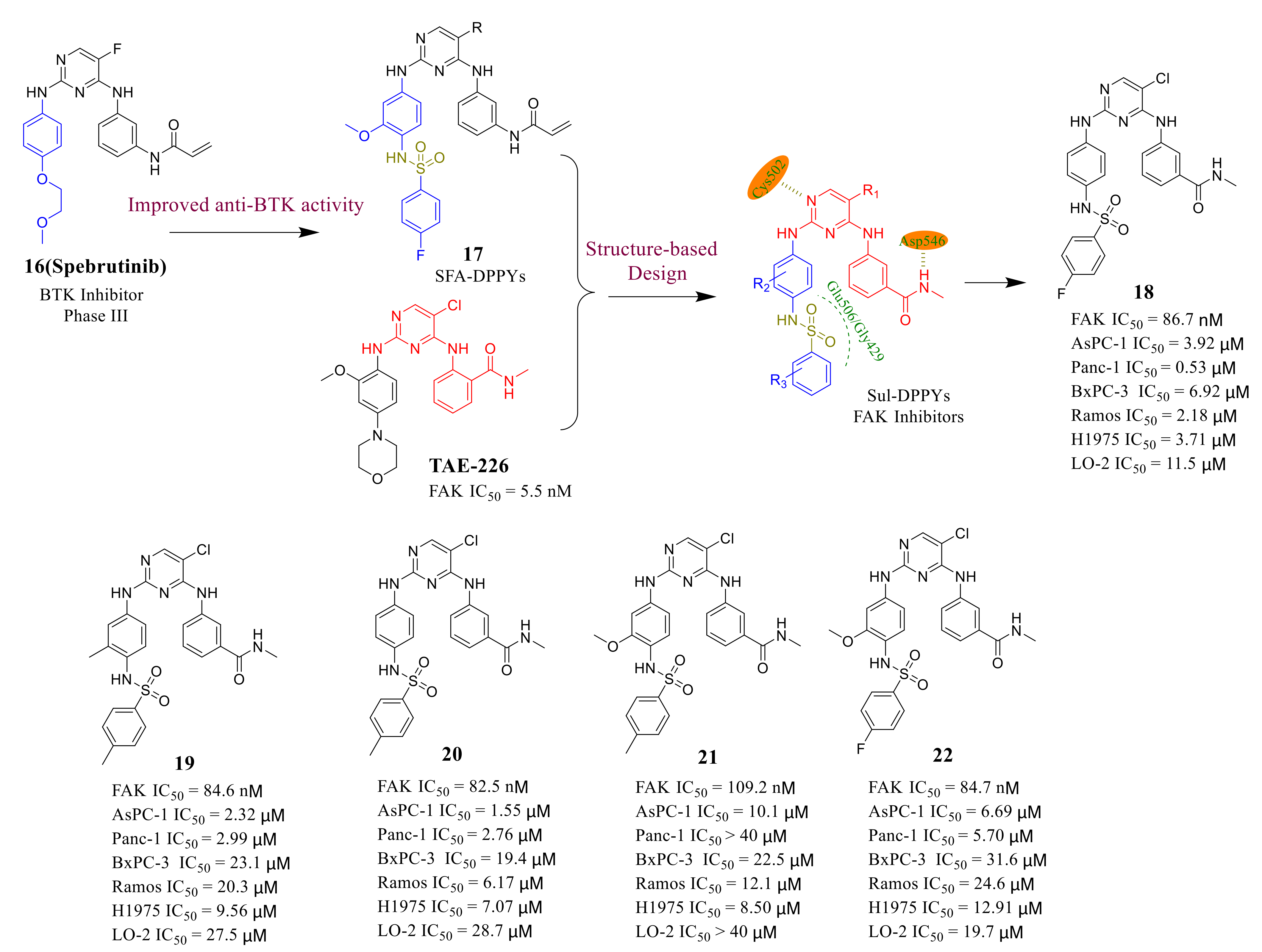
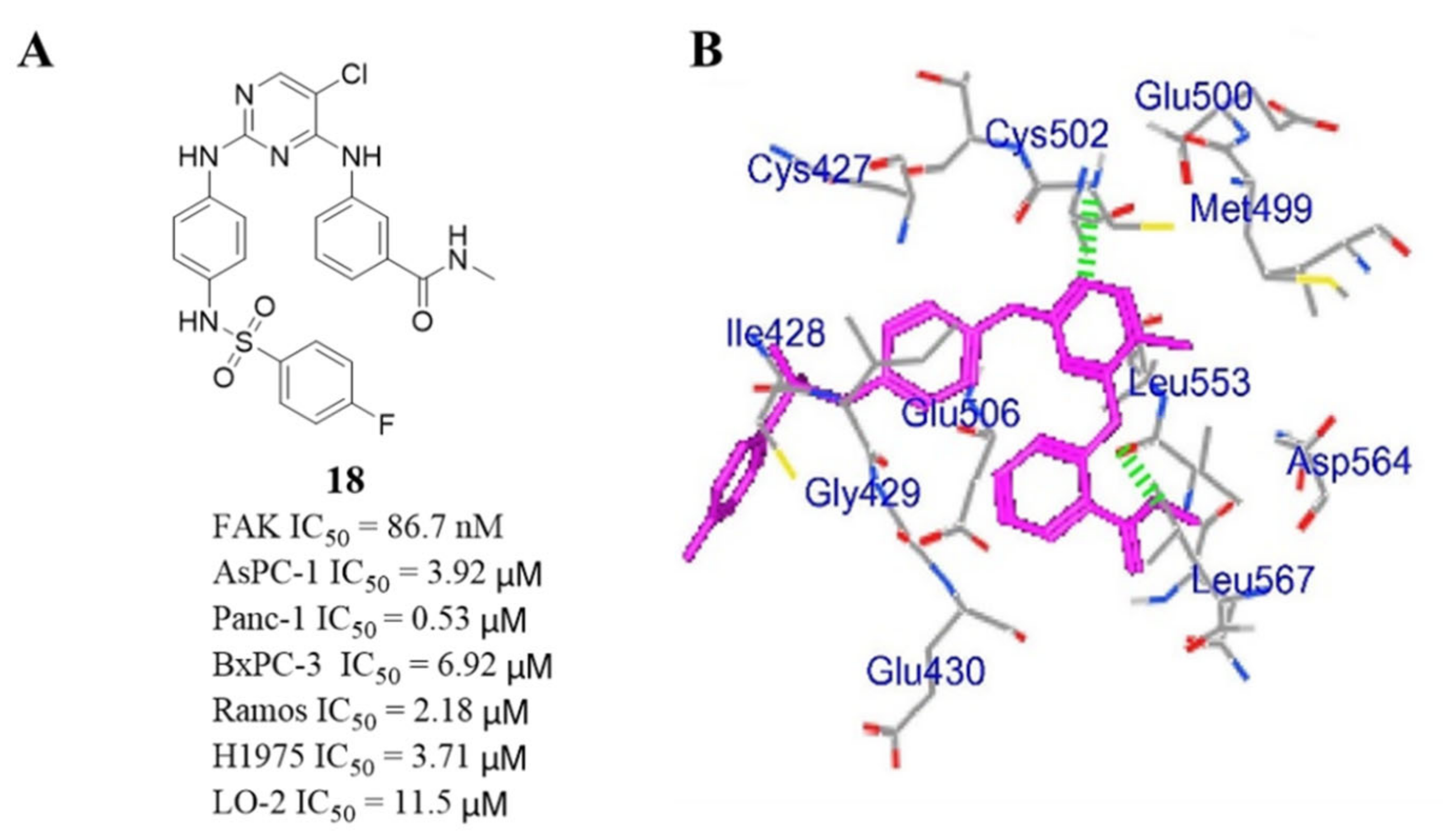
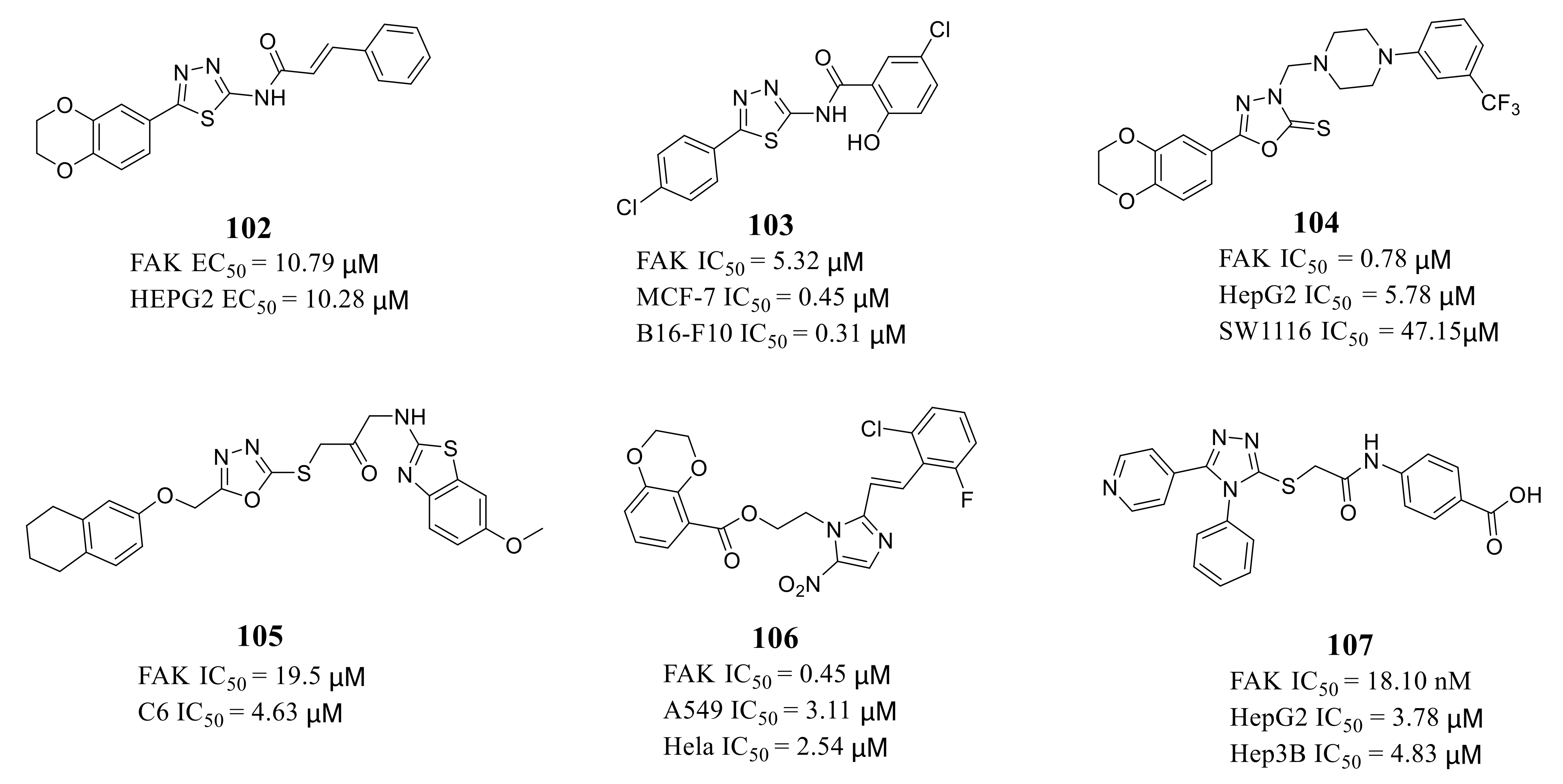
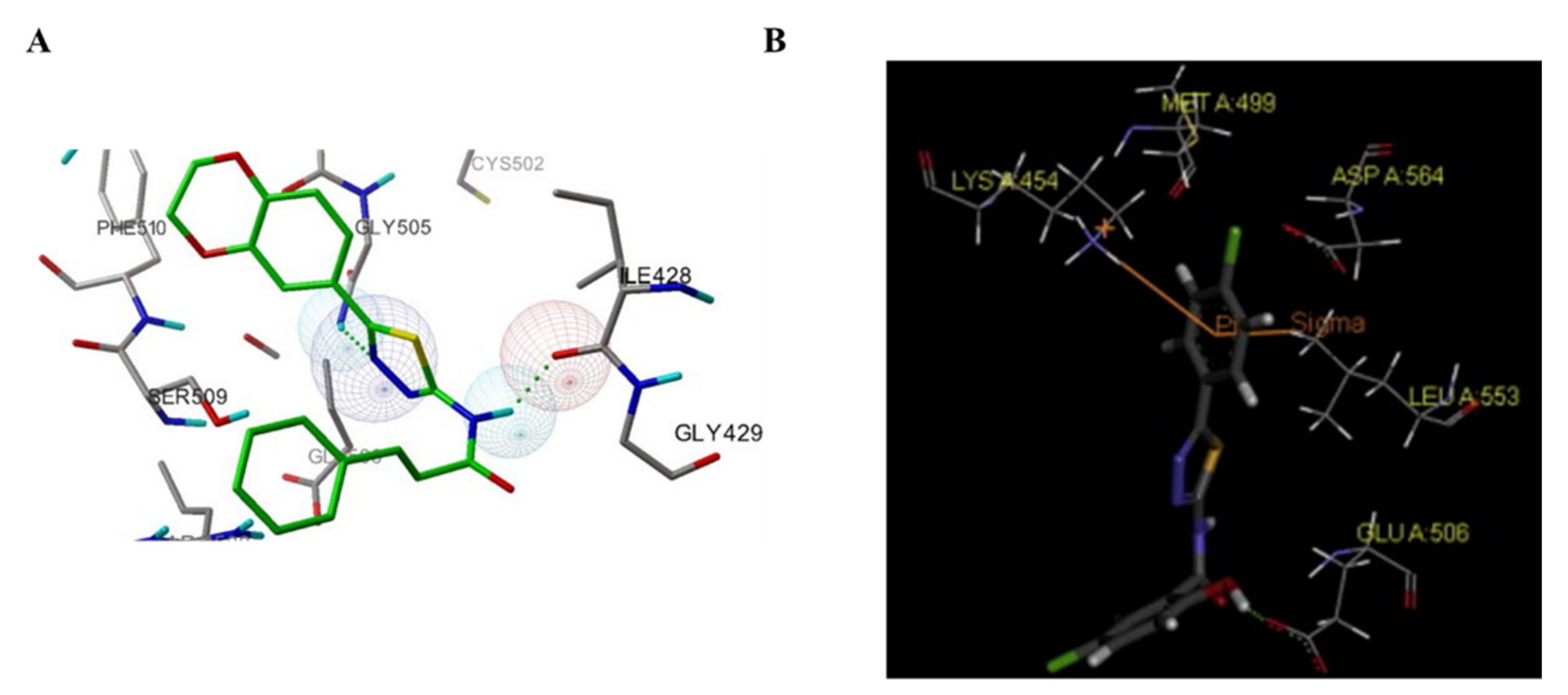
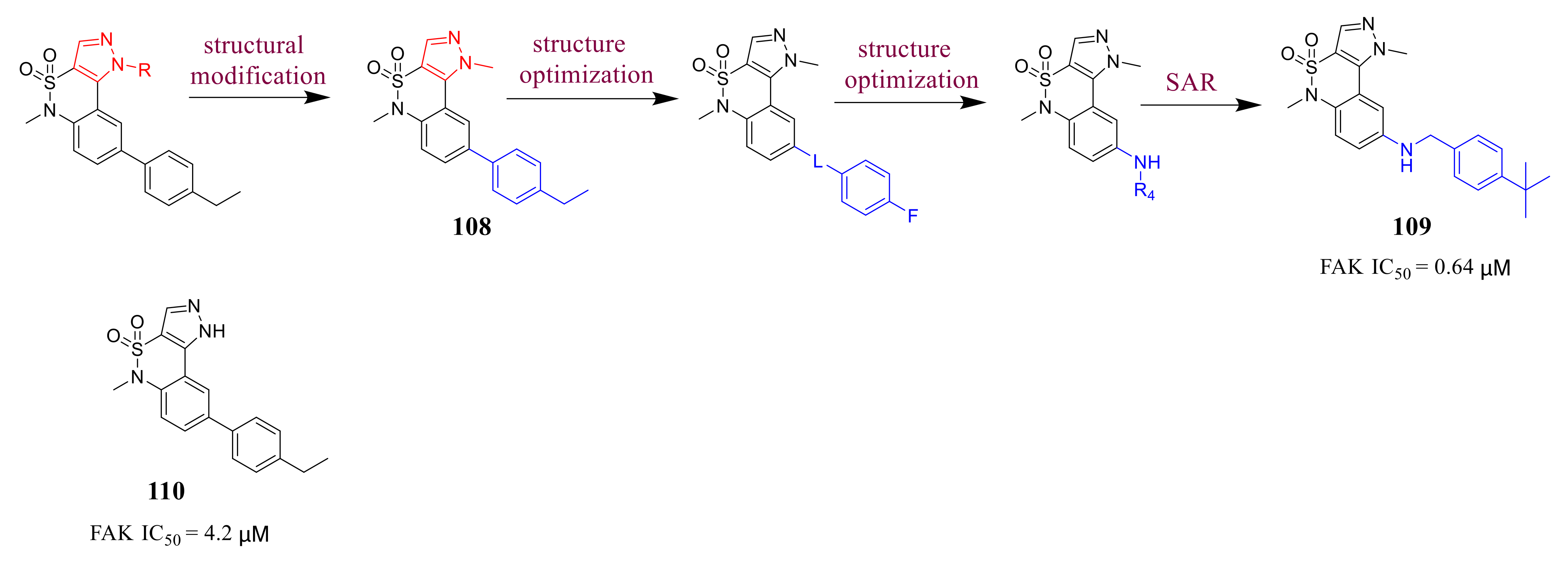
3.5. Other Types of FAK Inhibitors
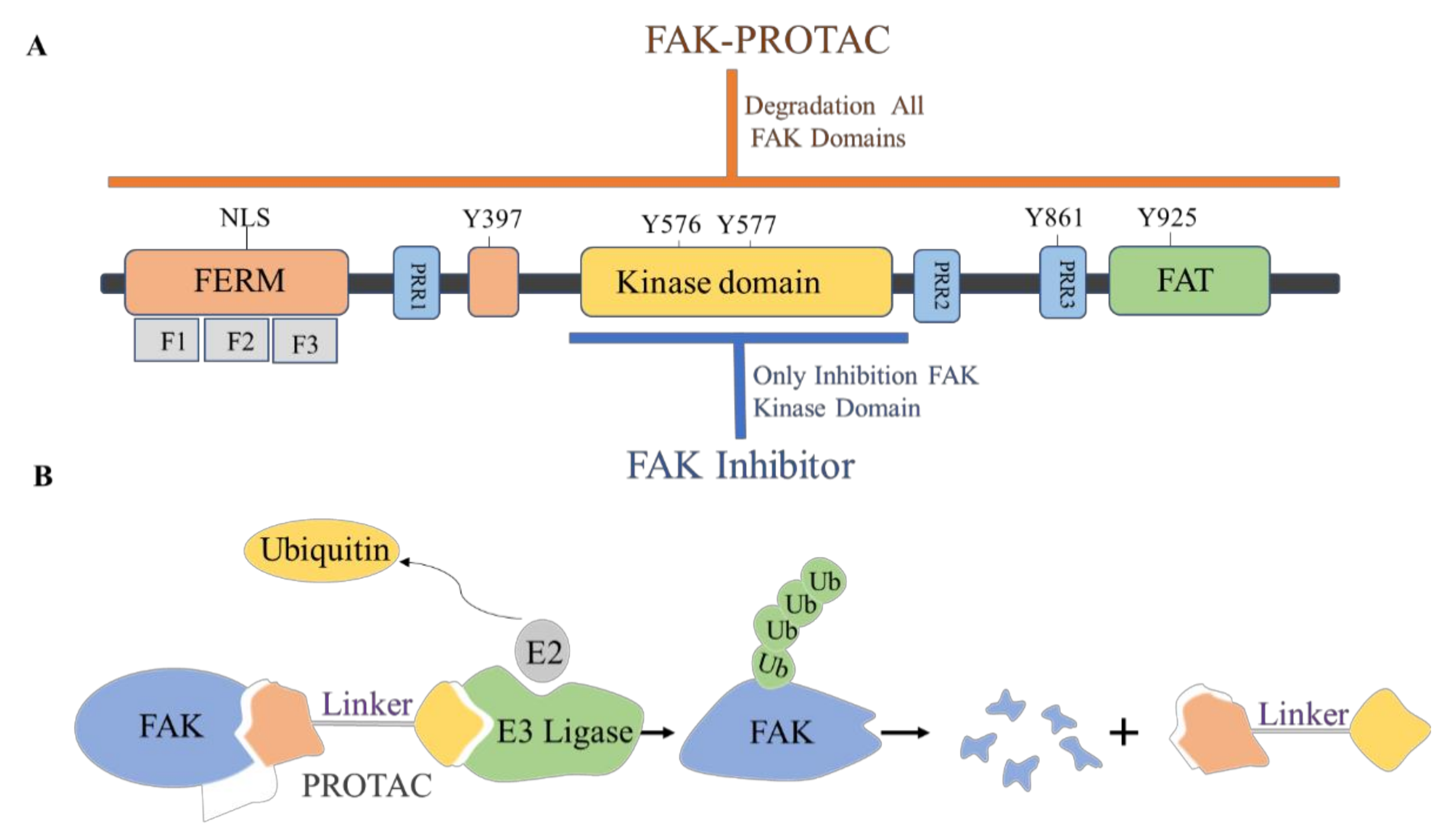
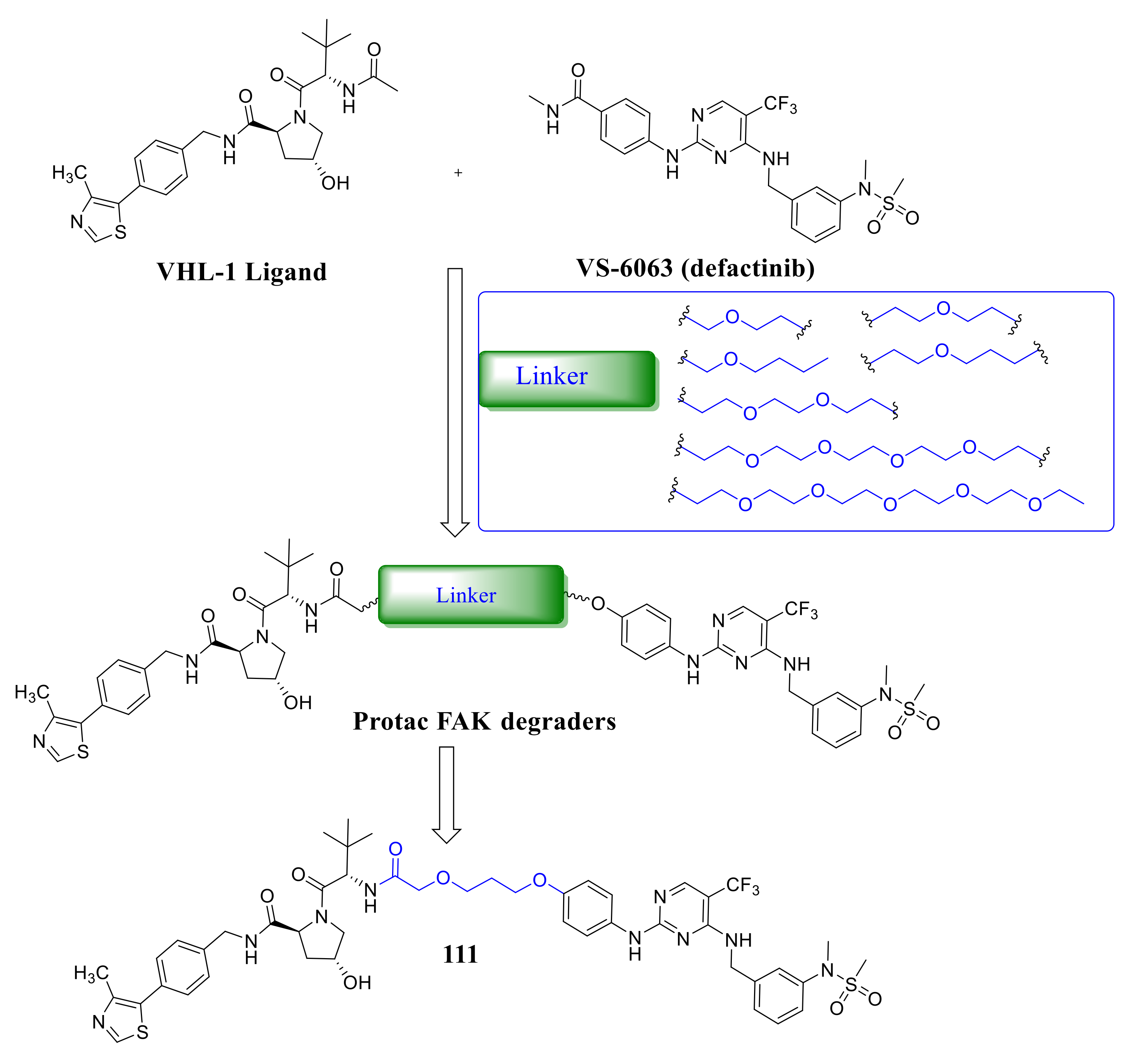
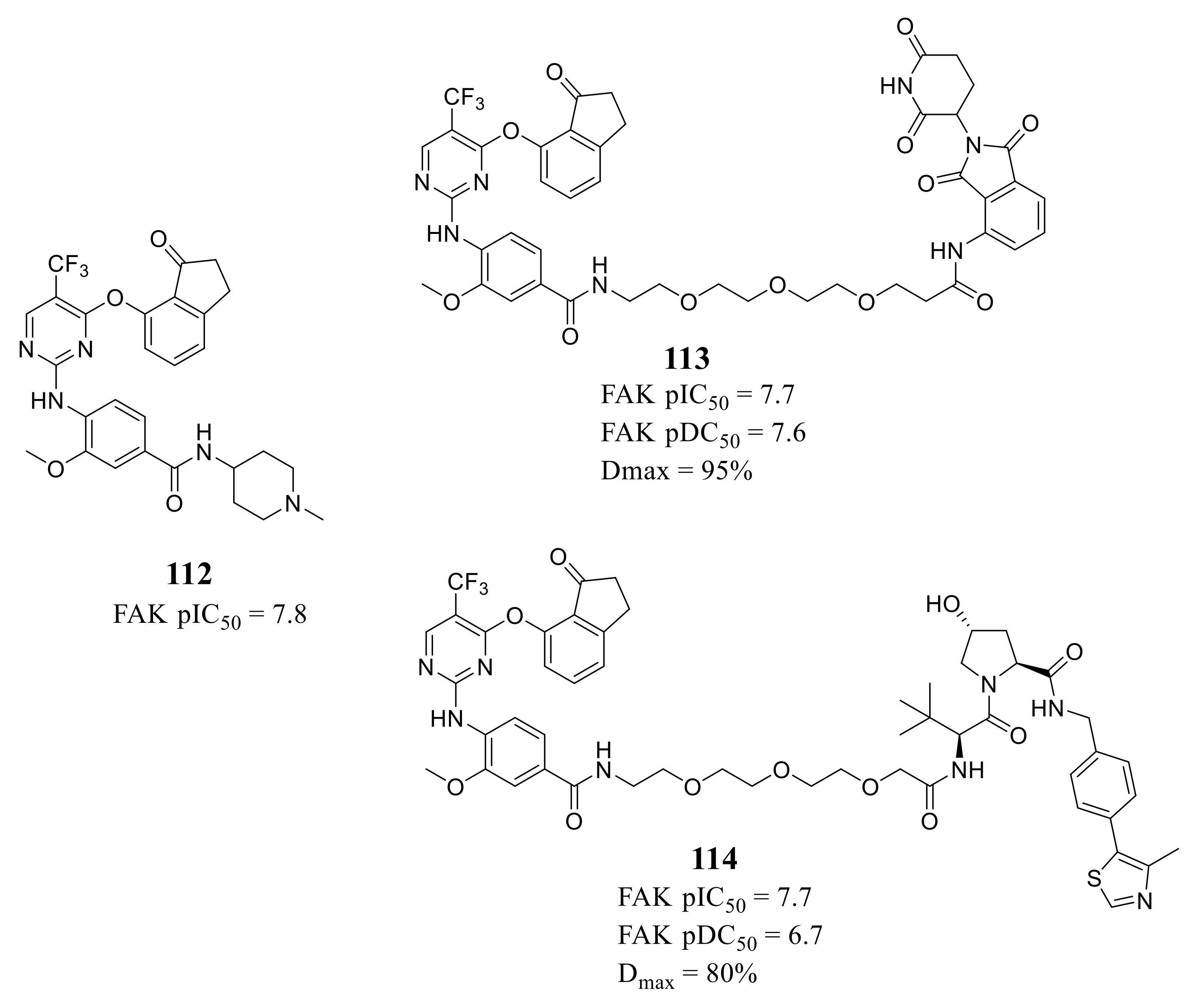
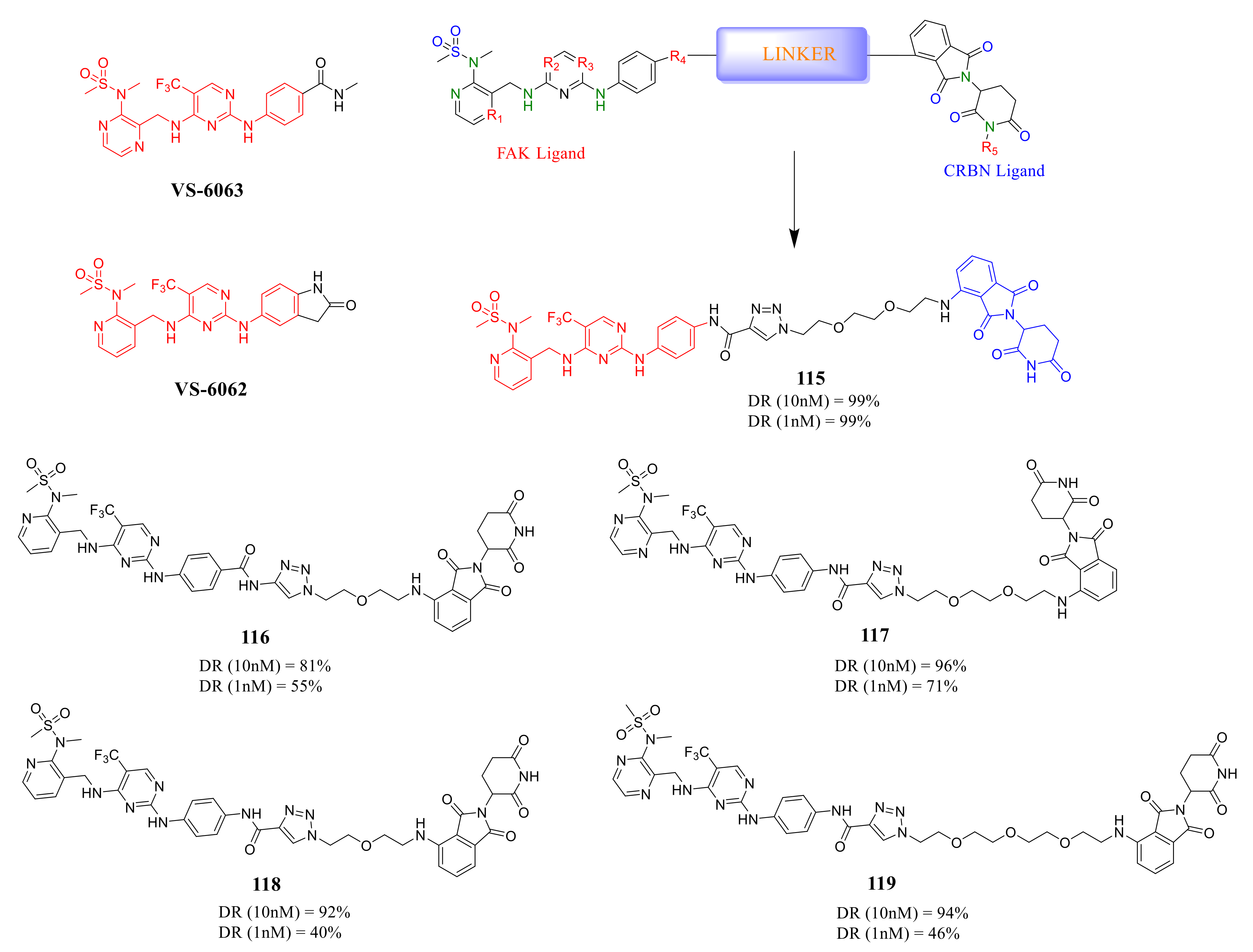
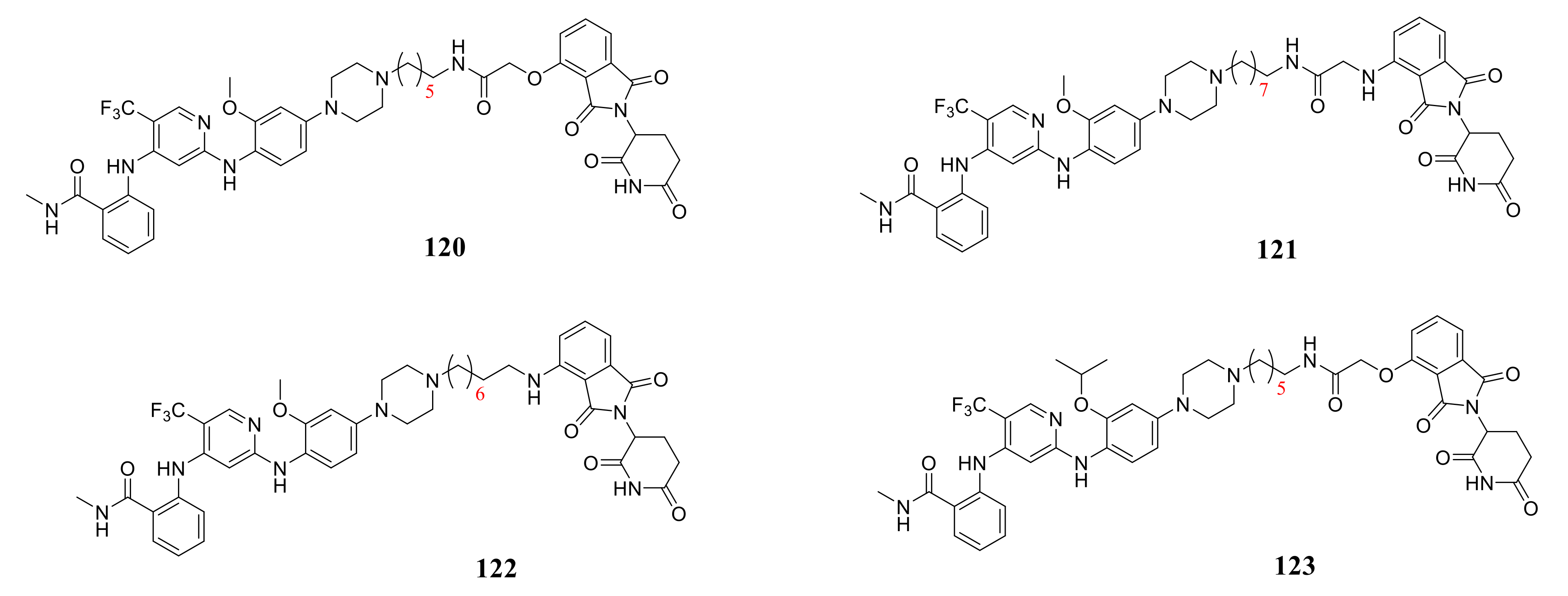
4. The Discovery of FAK-Targeting PROTACs and Their Therapeutic Significance
5. Concluding Remarks and Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- Cui, J.J. A new challenging and promising era of tyrosine kinase inhibitors. ACS Med. Chem. Lett. 2014, 5, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Pan, P.; Sun, H.; Xia, H.; Wang, X.; Li, Y.; Hou, T. Drug Discovery Targeting Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2019, 62, 10927–10954. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef]
- Schaller, M.D. The focal adhesion kinase. J. Endocrinol. 1996, 150, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116, 1409–1416. [Google Scholar] [CrossRef]
- Hildebrand, J.D.; Schaller, M.D.; Parsons, J.T. Identification of sequences required for the efficient localization of the Focal Adhesion Kinase, pp125(FAK), to cellular focal adhesions. J. Cell Biol. 1993, 123, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM domain: Organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef]
- Béraud, C.; Dormoy, V.; Danilin, S.; Lindner, V.; Béthry, A.; Hochane, M.; Coquard, C.; Barthelmebs, M.; Jacqmin, D.; Lang, H.; et al. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth. Int. J. Cancer 2015, 137, 1549–1559. [Google Scholar] [CrossRef]
- Pylayeva, Y.; Gillen, K.M.; Gerald, W.; Beggs, H.E.; Reichardt, L.F.; Giancotti, F.G. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J. Clin. Investig. 2009, 119, 252–266. [Google Scholar] [CrossRef]
- Siesser, P.M.F.; Hanks, S.K. The signaling and biological implications of FAK overexpression in cancer. Clin. Cancer Res. 2006, 12, 3233–3237. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Lechertier, T.; Hodivala-Dilke, K. Focal Adhesion Kinase and tumour angiogenesis. J. Pathol. 2012, 226, 404–412. [Google Scholar] [CrossRef]
- Smith, C.S.; Golubovskaya, V.M.; Peck, E.; Xu, L.-H.; Monia, B.P.; Yang, X.; Cance, W.G. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. Melanoma Res. 2005, 15, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Halder, J.; Landen, C.N., Jr.; Lutgendorf, S.K.; Li, Y.; Jennings, N.B.; Fan, D.; Nelkin, G.M.; Schmandt, R.; Schaller, M.D.; Sood, A.K. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin. Cancer Res. 2005, 11, 8829–8836. [Google Scholar] [CrossRef]
- Demirbas, A.; Sahin, D.; Demirbas, N.; Karaoglu, S.A. Synthesis of some new 1,3,4-thiadiazol-2-ylmethyl-1,2,4-triazole derivatives and investigation of their antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 2896–2903. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.W.; Armstrong, S.A. Designed to kill: Novel menin-MLL inhibitors target MLL-rearranged leukemia. Cancer Cell 2015, 27, 431–433. [Google Scholar] [CrossRef] [PubMed]
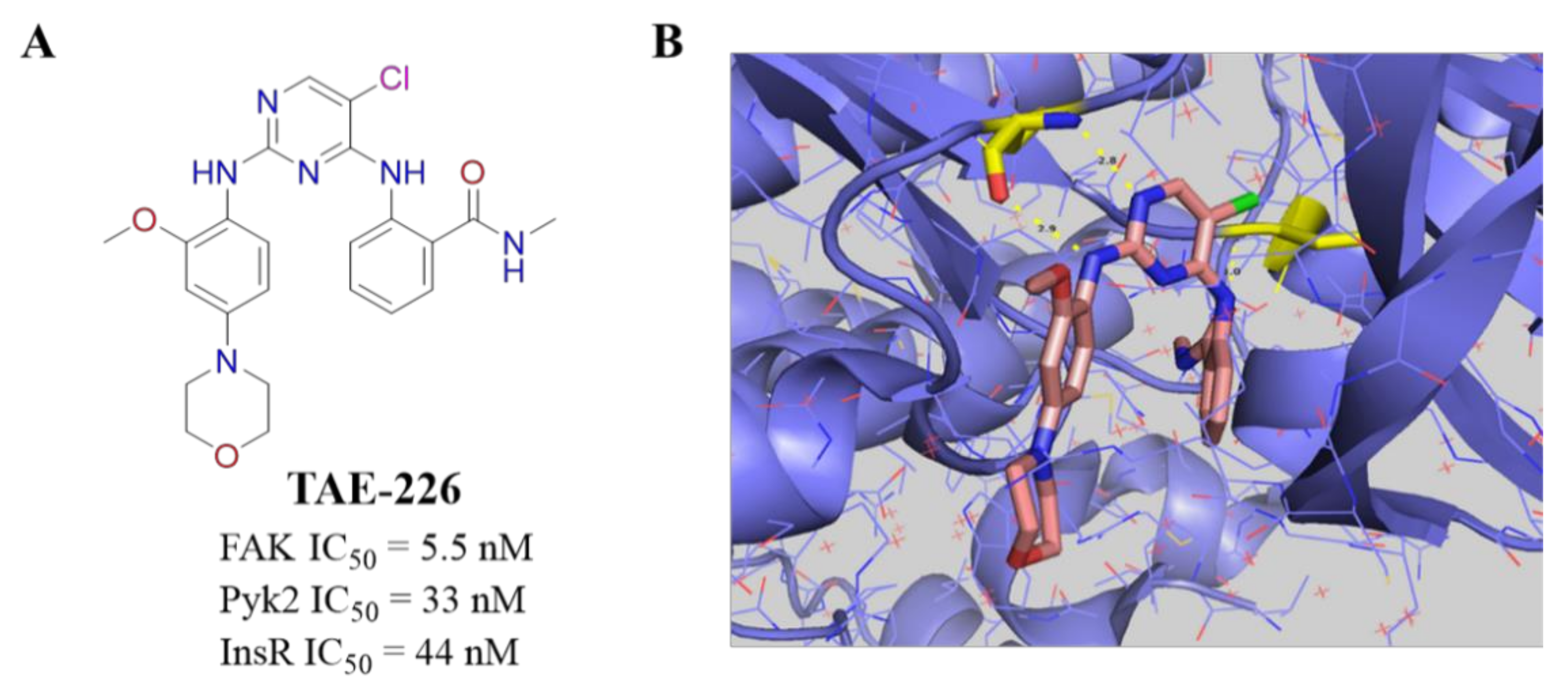
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [CrossRef]
- Ott, G.R.; Cheng, M.; Learn, K.S.; Wagner, J.; Gingrich, D.E.; Lisko, J.G.; Curry, M.; Mesaros, E.F.; Ghose, A.K.; Quail, M.R.; et al. Discovery of Clinical Candidate CEP-37440, a Selective Inhibitor of Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2016, 59, 7478–7496. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukuoka, K.; Takeda, M.; Iwasa, T.; Yoshida, T.; Horobin, J.; Keegan, M.; Vaickus, L.; Chavan, A.; Padval, M.; et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 77, 997–1003. [Google Scholar] [CrossRef]
