
Impact of Fluoroalkylation on the n-Type Charge Transport of Two Naphthodithiophene Diimide Derivatives




Abstract
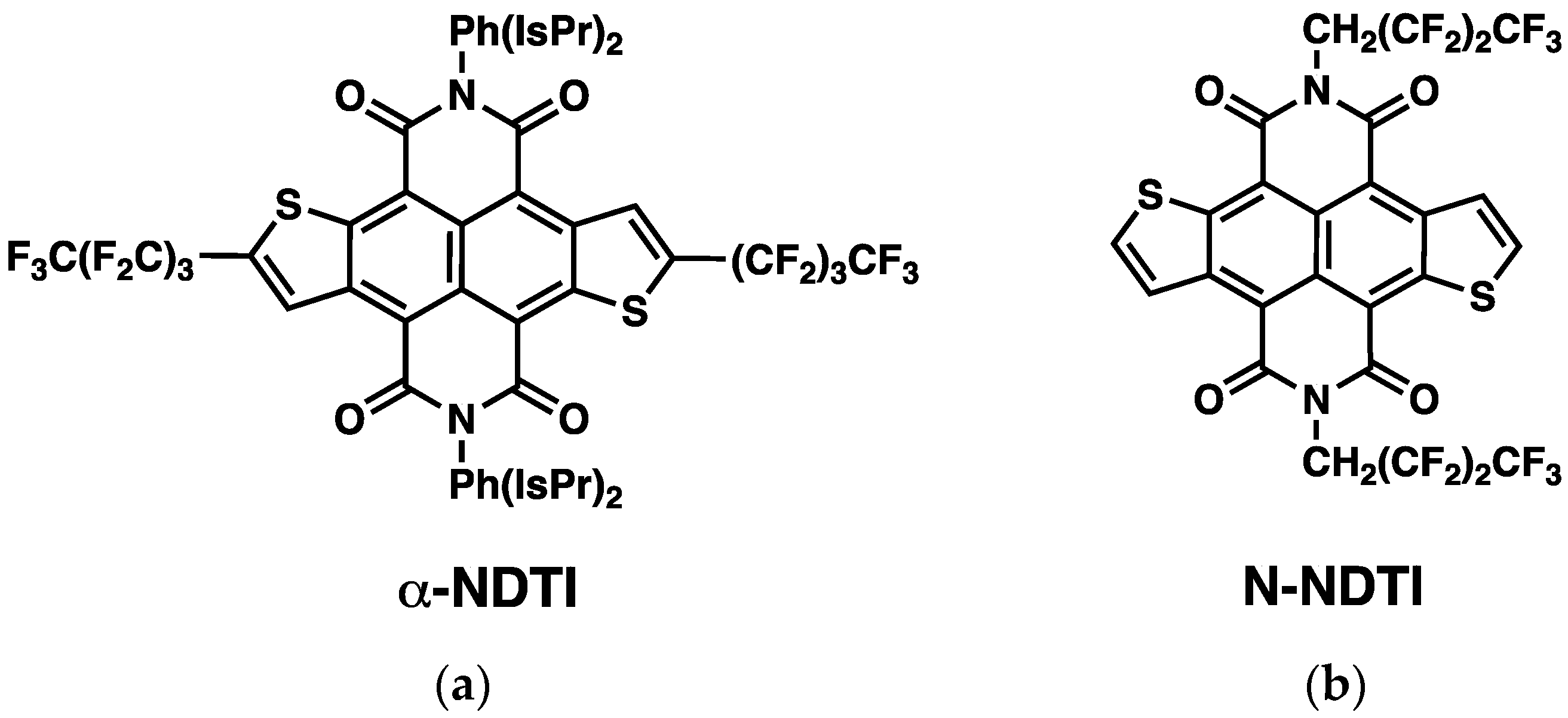
:1. Introduction
2. Results and Discussion
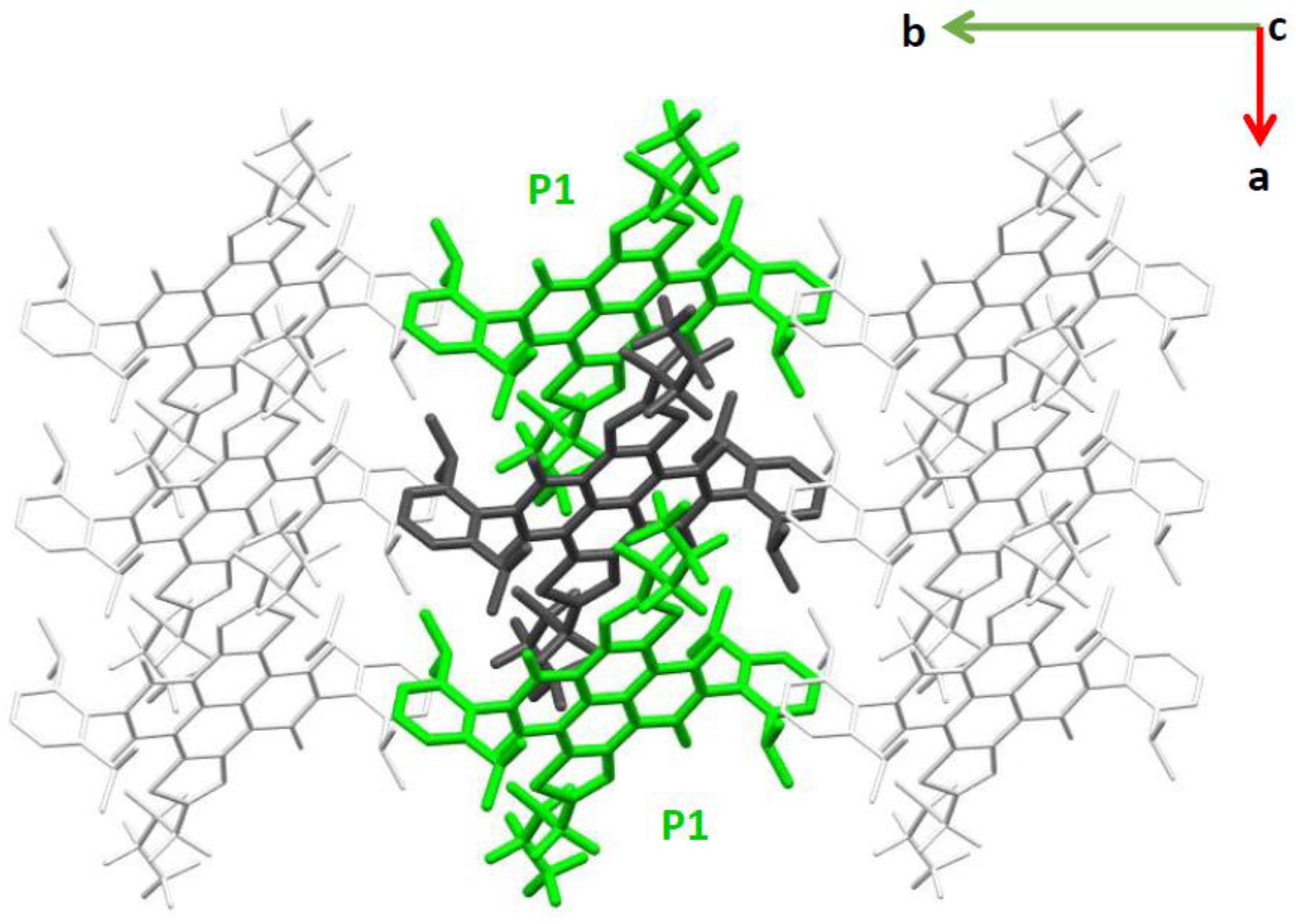
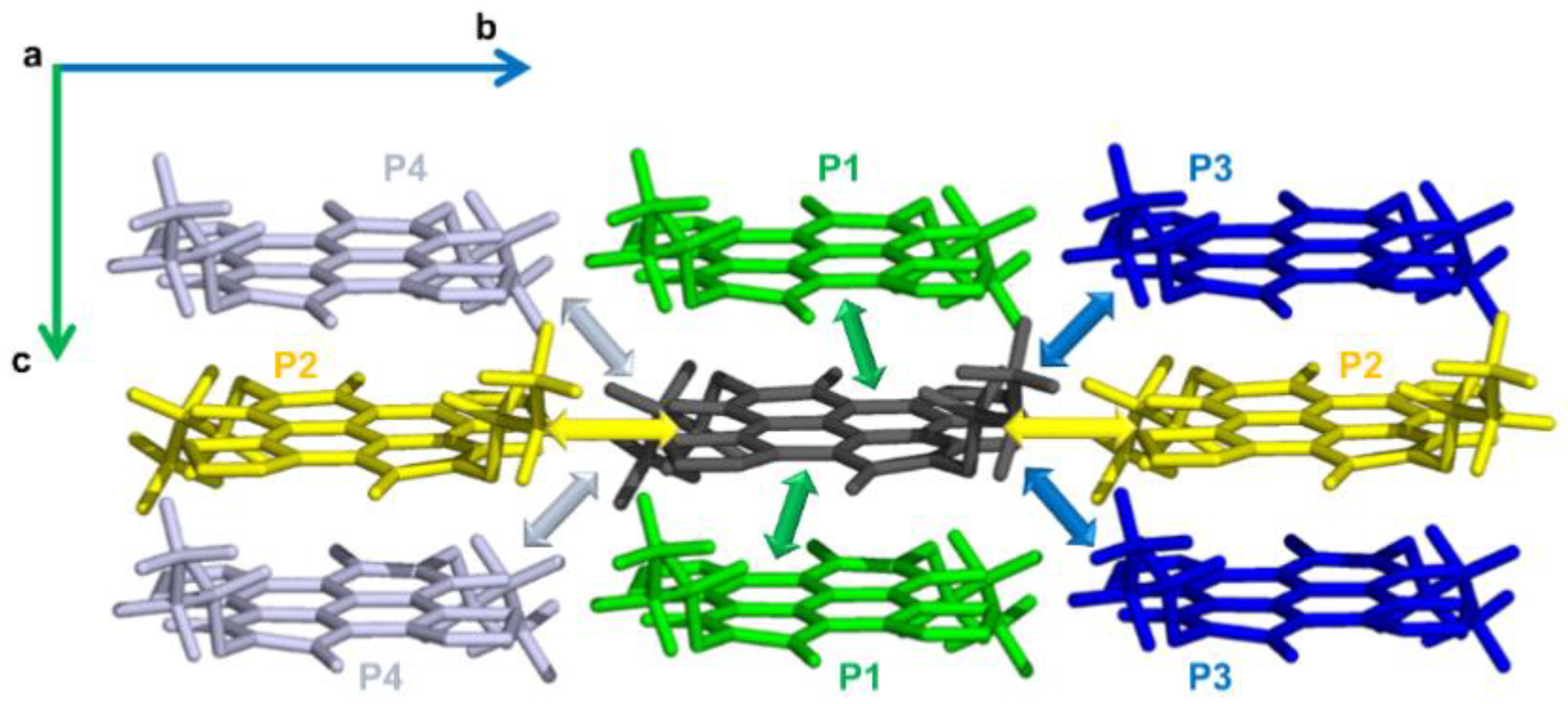
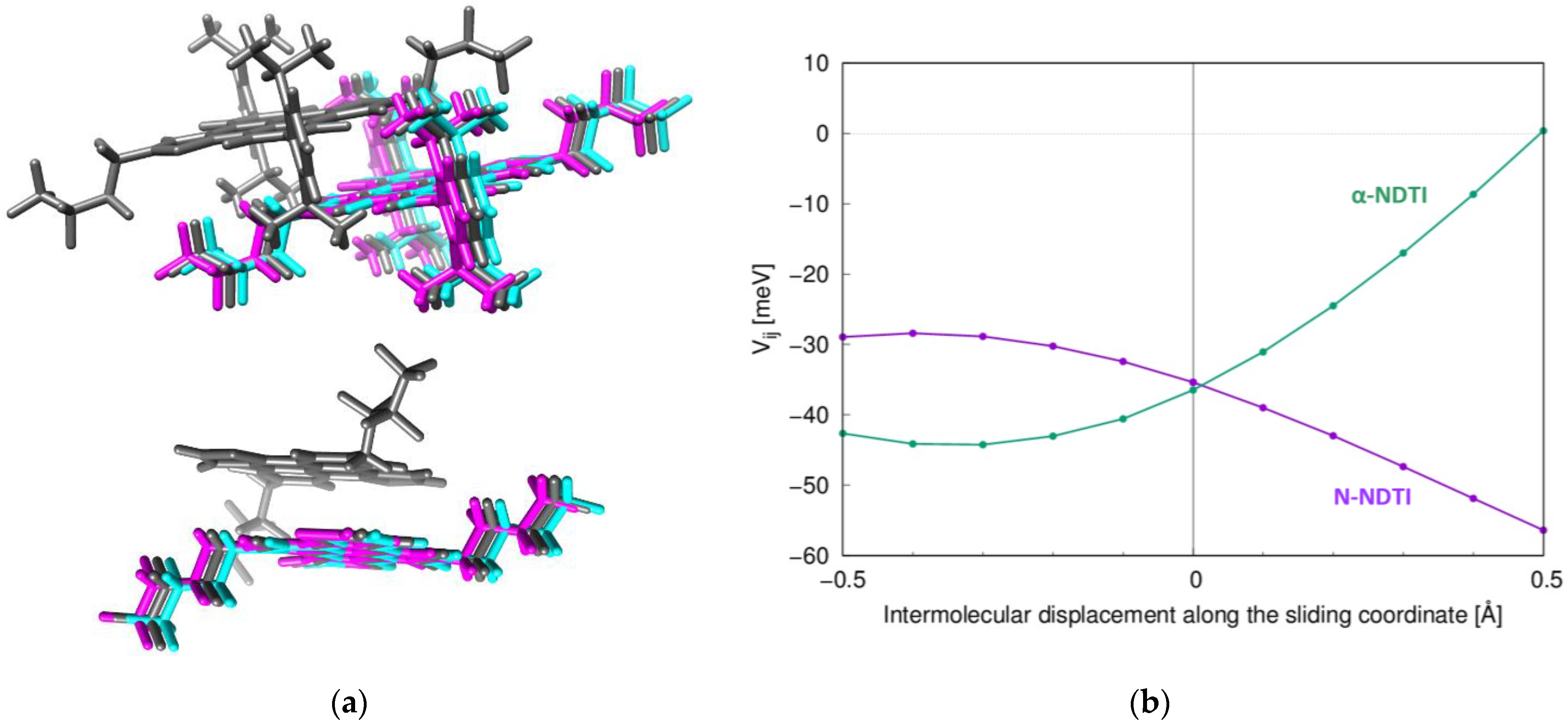
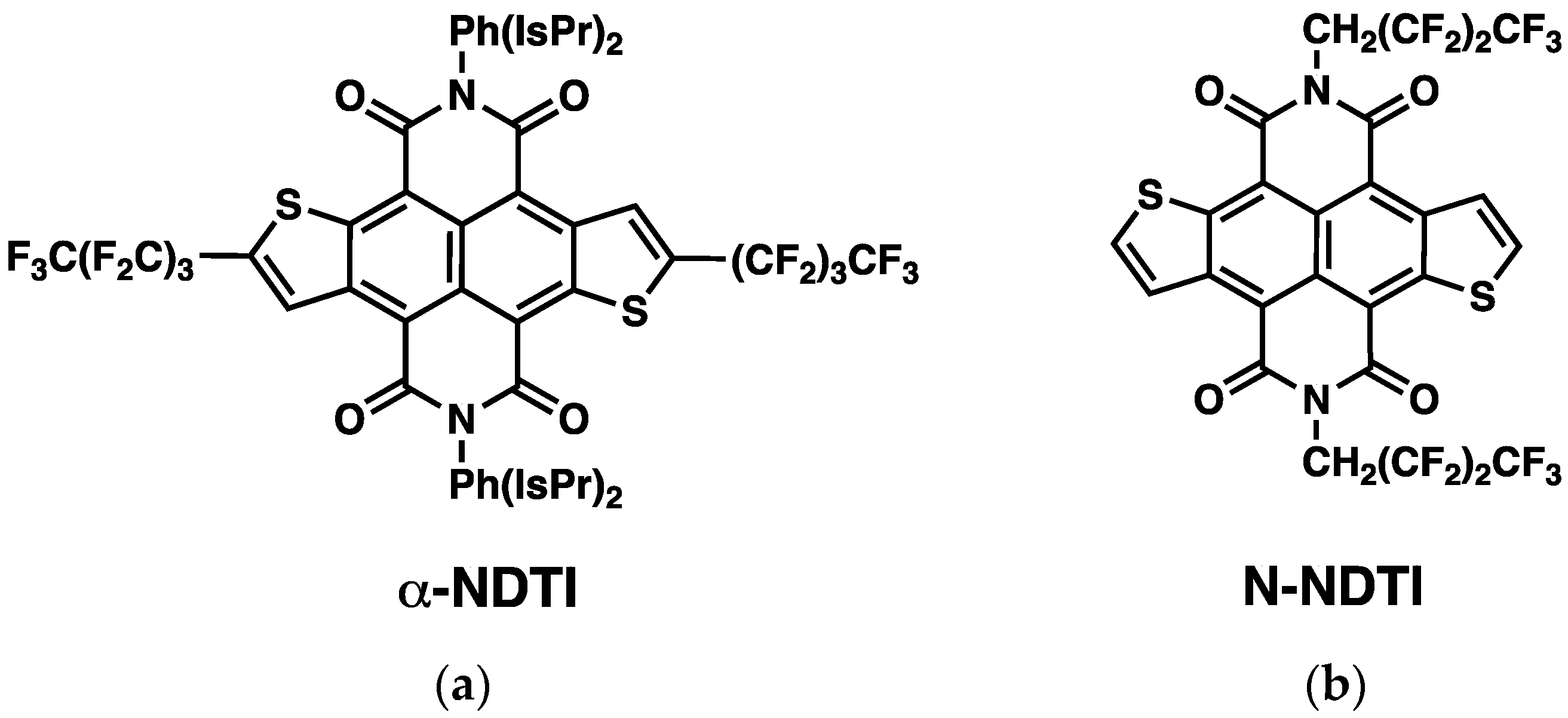
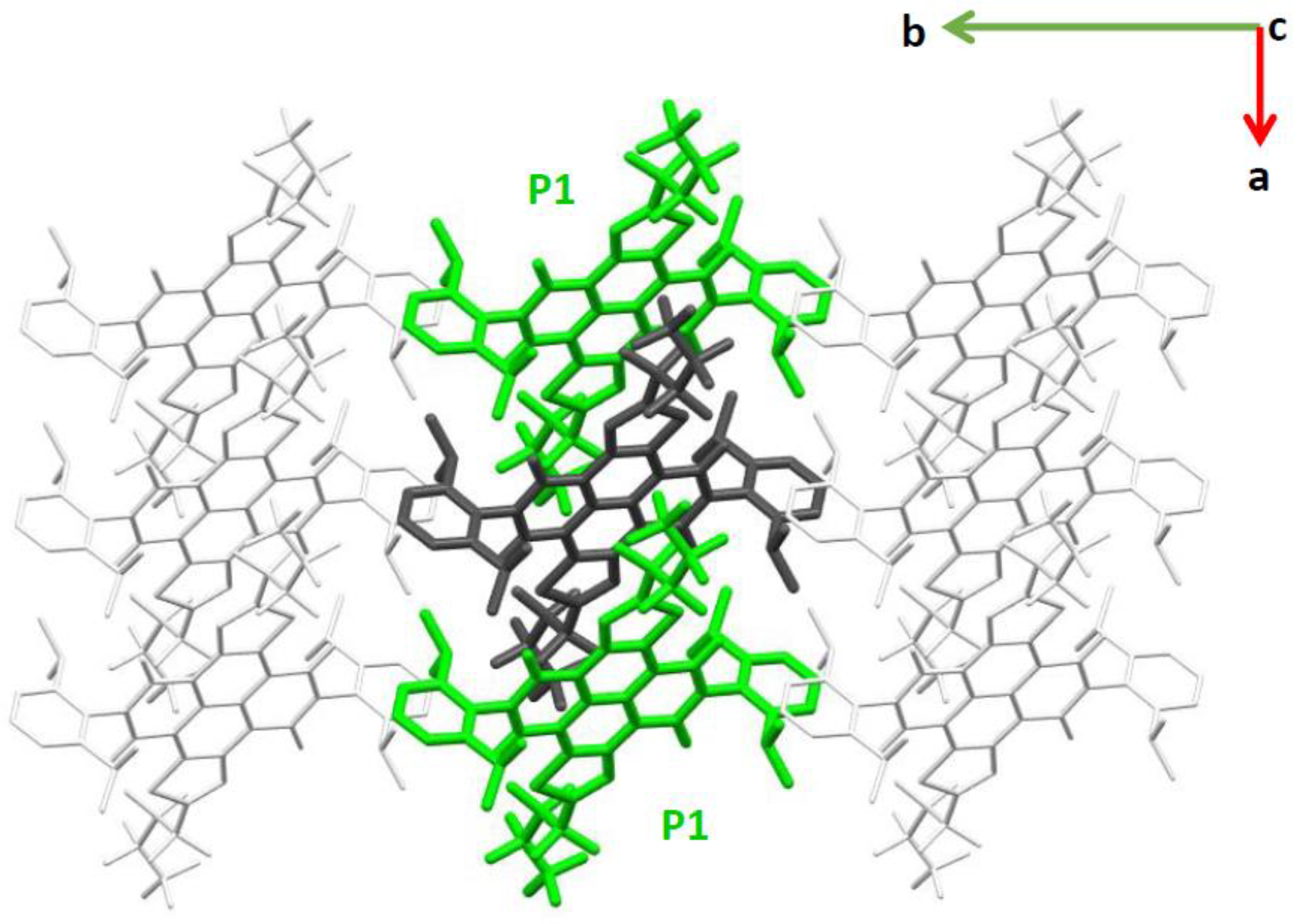
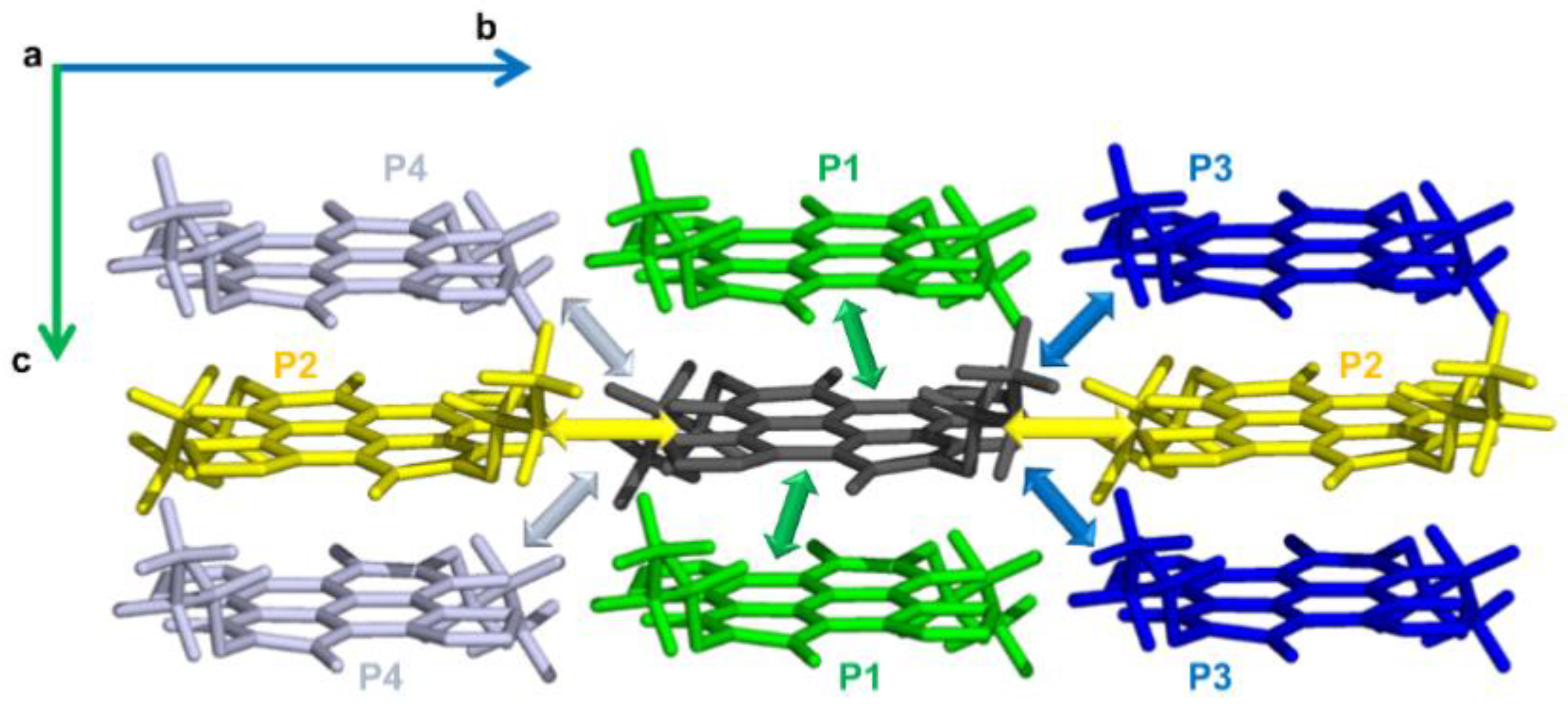
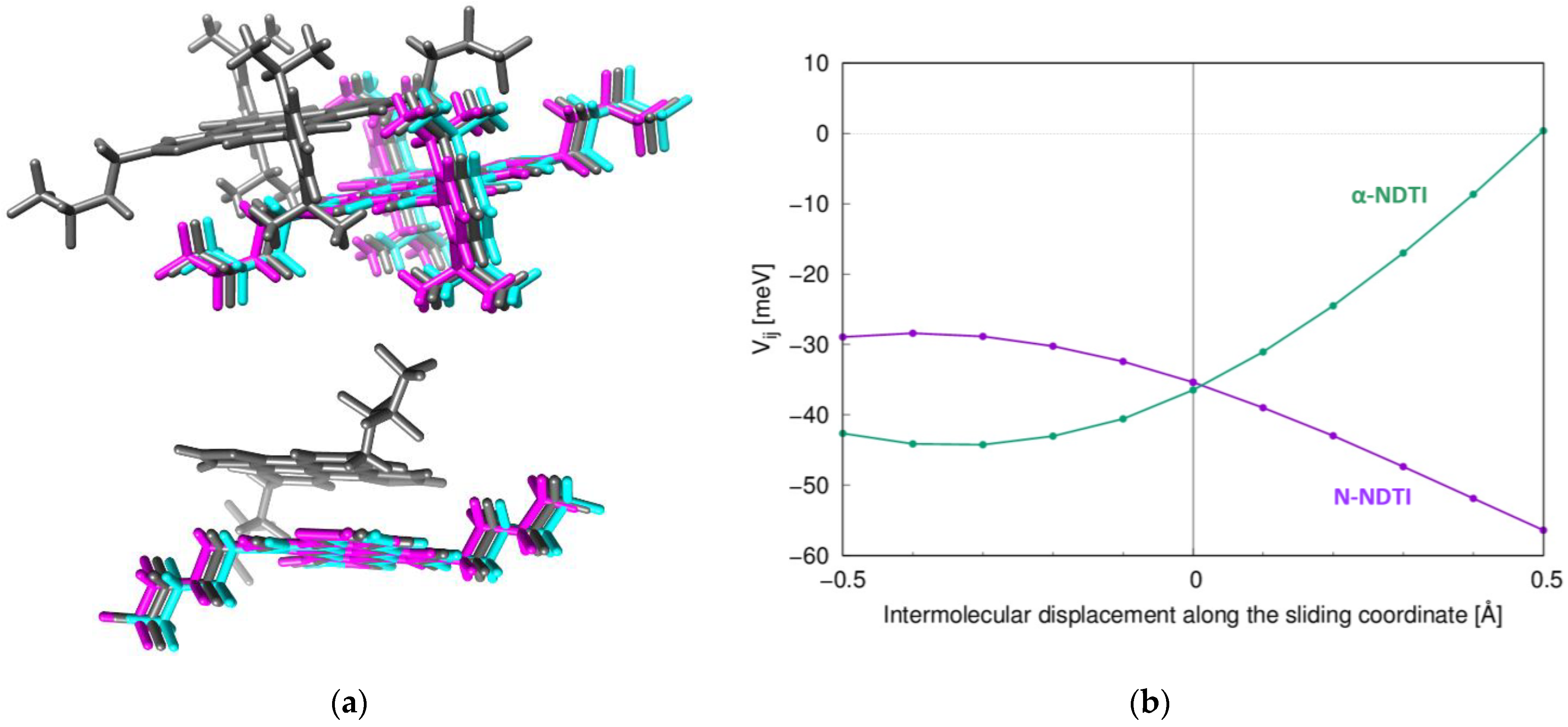
2.1. Electronic Couplings and Charge Transfer Pathways
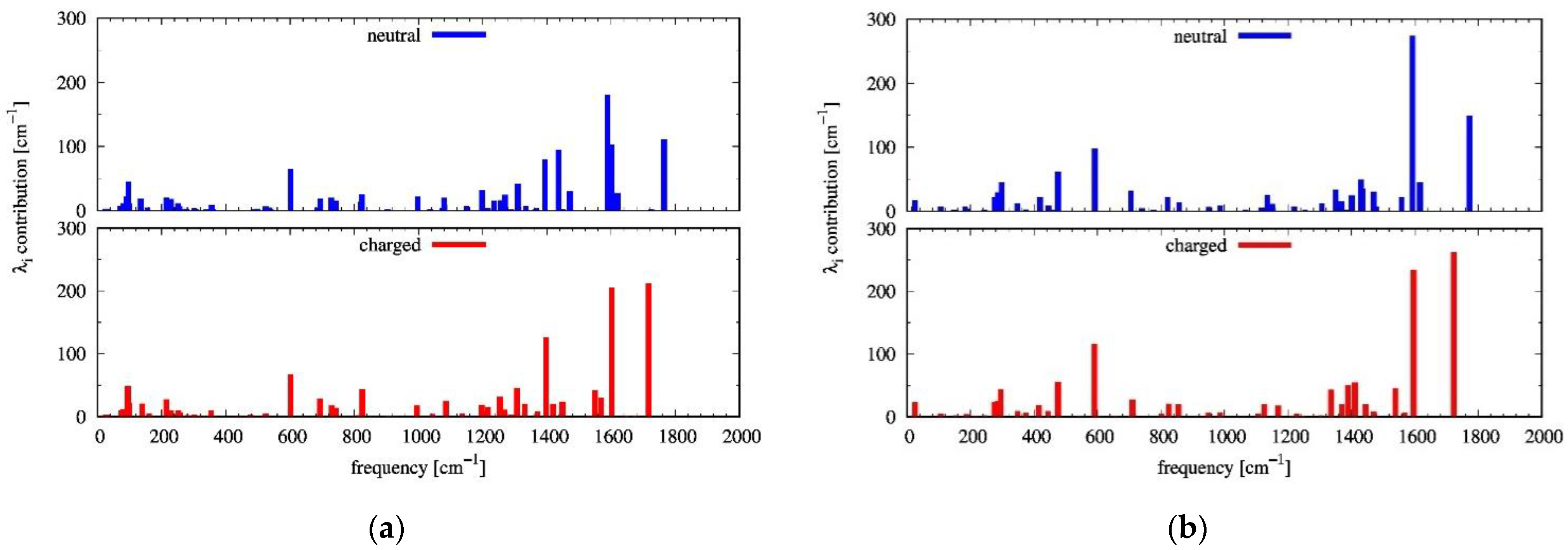
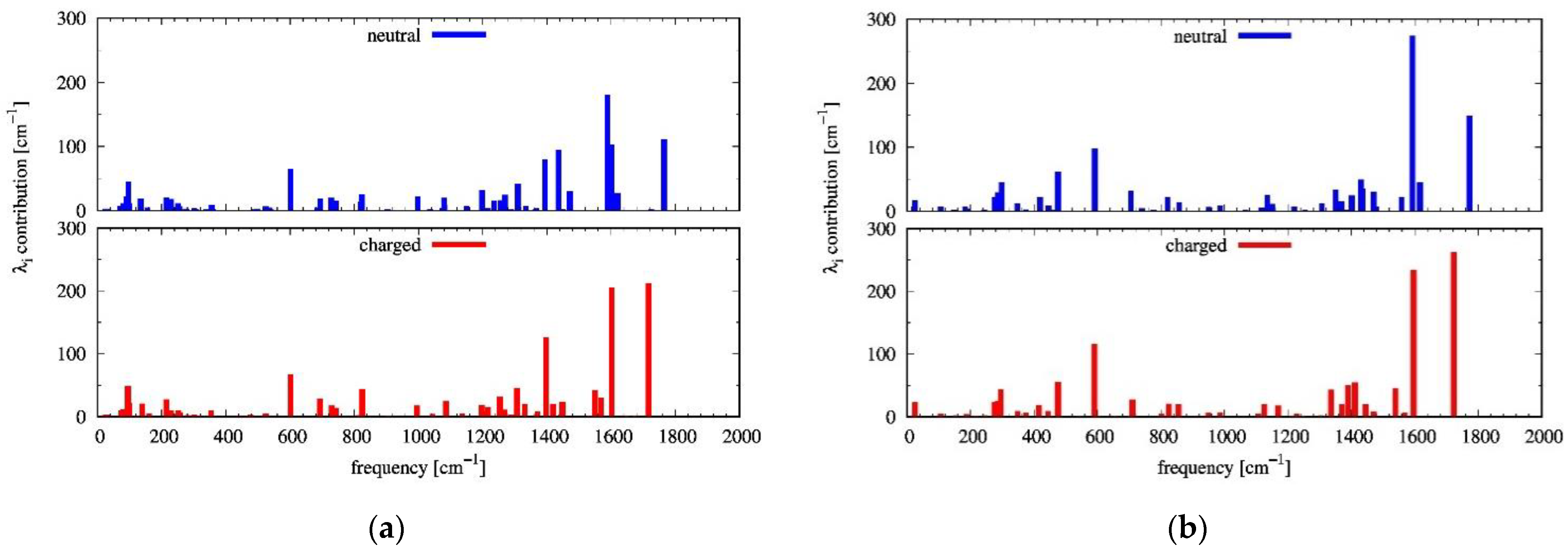
2.2. Reorganization Energies
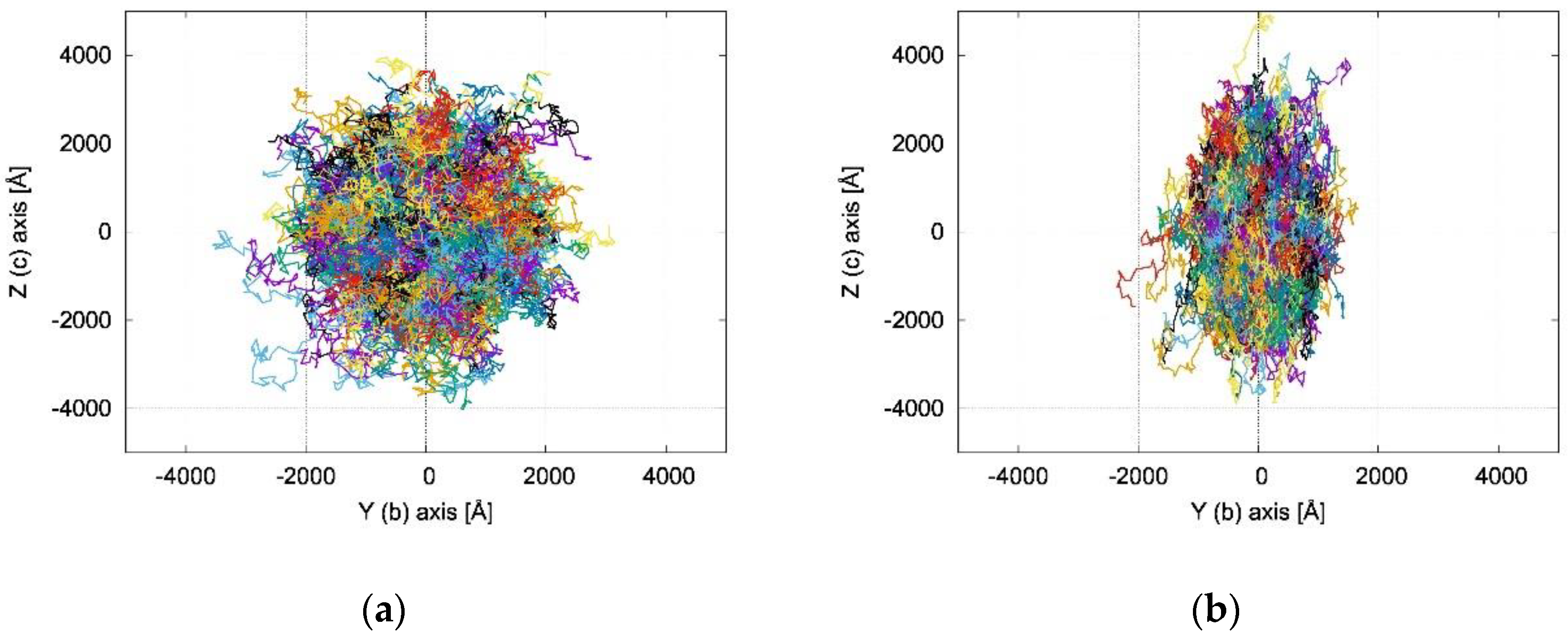
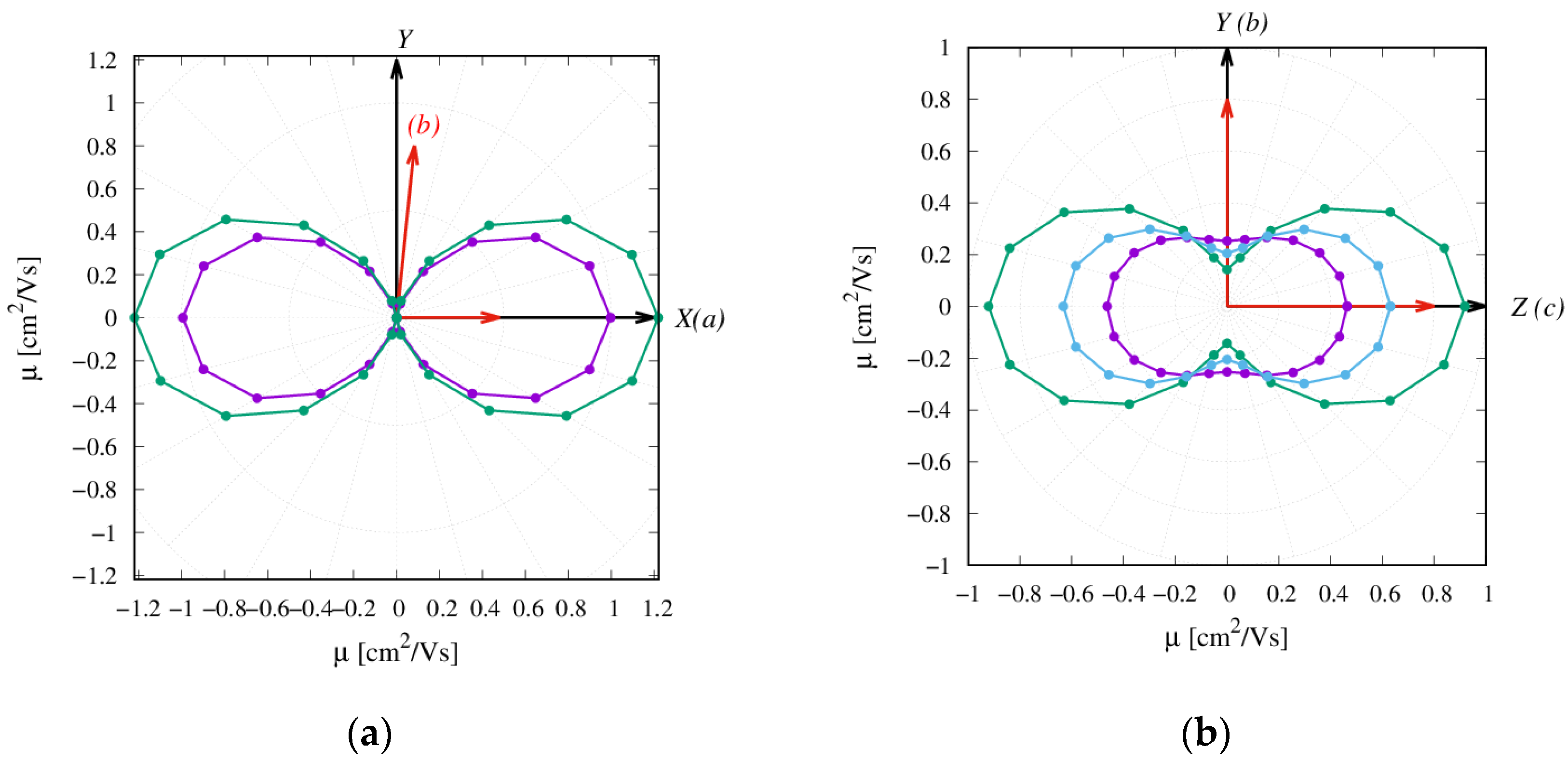
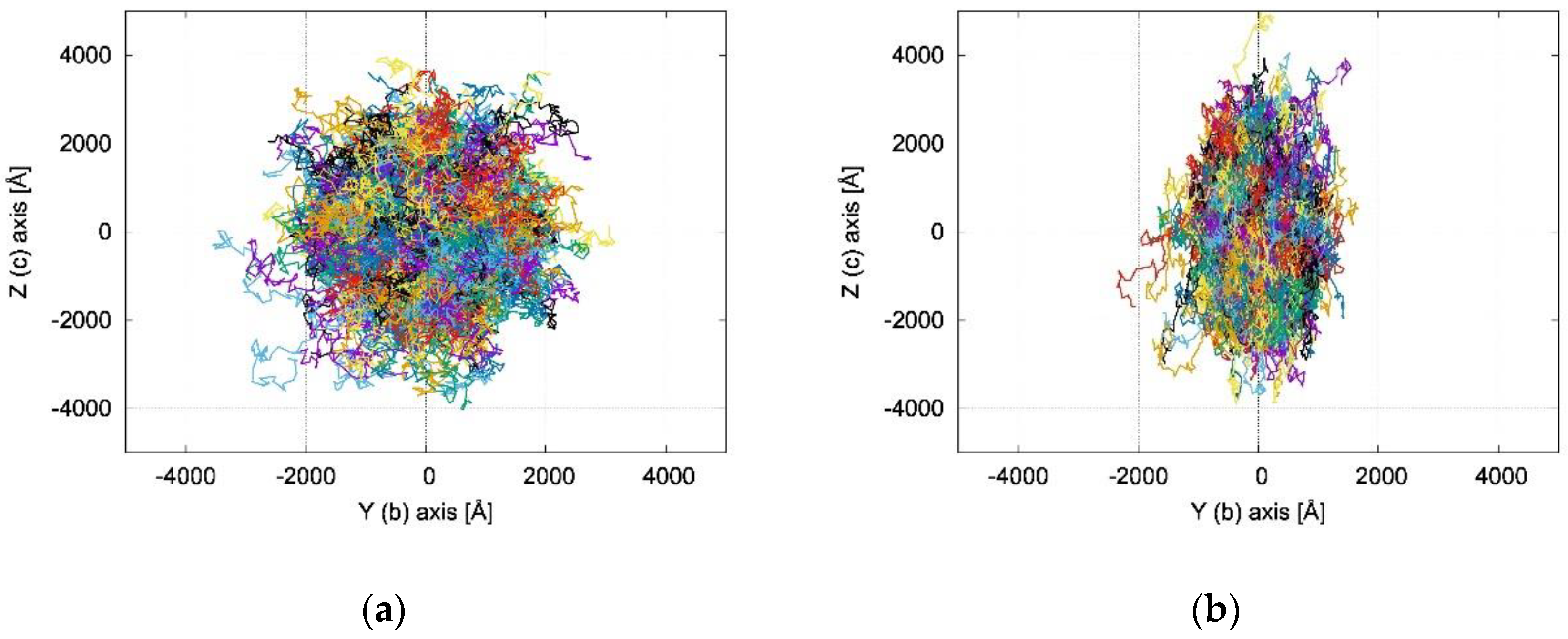
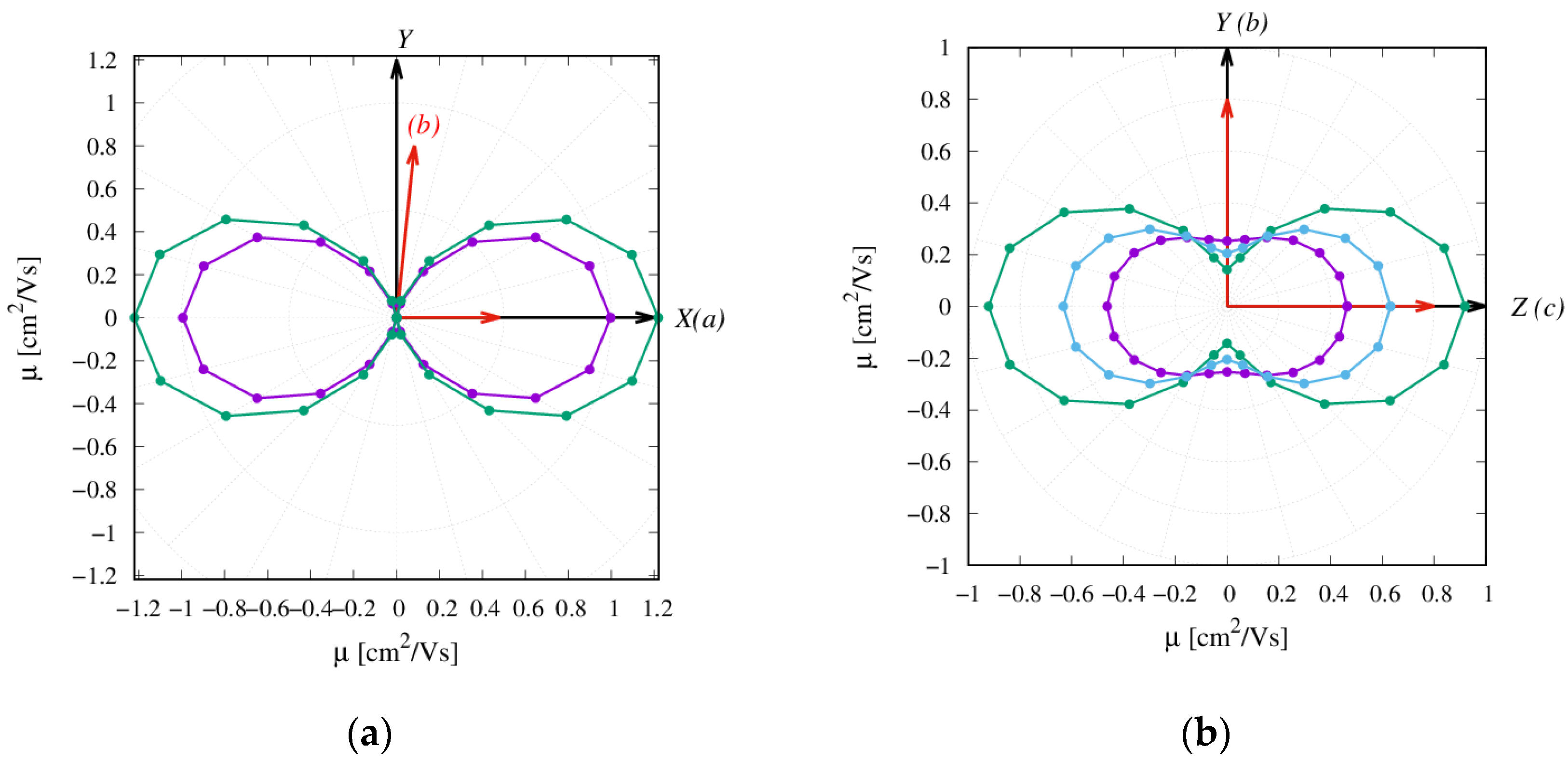
2.3. Charge Transfer Rate Constants and KMC Simulations
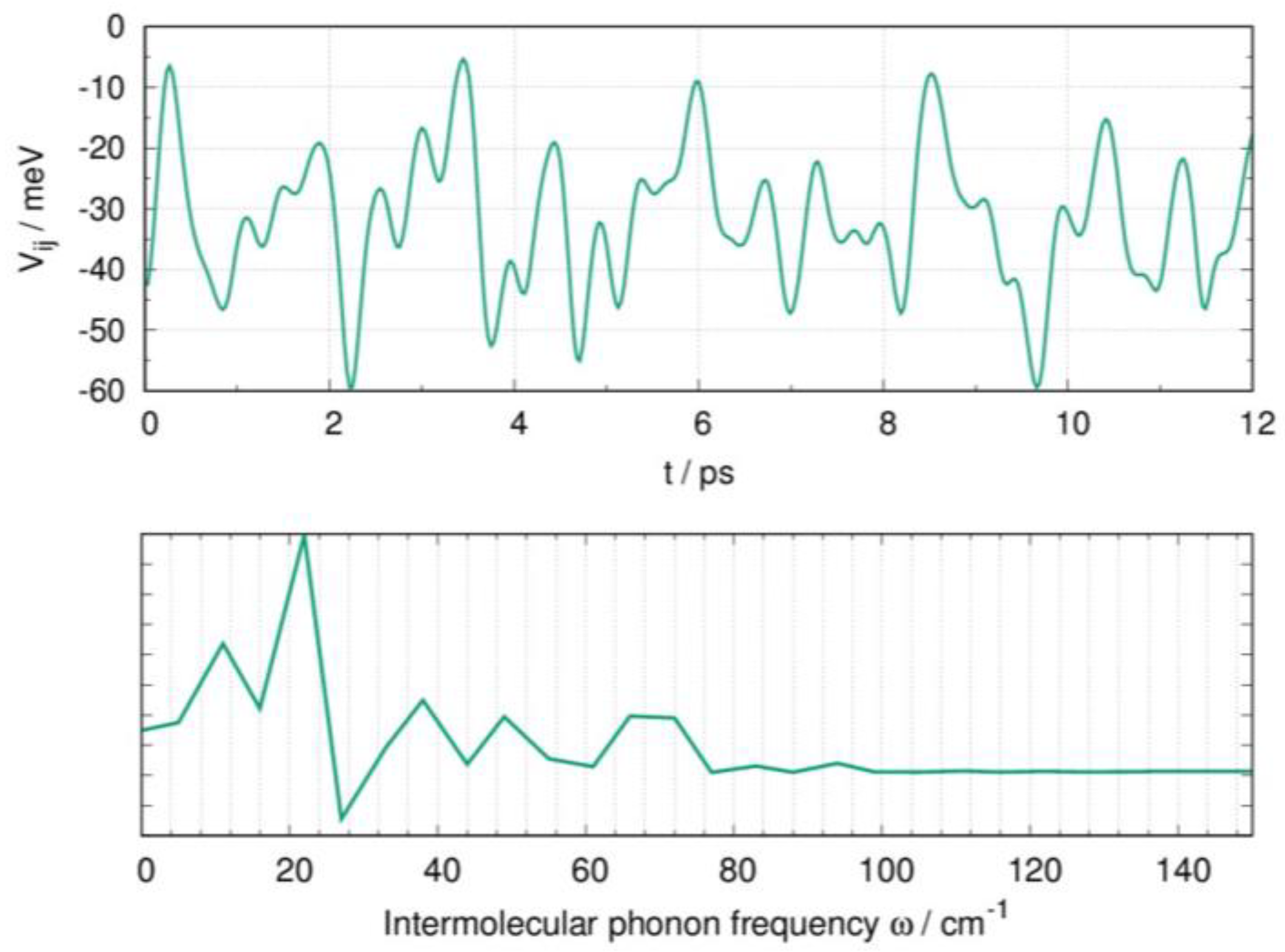
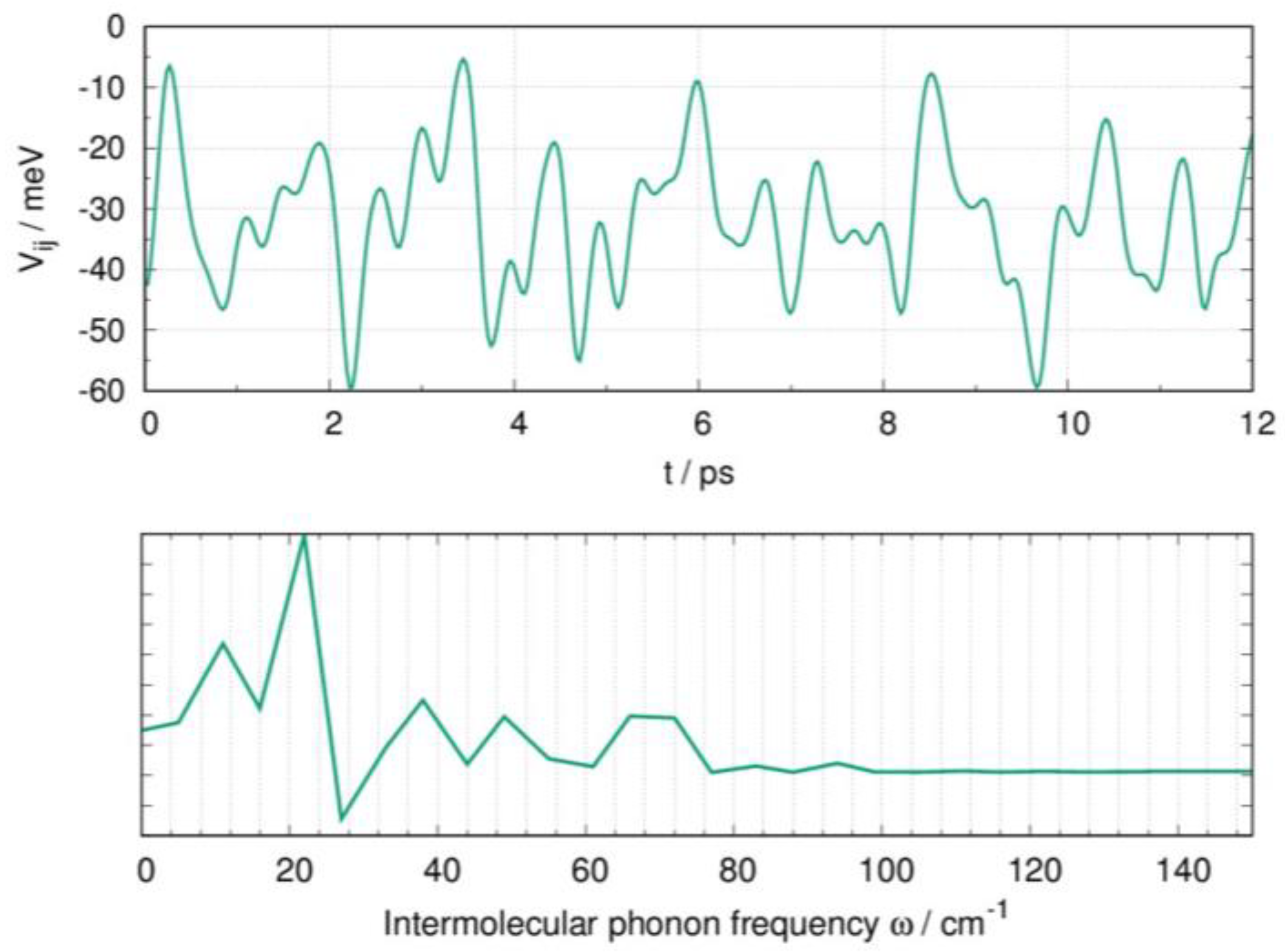
2.4. Influence of Dynamic Disorder
3. Computational Methods and Models
3.1. Reorganization Energy and Electronic Couplings
3.2. Charge Transfer Rate Constants and Kinetic Monte Carlo Simulations
3.3. Simulation of Thermally Induced Dynamic Disorder
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Usta, H.; Facchetti, A.; Marks, T.J. n-Channel semiconductor materials design for organic complementary circuits. Acc. Chem. Res. 2011, 44, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.R.; Frisbie, C.D.; da Silva Filho, D.A.; Brédas, J.-L.; Ewbank, P.C.; Mann, K.R. Introduction to drganic thin film transistors and design of n-channel organic semiconductors. Chem. Mater. 2004, 16, 4436–4451. [Google Scholar] [CrossRef]
- Okamoto, T.; Kumagai, S.; Fukuzaki, E.; Ishii, H.; Watanabe, G.; Niitsu, N.; Annaka, T.; Yamagishi, M.; Tani, Y.; Sugiura, H.; et al. Robust, high-performance n-type organic semiconductors. Sci. Adv. 2020, 6, eaaz0632. [Google Scholar] [CrossRef] [PubMed]
- Dhar, J.; Salzner, U.; Patil, S. Trends in molecular design strategies for ambient stable n-channel organic field effect transistors. J. Mater. Chem. C 2017, 5, 7404–7430. [Google Scholar] [CrossRef]
- Quinn, J.T.E.; Zhu, J.; Li, X.; Wang, J.; Li, Y. Recent progress in the development of n-type organic semiconductors for organic field effect transistors. J. Mater. Chem. C 2017, 5, 8654–8681. [Google Scholar] [CrossRef]
- Wang, S.; Sun, H.; Ail, U.; Vagin, M.; Persson, P.O.Å.; Andreasen, J.W.; Thiel, W.; Berggren, M.; Crispin, X.; Fazzi, D.; et al. Thermoelectric properties of solution-processed n-doped Ladder-type Conducting polymers. Adv. Mater. 2016, 28, 10764–10771. [Google Scholar] [CrossRef]
- Usta, H.; Newman, C.; Chen, Z.; Facchetti, A. Dithienocoronenediimide-based copolymers as novel ambipolar semiconductors for organic thin-film transistors. Adv. Mater. 2012, 24, 3678–3684. [Google Scholar] [CrossRef]
- Yue, W.; Nikolka, M.; Xiao, M.; Sadhanala, A.; Nielsen, C.B.; White, A.J.P.; Chen, H.-Y.; Onwubiko, A.; Sirringhaus, H.; McCulloch, I. Azaisoindigo conjugated polymers for high performance n-type and ambipolar thin film transistor applications. J. Mater. Chem. C 2016, 4, 9704–9710. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, B.; Tajima, K.; Nakano, M.; Takimiya, K.; Tang, A.; Zhou, E. Comparison among Perylene Diimide (PDI), Naphthalene Diimide (NDI), and Naphthodithiophene Diimide (NDTI) based n-type polymers for all-polymer solar cells application. Macromolecules 2017, 50, 3179–3185. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, B.; Tang, A.; Li, J.; Wang, X.; Zhou, E. Aromatic-Diimide-based n-type conjugated polymers for all–polymer solar cell applications. Adv. Mater. 2019, 31, 1804699. [Google Scholar] [CrossRef]
- Samanta, S.K.; Song, I.; Yoo, J.H.; Oh, J.H. Organic n-channel transistors based on [1]benzothieno[3,2-b]benzothiophene–rylene diimide donor-acceptor conjugated polymers. ACS Appl. Mater. Interfaces 2018, 10, 32444–32453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Jiang, W.; Li, X.; Hao, L.; Liu, C.; Wang, Z. Laterally Expanded Rylene Diimides with Uniform Branched Side Chains for Solution-Processed Air Stable n-Channel Thin Film Transistors. ACS Appl. Mater. Interfaces 2014, 6, 18098–18103. [Google Scholar] [CrossRef]