- Kang, Y.; Hu, W.; Ivan, C.; Dalton, H.J.; Miyake, T.; Pecot, C.V.; Zand, B.; Liu, T.; Huang, J.; Jennings, N.B.; et al. Role of Focal Adhesion Kinase in Regulating YB–1–Mediated Paclitaxel Resistance in Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.F.; Williams, M.; Arkenau, H.T.; Fleming, R.A.; Tolson, J.; Yan, L.; Zhang, J.; Singh, R.; Auger, K.R.; Lenox, L.; et al. A study of the focal adhesion kinase inhibitor GSK2256098 in patients with recurrent glioblastoma with evaluation of tumor penetration of [11C]GSK2256098. Neuro Oncol. 2018, 20, 1634–1642. [Google Scholar] [CrossRef]
- Mak, G.; Soria, J.C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Jones, S.F.; Siu, L.L.; Bendell, J.C.; Cleary, J.M.; Razak, A.R.A.; Infante, J.R.; Pandya, S.S.; Bedard, P.L.; Pierce, K.J.; Houk, B.; et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
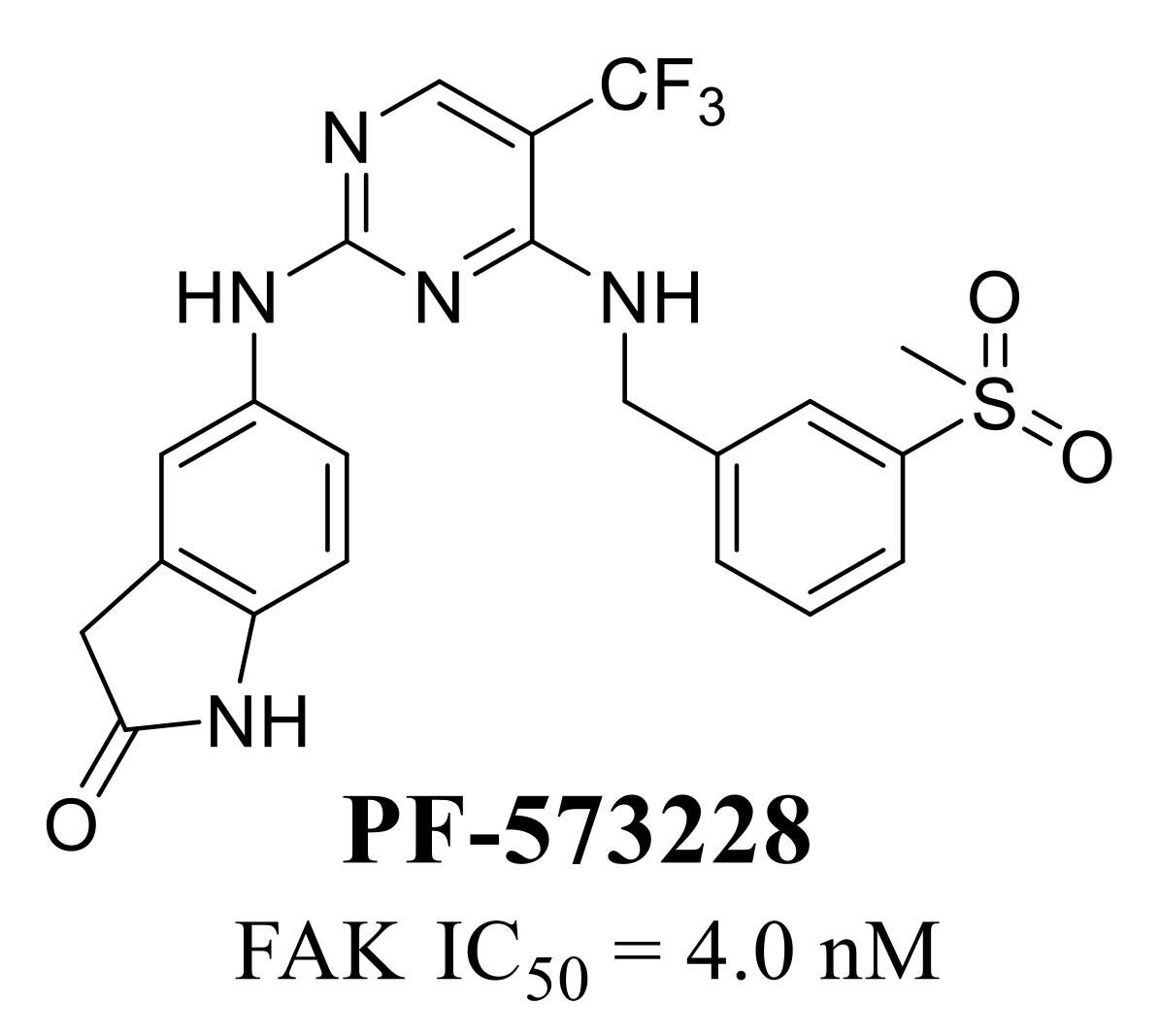
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 2020, 63, 14382–14403. [Google Scholar] [CrossRef]
- Fu, Q.; Socki, R.A.; Niles, P.B. Evaluating reaction pathways of hydrothermal abiotic organic synthesis at elevated temperatures and pressures using carbon isotopes. Geochim. Cosmochim. Acta 2015, 154, 1–17. [Google Scholar] [CrossRef]
- Rosado, P.; Lequerica-Fernández, P.; Peña, I.; Alonso-Durán, L.; de Vicente, J.C. In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression. Virchows Arch. 2012, 461, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gierut, J.; Wang, Z.; Miao, J.; Asara, J.M.; Tyner, A.L. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene 2013, 32, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.-T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Hamadi, A.; Bouali, M.; Dontenwill, M.; Stoeckel, H.; Takeda, K.; Rondé, P. Regulation of focal adhesion dynamics and disassembly by phosphorylation of FAK at tyrosine 397. J. Cell Sci. 2005, 118, 4415–4425. [Google Scholar] [CrossRef]
- Scheswohl, D.M.; Harrell, J.R.; Rajfur, Z.; Gao, G.; Campbell, S.L.; Schaller, M.D. Multiple paxillin binding sites regulate FAK function. J. Mol. Signal. 2008, 3, 1. [Google Scholar] [CrossRef]
- Lawson, C.; Lim, S.-T.; Uryu, S.; Chen, X.L.; Calderwood, D.A.; Schlaepfer, D.D. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012, 196, 223–232. [Google Scholar] [CrossRef]
- Subauste, M.C.; Pertz, O.; Adamson, E.D.; Turner, C.E.; Junger, S.; Hahn, K.M. Vinculin modulation of paxillin–FAK interactions regulates ERK to control survival and motility. J. Cell Biol. 2004, 165, 371–381. [Google Scholar] [CrossRef]
- Cho, J.-H.; Muralidharan, V.; Vila-Perello, M.; Raleigh, D.P.; Muir, T.W.; Palmer, A.G. Tuning protein autoinhibition by domain destabilization. Nat. Struct. Mol. Biol. 2011, 18, 550–555. [Google Scholar] [CrossRef][Green Version]
- Golubovskaya, V.M.; Kwen, F.A.; Cance, W.G. Focal adhesion kinase and cancer. Histol. Histopathol. 2009, 24, 503–510. [Google Scholar]
- Zhang, J.; He, D.-H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Spigel, D.R.; Jeong, W.; Scaglioni, P.P.; Zhang, S.; et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Roberts, W.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor Activity and Pharmacology of a Selective Focal Adhesion Kinase Inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Crompton, B.D.; Carlton, A.L.; Thorner, A.R.; Christie, A.L.; Du, J.; Calicchio, M.L.; Rivera, M.N.; Fleming, M.D.; Kohl, N.E.; Kung, A.L.; et al. High-Throughput Tyrosine Kinase Activity Profiling Identifies FAK as a Candidate Therapeutic Target in Ewing Sarcoma. Cancer Res. 2013, 73, 2873–2883. [Google Scholar] [CrossRef]