- Hecht, M.; Würthner, F. Supramolecularly Engineered J-Aggregates Based on Perylene Bisimide Dyes. Acc. Chem. Res. 2021, 54, 642–653. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Guo, Y.; Yu, C.; Zhang, F.; Xu, W.; Liu, Y.; Zhu, D. Diketopyrrolopyrrole-containing quinoidal small molecules for high-performance, air-stable, and solution-processable n-channel organic field-effect transistors. J. Am. Chem. Soc. 2012, 134, 4084–4087. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Qin, Y.; Zhang, J.; Guan, Y.-S.; Xu, H.; Xu, W.; Zhu, D. Ambipolar organic field-effect transistors based on diketopyrrolopyrrole derivatives containing different π-conjugating spacers. J. Mater. Chem. C 2016, 4, 4470–4477. [Google Scholar] [CrossRef]
- Katz, H.E.; Lovinger, A.J.; Johnson, J.; Kloc, C.; Siegrist, T.; Li, W.; Lin, Y.-Y.; Dodabalapur, A. A soluble and air-stable organic semiconductor with high electron mobility. Nature 2000, 404, 478–481. [Google Scholar] [CrossRef]
- Hu, Y.; Gao, X.; Di, C.; Yang, X.; Zhang, F.; Liu, Y.; Li, H.; Zhu, D. Core-expanded Naphthalene Diimides Fused with Sulfur Heterocycles and End-Capped with Electron-Withdrawing Groups for Air-Stable Solution-Processed n-Channel Organic Thin Film Transistors. Chem. Mater. 2011, 23, 1204–1215. [Google Scholar] [CrossRef]
- Gao, X.; Di, C.; Hu, Y.; Yang, X.; Fan, H.; Zhang, F.; Liu, Y.; Li, H.; Zhu, D. Core-Expanded Naphthalene Diimides Fused with 2-(1,3-Dithiol-2-Ylidene)Malonitrile Groups for High-Performance, Ambient-Stable, Solution-Processed n-Channel Organic Thin Film Transistors. J. Am. Chem. Soc. 2010, 132, 3697–3699. [Google Scholar] [CrossRef]
- Usta, H.; Risko, C.; Wang, Z.; Huang, H.; Deliomeroglu, M.K.; Zhukhovitskiy, A.; Facchetti, A.; Marks, T.J. Design, synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory. J. Am. Chem. Soc. 2009, 131, 5586–5608. [Google Scholar] [CrossRef]
- Lee, W.-Y.; Oh, J.H.; Suraru, S.-L.; Chen, W.-C.; Würthner, F.; Bao, Z. High-Mobility Air-Stable Solution-Shear-Processed n-Channel Organic Transistors Based on Core-Chlorinated Naphthalene Diimides. Adv. Funct. Mater. 2011, 21, 4173–4181. [Google Scholar] [CrossRef]
- Suraru, S.-L.; Würthner, F. Strategies for the Synthesis of Functional Naphthalene Diimides. Angew. Chem. Int. Ed. 2014, 53, 7428–7448. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.V.; Bhosale, S.V.; Bhargava, S.K. Recent progress of core-substituted naphthalenediimides: Highlights from 2010. Org. Biomol. Chem. 2012, 10, 6455–6468. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, Y.; Yan, H.; Facchetti, A. Naphthalenedicarboximide- vs Perylenedicarboximide-based copolymers. Synthesis and semiconducting properties in bottom-gate n-channel organic transistors. J. Am. Chem. Soc. 2009, 131, 8–9. [Google Scholar] [CrossRef]