- Bagi, C.M.; Christensen, J.; Cohen, D.P.; Roberts, W.G.; Wilkie, D.; Swanson, T.; Tuthill, T.; Andresen, C.J. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol. Ther. 2009, 8, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.L.; Baggerly, K.A.; Armaiz-Pena, G.N.; Kang, Y.; Sanguino, A.M.; Thanapprapasr, D.; Dalton, H.J.; Bottsford-Miller, J.; Zand, B.; Akbani, R.; et al. Focal adhesion kinase. Cancer Biol. Ther. 2014, 15, 919–929. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Infante, J.R.; Camidge, D.R.; Mileshkin, L.R.; Chen, E.X.; Hicks, R.J.; Rischin, D.; Fingert, H.; Pierce, K.J.; Xu, H.; Roberts, W.G.; et al. Safety, Pharmacokinetic, and Pharmacodynamic Phase I Dose-Escalation Trial of PF-00562271, an Inhibitor of Focal Adhesion Kinase, in Advanced Solid Tumors. J. Clin. Oncol. 2012, 30, 1527–1533. [Google Scholar] [CrossRef]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.-O.; Mielgo, A.; Lim, S.-T.; Liang, C.; Koenig, M.; Patel, N.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef]
- Kurmasheva, R.T.; Gorlick, R.; Kolb, E.A.; Keir, S.T.; Maris, J.M.; Lock, R.B.; Carol, H.; Kang, M.; Reynolds, C.P.; Wu, J.; et al. Initial testing of VS-4718, a novel inhibitor of focal adhesion kinase (FAK), against pediatric tumor models by the Pediatric Preclinical Testing Program. Pediatric Blood Cancer 2017, 64, e26304. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Eckford, P.D.W.; Sharom, F.J. ABC Efflux Pump-Based Resistance to Chemotherapy Drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Stavrovskaya, A.A.; Stromskaya, T.P. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry 2008, 73, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Yang, Y.; Cai, C.-Y.; Lei, Z.-N.; Wang, J.-Q.; Gupta, P.; Teng, Q.-X.; Chen, Z.-S.; Kong, D.; Yang, D.-H. VS-4718 Antagonizes Multidrug Resistance in ABCB1- and ABCG2-Overexpressing Cancer Cells by Inhibiting the Efflux Function of ABC Transporters. Front. Pharmacol. 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Frisch, S.M. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 791–793. [Google Scholar] [CrossRef][Green Version]
- Salem, I.; Alsalahi, M.; Chervoneva, I.; Aburto, L.D.; Addya, S.; Ott, G.R.; Ruggeri, B.A.; Cristofanilli, M.; Fernandez, S.V. The effects of CEP-37440, an inhibitor of focal adhesion kinase, in vitro and in vivo on inflammatory breast cancer cells. Breast Cancer Res. 2016, 18, 37. [Google Scholar] [CrossRef]
- Song, Z.; Yang, Y.; Liu, Z.; Peng, X.; Guo, J.; Yang, X.; Wu, K.; Ai, J.; Ding, J.; Geng, M.; et al. Discovery of Novel 2,4-Diarylaminopyrimidine Analogues (DAAPalogues) Showing Potent Inhibitory Activities against Both Wild-type and Mutant ALK Kinases. J. Med. Chem. 2015, 58, 197–211. [Google Scholar] [CrossRef]
- Huang, W.-S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef] [PubMed]
- Ott, G.R.; Tripathy, R.; Cheng, M.; McHugh, R.; Anzalone, A.V.; Underiner, T.L.; Curry, M.A.; Quail, M.R.; Lu, L.; Wan, W.; et al. Discovery of a potent inhibitor of anaplastic lymphoma kinase with in vivo antitumor activity. ACS Med. Chem. Lett. 2010, 1, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal Structures of Anaplastic Lymphoma Kinase in Complex with ATP Competitive Inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.G.; Fukazawa, T.; Nishikawa, T.; Watanabe, N.; Sakurama, K.; Motoki, T.; Takaoka, M.; Hatakeyama, S.; Omori, O.; Ohara, T.; et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol. Rep. 2008, 20, 1473–1477. [Google Scholar]
- Fukami, S.; Tomioka, D.; Murakami, Y.; Honda, T.; Hatakeyama, S. Pharmacological profiling of a dual FAK/IGF-1R kinase inhibitor TAE226 in cellular and in vivo tumor models. BMC Res. Notes 2019, 12, 347. [Google Scholar] [CrossRef]
- Hao, H.F.; Naomoto, Y.; Bao, X.H.; Watanabe, N.; Sakurama, K.; Noma, K.; Tomono, Y.; Fukazawa, T.; Shirakawa, Y.; Yamatsuji, T.; et al. Progress in researches about focal adhesion kinase in gastrointestinal tract. World J. Gastroenterol. 2009, 15, 5916–5923. [Google Scholar] [CrossRef]
- Tai, Y.-L.; Chen, L.-C.; Shen, T.-L. Emerging Roles of Focal Adhesion Kinase in Cancer. BioMed Res. Int. 2015, 2015, 690690. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Eck, M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE 2008, 3, e3800. [Google Scholar] [CrossRef]
- Alexander, S.; Walter, F. Clinical Importance and Potential Use of Small Molecule Inhibitors of Focal Adhesion Kinase. Anti Cancer Agents Med. Chem. 2011, 11, 593–599. [Google Scholar]
- Schultze, A.; Fiedler, W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 777–788. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Martin, K.H.; Tilghman, R.W.; Iwanicki, M.; Ung, E.J.; Autry, C.; Luzzio, M.J.; Cooper, B.; Kath, J.C.; Roberts, W.G.; et al. Cellular Characterization of a Novel Focal Adhesion Kinase Inhibitor. J. Biol. Chem. 2007, 282, 14845–14852. [Google Scholar] [CrossRef] [PubMed]
- Determann, R.; Dreher, J.; Baumann, K.; Preu, L.; Jones, P.G.; Totzke, F.; Schachtele, C.; Kubbutat, M.H.; Kunick, C. 2-Anilino-4-(benzimidazol-2-yl)pyrimidines-a multikinase inhibitor scaffold with antiproliferative activity toward cancer cell lines. Eur. J. Med. Chem. 2012, 53, 254–263. [Google Scholar] [CrossRef]
- Farand, J.; Mai, N.; Chandrasekhar, J.; Newby, Z.E.; van Veldhuizen, J.; Loyer-Drew, J.; Venkataramani, C.; Guerrero, J.; Kwok, A.; Li, N.; et al. Selectivity switch between FAK and Pyk2: Macrocyclization of FAK inhibitors improves Pyk2 potency. Bioorg. Med. Chem. Lett. 2016, 26, 5926–5930. [Google Scholar] [CrossRef] [PubMed]
- Winum, J.-Y.; Scozzafava, A.; Montero, J.-L.; Supuran, C.T. Therapeutic potential of sulfamides as enzyme inhibitors. Med. Res. Rev. 2006, 26, 767–792. [Google Scholar] [CrossRef]
- Scozzafava, A.; Supuran, C.T. Carbonic anhydrase and matrix metalloproteinase inhibitors: Sulfonylated amino acid hydroxamates with MMP inhibitory properties act as efficient inhibitors of CA isozymes I, II, and IV, and N-hydroxysulfonamides inhibit both these zinc enzymes. J. Med. Chem. 2000, 43, 3677–3687. [Google Scholar] [CrossRef]
- Fukuoka, K.; Usuda, J.; Iwamoto, Y.; Fukumoto, H.; Nakamura, T.; Yoneda, T.; Narita, N.; Saijo, N.; Nishio, K. Mechanisms of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non-small cell lung cancer cells. Investig. New Drugs 2001, 19, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qu, M.; Xu, L.; Han, X.; Wang, C.; Shu, X.; Yao, J.; Liu, K.; Peng, J.; Li, Y.; et al. Design and synthesis of sulfonamide-substituted diphenylpyrimidines (SFA-DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors with improved activity toward B-cell lymphoblastic leukemia. Eur. J. Med. Chem. 2017, 135, 60–69. [Google Scholar] [CrossRef]
- Qu, M.; Liu, Z.; Zhao, D.; Wang, C.; Zhang, J.; Tang, Z.; Liu, K.; Shu, X.; Yuan, H.; Ma, X. Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (Sul-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with antitumor activity. Bioorg. Med. Chem. 2017, 25, 3989–3996. [Google Scholar] [CrossRef] [PubMed]
- Shanthi, E.; Krishna, M.H.; Arunesh, G.M.; Reddy, K.V.; Kumar, J.S.; Viswanadhan, V.N. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: A patent review. Expert Opin. Ther. Pat. 2014, 24, 1077–1100. [Google Scholar] [CrossRef]