- Fukutomi, Y.; Nakano, M.; Hu, J.; Osaka, I.; Takimiya, K. Naphthodithiophenediimide (NDTI): Synthesis, structure, and applications. J. Am. Chem. Soc. 2013, 135, 11445–11448. [Google Scholar] [CrossRef]
- Yue, W.; Gao, J.; Li, Y.; Jiang, W.; Di Motta, S.; Negri, F.; Wang, Z. One-pot synthesis of stable NIR tetracene diimides via double cross-coupling. J. Am. Chem. Soc. 2011, 133, 18054–18057. [Google Scholar] [CrossRef]
- Zhang, F.; Hu, Y.; Schuettfort, T.; Di, C.; Gao, X.; McNeill, C.R.; Thomsen, L.; Mannsfeld, S.C.B.; Yuan, W.; Sirringhaus, H.; et al. Critical role of alkyl chain branching of organic semiconductors in enabling solution-processed n-channel organic thin-film Transistors with Mobility of up to 3.50 cm2 V–1 s–1. J. Am. Chem. Soc. 2013, 135, 2338–2349. [Google Scholar] [CrossRef]
- Nakano, M.; Osaka, I.; Hashizume, D.; Takimiya, K. α-Modified naphthodithiophene diimides—molecular design strategy for air-stable n-channel organic semiconductors. Chem. Mater. 2015, 27, 6418–6425. [Google Scholar] [CrossRef]
- Takimiya, K.; Osaka, I. Naphthodithiophenes: Emerging building blocks for organic electronics. Chem. Rec. 2015, 15, 175–188. [Google Scholar] [CrossRef]
- Ran, H.; Chen, L.; Yang, X.; Zhang, J.; Zhao, Z.; Han, R.; Duan, X.; Hu, J.Y. Arylacetylene end capped naphthodithiophene diimide (NDTI)-based semiconductors for air-stable, solution-processed n-channel organic field-effect transistors: Effect of terminal aryl groups on charge transport. Dyes Pigments 2019, 169, 7–14. [Google Scholar] [CrossRef]
- Ran, H.; Duan, X.; Zheng, R.; Xie, F.; Chen, L.; Zhao, Z.; Han, R.; Lei, Z.; Hu, J.-Y. Two isomeric azulene-decorated naphthodithiophene diimide-based triads: Molecular orbital distribution controls polarity change of OFETs through connection position. ACS Appl. Mater. Interfaces 2020, 12, 23225–23235. [Google Scholar] [CrossRef]