- Cao, F.Y.; Zhou, X.P.; Su, J.; Yang, X.H.; Mu, F.H.; Shen, J.Y.; Sun, W. Chemical Structure Characteristics and Bioactivity of Small Molecule FAK Inhibitors. Anti Cancer Agents Med. Chem. 2016, 16, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Lee, M.Y.; Kim, M.H.; Kim, J.; Kim, S.H.; Kim, B.T.; Jeong, I.H.; Chang, S.; Kim, S.H.; Chang, S.-Y. Synthesis and SAR of sulfonyl- and phosphoryl amidine compounds as anti-resorptive agents. Bioorg. Med. Chem. Lett. 2010, 20, 541–545. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Rautio, J. Prodrugs—An efficient way to breach delivery and targeting barriers. Curr. Top. Med. Chem. 2011, 11, 2265–2287. [Google Scholar] [CrossRef]
- Kamiyama, A.; Nakajima, M.; Han, L.; Wada, K.; Mizutani, M.; Tabuchi, Y.; Kojima-Yuasa, A.; Matsui-Yuasa, I.; Suzuki, H.; Fukuyama, K.; et al. Phosphonate-based irreversible inhibitors of human γ-glutamyl transpeptidase (GGT). GGsTop is a non-toxic and highly selective inhibitor with critical electrostatic interaction with an active-site residue Lys562 for enhanced inhibitory activity. Bioorg. Med. Chem. 2016, 24, 5340–5352. [Google Scholar] [CrossRef]
- Ge, Y.; Yang, H.; Wang, C.; Meng, Q.; Li, L.; Sun, H.; Zhen, Y.; Liu, K.; Li, Y.; Ma, X. Design and synthesis of phosphoryl-substituted diphenylpyrimidines (Pho-DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors: Targeted treatment of B lymphoblastic leukemia cell lines. Bioorg. Med. Chem. 2017, 25, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Huang, S.; Qu, M.; Wang, C.; Liu, Z.; Li, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X.; et al. Structural optimization of diphenylpyrimidine derivatives (DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors with improved activity toward B leukemia cell lines. Eur. J. Med. Chem. 2017, 126, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Jin, Y.; Wang, C.; Zhang, J.; Tang, Z.; Peng, J.; Liu, K.; Li, Y.; Zhou, Y.; Ma, X. Discovery of Novel Bruton’s Tyrosine Kinase (BTK) Inhibitors Bearing a N,9-Diphenyl-9H-purin-2-amine Scaffold. ACS Med. Chem. Lett. 2016, 7, 1050–1055. [Google Scholar] [CrossRef]
- Liu, H.-M.; Lee, C.-W. Reply to Letter to the Editor “Concern on article “Predicting procedure successful rate and 1-year patency after endovascular recanalization for chronic carotid artery occlusion by CT angiography”. Int. J. Cardiol. 2017, 229, 60. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, B.; Ge, Y.; Huang, J.; Song, S.; Wang, C.; Yao, J.; Liu, K.; Li, Y.; Li, Y.; et al. Phosphamide-containing diphenylpyrimidine analogues (PA-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with enhanced activity against pancreatic cancer cell lines. Bioorg. Med. Chem. 2017, 25, 6313–6321. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Xu, T.; He, G.; Ouyang, L.; Han, B.; Peng, C.; Song, X.; Xiang, M. Discovery of Novel Focal Adhesion Kinase Inhibitors Using a Hybrid Protocol of Virtual Screening Approach Based on Multicomplex-Based Pharmacophore and Molecular Docking. Int. J. Mol. Sci. 2012, 13, 15668–15678. [Google Scholar] [CrossRef]
- Wang, L.; Ai, M.; Yu, J.; Jin, L.; Wang, C.; Liu, Z.; Shu, X.; Tang, Z.; Liu, K.; Luo, H.; et al. Structure-based modification of carbonyl-diphenylpyrimidines (Car-DPPYs) as a novel focal adhesion kinase (FAK) inhibitor against various stubborn cancer cells. Eur. J. Med. Chem. 2019, 172, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, K. Irreversible kinase inhibitors gain traction. Nat. Rev. Drug Discov. 2013, 12, 649–651. [Google Scholar] [CrossRef]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Barf, T.; Kaptein, A. Irreversible protein kinase inhibitors: Balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef]
- Yen-Pon, E.; Li, B.; Acebrón-Garcia-de-Eulate, M.; Tomkiewicz-Raulet, C.; Dawson, J.; Lietha, D.; Frame, M.C.; Coumoul, X.; Garbay, C.; Etheve-Quelquejeu, M.; et al. Structure-Based Design, Synthesis, and Characterization of the First Irreversible Inhibitor of Focal Adhesion Kinase. ACS Chem. Biol. 2018, 13, 2067–2073. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; Bolzani, V.d.; Barreiro, E.J.; Fraga, C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Fu, D.-J.; Zhang, L.; Song, J.; Mao, R.-W.; Zhao, R.-H.; Liu, Y.-C.; Hou, Y.-H.; Li, J.-H.; Yang, J.-J.; Jin, C.-Y.; et al. Design and synthesis of formononetin-dithiocarbamate hybrids that inhibit growth and migration of PC-3 cells via MAPK/Wnt signaling pathways. Eur. J. Med. Chem. 2017, 127, 87–99. [Google Scholar] [CrossRef]
- Fu, D.-J.; Zhang, S.-Y.; Liu, Y.-C.; Zhang, L.; Liu, J.-J.; Song, J.; Zhao, R.-H.; Li, F.; Sun, H.-H.; Liu, H.-M.; et al. Design, synthesis and antiproliferative activity studies of novel dithiocarbamate–chalcone derivates. Bioorg. Med. Chem. Lett. 2016, 26, 3918–3922. [Google Scholar] [CrossRef]
- Su, Y.; Li, R.; Ning, X.; Lin, Z.; Zhao, X.; Zhou, J.; Liu, J.; Jin, Y.; Yin, Y. Discovery of 2,4-diarylaminopyrimidine derivatives bearing dithiocarbamate moiety as novel FAK inhibitors with antitumor and anti-angiogenesis activities. Eur. J. Med. Chem. 2019, 177, 32–46. [Google Scholar] [CrossRef]
- Ai, M.; Wang, C.; Tang, Z.; Liu, K.; Sun, X.; Ma, T.; Li, Y.; Ma, X.; Li, L.; Chen, L. Design and synthesis of diphenylpyrimidine derivatives (DPPYs) as potential dual EGFR T790M and FAK inhibitors against a diverse range of cancer cell lines. Bioorg. Chem. 2020, 94, 103408. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Wang, Z. EGFR mutations as a prognostic and predictive marker in non-small-cell lung cancer. Drug Des. Dev. Ther. 2014, 8, 1595–1611. [Google Scholar]
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J.; et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, Y.; Tomkiewicz-Raulet, C.; Dao, P.; Lietha, D.; Yen-Pon, E.; Du, Z.; Coumoul, X.; Garbay, C.; Etheve-Quelquejeu, M.; et al. Design, Synthesis, and Biological Evaluation of Covalent Inhibitors of Focal Adhesion Kinase (FAK) against Human Malignant Glioblastoma. J. Med. Chem. 2020, 63, 12707–12724. [Google Scholar] [CrossRef]
- Qi, Y.; Li, Y.; Fang, Y.; Qiang, B.; Gao, H.; Wang, S.; Zhang, H. Design, synthesis, and biological evaluation of F-18-labelled 2, 4-diaminopyrimidine-type FAK-targeted inhibitors as potential tumour imaging agents. Bioorg. Med. Chem. Lett. 2020, 30, 127452. [Google Scholar] [CrossRef]
- Qi, Y.; Li, Y.; Fang, Y.; Gao, H.; Qiang, B.; Wang, S.; Zhang, H. Design, Synthesis, Biological Evaluation, and Molecular Docking of 2,4-Diaminopyrimidine Derivatives Targeting Focal Adhesion Kinase as Tumor Radiotracers. Mol. Pharm. 2021, 18, 1634–1642. [Google Scholar] [CrossRef]
- Wang, R.; Chen, Y.; Zhao, X.; Yu, S.; Yang, B.; Wu, T.; Guo, J.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis and biological evaluation of novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potential FAK inhibitors and anticancer agents. Eur. J. Med. Chem. 2019, 183, 111716. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, S.; Zhao, X.; Chen, Y.; Yang, B.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 2020, 188, 112024. [Google Scholar] [CrossRef] [PubMed]
- Groendyke, B.J.; Nabet, B.; Mohardt, M.L.; Zhang, H.; Peng, K.; Koide, E.; Coffey, C.R.; Che, J.; Scott, D.A.; Bass, A.J.; et al. Discovery of a Pyrimidothiazolodiazepinone as a Potent and Selective Focal Adhesion Kinase (FAK) Inhibitor. ACS Med. Chem. Lett. 2021, 12, 30–38. [Google Scholar] [CrossRef]