- Ji, L.F.; Fan, J.X.; Zhang, S.F.; Ren, A.M. Theoretical investigations into the charge transfer properties of thiophene α-substituted naphthodithiophene diimides: Excellent n-channel and ambipolar organic semiconductors. Phys. Chem. Chem. Phys. 2017, 19, 13978–13993. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liu, C.; Li, Y.; Wang, Z. Fluoroalkyl-modified naphthodithiophene diimides. Chem. Commun. 2017, 53, 188–191. [Google Scholar] [CrossRef]
- Di Donato, E.; Fornari, R.P.; Di Motta, S.; Li, Y.; Wang, Z.; Negri, F. n-Type charge transport and mobility of fluorinated perylene bisimide semiconductors. J. Phys. Chem. B 2010, 114, 5327–5334. [Google Scholar] [CrossRef] [PubMed]
- Di Motta, S.; Siracusa, M.; Negri, F. Structural and thermal effects on the charge transport of core-twisted chlorinated perylene bisimide semiconductors. J. Phys. Chem. C 2011, 115, 20754–20764. [Google Scholar] [CrossRef]
- Canola, S.; Negri, F. Anisotropy of the n-type charge transport and thermal effects in crystals of a fluoro-alkylated naphthalene diimide: A computational investigation. Phys. Chem. Chem. Phys. 2014, 16, 21550–21558. [Google Scholar] [CrossRef]
- Canola, S.; Graham, C.; Pérez-Jiménez, Á.J.; Sancho-García, J.C.; Negri, F. Charge transport parameters for carbon based nanohoops and donor-acceptor derivatives. Phys. Chem. Chem. Phys. 2019, 21, 2057–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratini, S.; Mayou, D.; Ciuchi, S. The transient localization scenario for charge transport in crystalline organic materials. Adv. Funct. Mater. 2016, 26, 2292–2315. [Google Scholar] [CrossRef] [Green Version]
- Troisi, A. Prediction of the absolute charge mobility of molecular semiconductors: The case of rubrene. Adv. Mater. 2007, 19, 2000–2004. [Google Scholar] [CrossRef]
- Troisi, A. Charge transport in high mobility molecular semiconductors: Classical models and new theories. Chem. Soc. Rev. 2011, 40, 2347. [Google Scholar] [CrossRef]
- Coropceanu, V.; Sánchez-Carrera, R.S.; Paramonov, P.; Day, G.M.; Brédas, J.-L. Interaction of charge carriers with lattice vibrations in organic molecular semiconductors: Naphthalene as a case study. J. Phys. Chem. C 2009, 113, 4679–4686. [Google Scholar] [CrossRef]