- Groendyke, B.J.; Powell, C.E.; Feru, F.; Gero, T.W.; Li, Z.; Szabo, H.; Pang, K.; Feutrill, J.; Chen, B.; Li, B.; et al. Benzopyrimidodiazepinone inhibitors of TNK2. Bioorg. Med. Chem. Lett. 2020, 30, 126948. [Google Scholar] [CrossRef]
- Song, J.; Cui, X.-X.; Wu, B.-W.; Li, D.; Wang, S.-H.; Shi, L.; Zhu, T.; Zhang, Y.-B.; Zhang, S.-Y. Discovery of 1,2,4-triazine-based derivatives as novel neddylation inhibitors and anticancer activity studies against gastric cancer MGC-803 cells. Bioorg. Med. Chem. Lett. 2020, 30, 126791. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Song, J.; Hou, Y.-H.; Zhao, R.-H.; Li, J.-H.; Mao, R.-W.; Yang, J.-J.; Li, P.; Zi, X.-L.; Li, Z.-H.; et al. Discovery of 5,6-diaryl-1,2,4-triazines hybrids as potential apoptosis inducers. Eur. J. Med. Chem. 2017, 138, 1076–1088. [Google Scholar] [CrossRef]
- Zificsak, C.A.; Gingrich, D.E.; Breslin, H.J.; Dunn, D.D.; Milkiewicz, K.L.; Theroff, J.P.; Thieu, T.V.; Underiner, T.L.; Weinberg, L.R.; Aimone, L.D.; et al. Optimization of a novel kinase inhibitor scaffold for the dual inhibition of JAK2 and FAK kinases. Bioorg. Med. Chem. Lett. 2012, 22, 133–137. [Google Scholar] [CrossRef]
- Dao, P.; Jarray, R.; le Coq, J.; Lietha, D.; Loukaci, A.; Lepelletier, Y.; Hadj-Slimane, R.; Garbay, C.; Raynaud, F.; Chen, H. Synthesis of novel diarylamino-1,3,5-triazine derivatives as FAK inhibitors with anti-angiogenic activity. Bioorg. Med. Chem. Lett. 2013, 23, 4552–4556. [Google Scholar] [CrossRef]
- Dao, P.; Smith, N.; Tomkiewicz-Raulet, C.; Yen-Pon, E.; Camacho-Artacho, M.; Lietha, D.; Herbeuval, J.P.; Coumoul, X.; Garbay, C.; Chen, H. Design, synthesis, and evaluation of novel imidazo[1,2-a][1,3,5]triazines and their derivatives as focal adhesion kinase inhibitors with antitumor activity. J. Med. Chem. 2015, 58, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Dao, P.; Lietha, D.; Etheve-Quelquejeu, M.; Garbay, C.; Chen, H. Synthesis of novel 1,2,4-triazine scaffold as FAK inhibitors with antitumor activity. Bioorg. Med. Chem. Lett. 2017, 27, 1727–1730. [Google Scholar] [CrossRef]
- Yang, X.-H.; Xiang, L.; Li, X.; Zhao, T.-T.; Zhang, H.; Zhou, W.-P.; Wang, X.-M.; Gong, H.-B.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of 1,3,4-thiadiazol-2-amide derivatives as novel anticancer agents. Bioorg. Med. Chem. 2012, 20, 2789–2795. [Google Scholar] [CrossRef]
- Sun, J.; Yang, Y.-S.; Li, W.; Zhang, Y.-B.; Wang, X.-L.; Tang, J.-F.; Zhu, H.-L. Synthesis, biological evaluation and molecular docking studies of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan as potential antitumor agents. Bioorg. Med. Chem. Lett. 2011, 21, 6116–6121. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ren, S.-Z.; Lu, X.-Y.; Li, J.-J.; Shen, F.-Q.; Xu, C.; Zhu, H.-L. Discovery of a series of 1,3,4-oxadiazole-2(3H)-thione derivatives containing piperazine skeleton as potential FAK inhibitors. Bioorg. Med. Chem. 2017, 25, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Sever, B.; Çiftçi, G.A.; Turan-Zitouni, G.; Kaplancıklı, Z.A.; Özdemir, A. Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors. Eur. J. Med. Chem. 2018, 155, 905–924. [Google Scholar] [CrossRef]
- Duan, Y.-T.; Yao, Y.-F.; Huang, W.; Makawana, J.A.; Teraiya, S.B.; Thumar, N.j.; Tang, D.-J.; Tao, X.-X.; Wang, Z.-C.; Jiang, A.-Q.; et al. Synthesis, biological evaluation, and molecular docking studies of novel 2-styryl-5-nitroimidazole derivatives containing 1,4-benzodioxan moiety as FAK inhibitors with anticancer activity. Bioorg. Med. Chem. 2014, 22, 2947–2954. [Google Scholar] [CrossRef]
- Mustafa, M.; Abuo-Rahma, G.E.-D.A.; El-Hafeez, A.A.A.; Ahmed, E.R.; Abdelhamid, D.; Ghosh, P.; Hayallah, A.M. Discovery of antiproliferative and anti-FAK inhibitory activity of 1,2,4-triazole derivatives containing acetamido carboxylic acid skeleton. Bioorg. Med. Chem. Lett. 2021, 40, 127965. [Google Scholar] [CrossRef]
- Tomita, N.; Hayashi, Y.; Suzuki, S.; Oomori, Y.; Aramaki, Y.; Matsushita, Y.; Iwatani, M.; Iwata, H.; Okabe, A.; Awazu, Y.; et al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg. Med. Chem. Lett. 2013, 23, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781. [Google Scholar] [CrossRef]
- Raina, K.; Crews, C.M. Chemical Inducers of Targeted Protein Degradation*. J. Biol. Chem. 2010, 285, 11057–11060. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Bifunctional Pyrimidines as Modulators of Focal Adhesion Kinase. ACS Med. Chem. Lett. 2020, 11, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Popow, J.; Arnhof, H.; Bader, G.; Berger, H.; Ciulli, A.; Covini, D.; Dank, C.; Gmaschitz, T.; Greb, P.; Karolyi-Özguer, J.; et al. Highly Selective PTK2 Proteolysis Targeting Chimeras to Probe Focal Adhesion Kinase Scaffolding Functions. J. Med. Chem. 2019, 62, 2508–2520. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wu, Y.; Sun, Y.; Yang, Y.; Zhou, G.; Rao, Y. Design, Synthesis, and Evaluation of Highly Potent FAK-Targeting PROTACs. ACS Med. Chem. Lett. 2020, 11, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Kargbo, R.B. Chemically Induced Degradation of FAK-ALK for Application in Cancer Therapeutics. ACS Med. Chem. Lett. 2020, 11, 1367–1368. [Google Scholar] [CrossRef] [PubMed]









































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | Target (IC50) | Company | Clinical Trial | Status | Identifier |
|---|---|---|---|---|---|---|
| GSK-2256098 |  | FAK IC50 = 0.4 nM | Glaxo Smith Kline | phase I | completed completed completed | NCT01138033 NCT00996671 NCT02551653 |
| VS-6063 (PF-04554878) (Defactinib) |  | FAK IC50 = 0.6 nM Pyk2 IC50 = 0.6 nM | Verastem | phase II | completed completed completed | NCT01943292 NCT00787033 NCT01951690 |
| VS-6062 (PF-00562271) |  | FAK IC50 = 1.5 nM Pyk2 IC50 = 14 nM | Verastem | phase I | completed | NCT00666926 |
| VS-4718 (PND-1186) |  | FAK IC50 = 1.5 nM | Verastem | phase I | terminated withdrawn | NCT01849744 NCT02215629 |
| CEP-37440 |  | FAK IC50 = 2 nM ALK IC50 = 3.1 nM | Teva Branded | phase I | completed | NCT01922752 |
| BI-853520 (IN-10018) | Unknown | FAK | Boehringer Ingelheim | phase I | completed completed | NCT01335269 NCT01905111 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, X.-J.; Liu, X.-J.; Liu, Y.; Liu, W.-B.; Li, Y.-R.; Yu, G.-X.; Tian, X.-Y.; Zhang, Y.-B.; Song, J.; Jin, C.-Y.; et al. Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy. Molecules 2021, 26, 4250. https://doi.org/10.3390/molecules26144250
Pang X-J, Liu X-J, Liu Y, Liu W-B, Li Y-R, Yu G-X, Tian X-Y, Zhang Y-B, Song J, Jin C-Y, et al. Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy. Molecules. 2021; 26(14):4250. https://doi.org/10.3390/molecules26144250
Chicago/Turabian StylePang, Xiao-Jing, Xiu-Juan Liu, Yuan Liu, Wen-Bo Liu, Yin-Ru Li, Guang-Xi Yu, Xin-Yi Tian, Yan-Bing Zhang, Jian Song, Cheng-Yun Jin, and et al. 2021. "Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy" Molecules 26, no. 14: 4250. https://doi.org/10.3390/molecules26144250
APA StylePang, X.-J., Liu, X.-J., Liu, Y., Liu, W.-B., Li, Y.-R., Yu, G.-X., Tian, X.-Y., Zhang, Y.-B., Song, J., Jin, C.-Y., & Zhang, S.-Y. (2021). Drug Discovery Targeting Focal Adhesion Kinase (FAK) as a Promising Cancer Therapy. Molecules, 26(14), 4250. https://doi.org/10.3390/molecules26144250