- Harrelson, T.F.; Dantanarayana, V.; Xie, X.; Koshnick, C.; Nai, D.; Fair, R.; Nuñez, S.A.; Thomas, A.K.; Murrey, T.L.; Hickner, M.A.; et al. Direct probe of the nuclear modes limiting charge mobility in molecular semiconductors. Mater. Horiz. 2019, 6, 182–191. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Yi, Y.; Coropceanu, V.; Brédas, J.-L. Impact of phonon dispersion on nonlocal electron-phonon couplings in organic semiconductors: The naphthalene crystal as a case study. J. Phys. Chem. C 2018, 122, 44–49. [Google Scholar] [CrossRef]
- Di Motta, S.; Di Donato, E.; Negri, F.; Orlandi, G.; Fazzi, D.; Castiglioni, C. Resistive molecular memories: Influence of molecular parameters on the electrical bistability. J. Am. Chem. Soc. 2009, 131, 6591–6598. [Google Scholar] [CrossRef]
- Bronstein, H.; Nielsen, C.B.; Schroeder, B.C.; McCulloch, I. The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 2020, 4, 66–77. [Google Scholar] [CrossRef]
- Shimanouchi, T. Local and overall vibrations of polymer chains. Pure Appl. Chem. 1973, 36, 93–108. [Google Scholar] [CrossRef]
- McMahon, D.P.; Troisi, A. Evaluation of the external reorganization energy of polyacenes. J. Phys. Chem. Lett. 2010, 1, 941–946. [Google Scholar] [CrossRef]
- Norton, J.E.; Brédas, J.-L. Polarization energies in oligoacene semiconductor crystals. J. Am. Chem. Soc. 2008, 130, 12377–12384. [Google Scholar] [CrossRef] [PubMed]
- Giannini, S.; Carof, A.; Ellis, M.; Yang, H.; Ziogos, O.G.; Ghosh, S.; Blumberger, J. Quantum localization and delocalization of charge carriers in organic semiconducting crystals. Nat. Commun. 2019, 10, 3843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Brédas, J.-L. Developing molecular-level models for organic field-effect transistors. Natl. Sci. Rev. 2021, 8, 1–14. [Google Scholar] [CrossRef]
- Schweicher, G.; Garbay, G.; Jouclas, R.; Vibert, F.; Devaux, F.; Geerts, Y.H. Molecular Semiconductors for Logic Operations: Dead–End or Bright Future? Adv. Mater. 2020, 32, 1905909. [Google Scholar] [CrossRef]
- Schweicher, G.; Olivier, Y.; Lemaur, V.; Geerts, Y.H. What currently limits charge carrier mobility in crystals of molecular semiconductors? Isr. J. Chem. 2014, 54, 595–620. [Google Scholar] [CrossRef]
- Bittle, E.G.; Basham, J.I.; Jackson, T.N.; Jurchescu, O.D.; Gundlach, D.J. Mobility overestimation due to gated contacts in organic field-effect transistors. Nat. Commun. 2016, 7, 10908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, A.F.; Singh, S.; Fallon, K.J.; Hodsden, T.; Han, Y.; Schroeder, B.C.; Bronstein, H.; Heeney, M.; McCulloch, I.; Anthopoulos, T.D. Recent progress in high-mobility organic transistors: A reality check. Adv. Mater. 2018, 30, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Ciuchi, S.; Fratini, S. Electronic transport and quantum localization effects in organic semiconductors. Phys. Rev. B 2012, 86, 245201. [Google Scholar] [CrossRef] [Green Version]
- Minder, N.A.; Lu, S.; Fratini, S.; Ciuchi, S.; Facchetti, A.; Morpurgo, A.F. Tailoring the molecular structure to suppress extrinsic disorder in organic transistors. Adv. Mater. 2014, 26, 1254–1260. [Google Scholar] [CrossRef] [Green Version]
- Nematiaram, T.; Troisi, A. Strategies to reduce the dynamic disorder in molecular semiconductors. Mater. Horiz. 2020, 7, 2922–2928. [Google Scholar] [CrossRef]
- Martinelli, N.G.; Olivier, Y.; Athanasopoulos, S.; Ruiz Delgado, M.-C.; Pigg, K.R.; da Silva Filho, D.; Sánchez-Carrera, R.S.; Venuti, E.; Della Valle, R.G.; Brédas, J.-L.; et al. Influence of intermolecular vibrations on the electronic coupling in organic semiconductors: The case of anthracene and perfluoropentacene. Chemphyschem 2009, 10, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Gosar, P.; Vilfan, I. Phonon-assisted current in organic molecular crystals. Mol. Phys. 1970, 18, 49–61. [Google Scholar] [CrossRef]
- Munn, R.W.; Silbey, R. Theory of electronic transport in molecular crystals. III. Diffusion coefficient incorporating nonlocal linear electron–phonon coupling. J. Chem. Phys. 1985, 83, 1854–1864. [Google Scholar] [CrossRef] [Green Version]
- Girlando, A.; Grisanti, L.; Masino, M.; Brillante, A.; Della Valle, R.G.; Venuti, E. Interaction of charge carriers with lattice and molecular phonons in crystalline pentacene. J. Chem. Phys. 2011. [Google Scholar] [CrossRef]
- Xie, X.; Santana-Bonilla, A.; Troisi, A. Nonlocal electron–phonon coupling in prototypical molecular semiconductors from first principles. J. Chem. Theory Comput. 2018, 14, 3752–3762. [Google Scholar] [CrossRef]
- Schweicher, G.; D’Avino, G.; Ruggiero, M.T.; Harkin, D.J.; Broch, K.; Venkateshvaran, D.; Liu, G.; Richard, A.; Ruzié, C.; Armstrong, J.; et al. Chasing the “killer” phonon mode for the rational design of low-disorder, high-mobility molecular semiconductors. Adv. Mater. 2019, 31, 1902407. [Google Scholar] [CrossRef] [Green Version]
- Skourtis, S.S.; Balabin, I.A.; Kawatsu, T.; Beratan, D.N. Protein dynamics and electron transfer: Electronic decoherence and non-Condon effects. Proc. Natl. Acad. Sci. USA 2005, 102, 3552–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troisi, A.; Orlandi, G. Dynamics of the intermolecular transfer integral in crystalline organic semiconductors. J. Phys. Chem. A 2006, 110, 4065–4070. [Google Scholar] [CrossRef] [PubMed]
- Troisi, A.; Orlandi, G.; Anthony, J.E. Electronic interactions and thermal disorder in molecular crystals containing cofacial pentacene units. Chem. Mater. 2005, 17, 5024–5031. [Google Scholar] [CrossRef]
- Shuai, Z.; Geng, H.; Xu, W.; Liao, Y.; André, J.-M. From charge transport parameters to charge mobility in organic semiconductors through multiscale simulation. Chem. Soc. Rev. 2014, 43, 2662. [Google Scholar] [CrossRef] [PubMed]
- Oberhofer, H.; Reuter, K.; Blumberger, J. Charge transport in molecular materials: An assessment of computational methods. Chem. Rev. 2017, 117, 10319–10357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, D.L.; Troisi, A. Modelling charge transport in organic semiconductors: From quantum dynamics to soft matter. Phys. Chem. Chem. Phys. 2008, 10, 5941–5952. [Google Scholar] [CrossRef] [Green Version]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-transfer and energy-transfer processes in π-conjugated oligomers and polymers: A molecular picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.-L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Negri, F.; Zgierski, M.Z. Theoretical analysis of vibronic structure of absorption spectrum of fulvene. J. Chem. Phys. 1995, 102, 5165–5173. [Google Scholar] [CrossRef]
- Negri, F.; Orlandi, G. The T 1 resonance Raman spectra of 1,3,5–hexatriene and its deuterated isotopomers: An ab initio re-investigation. J. Chem. Phys. 1995, 103, 2412–2419. [Google Scholar] [CrossRef]
- Baumeier, B.; Kirkpatrick, J.; Andrienko, D. Density-functional based determination of intermolecular charge transfer properties for large-scale morphologies. Phys. Chem. Chem. Phys. 2010, 12, 11103. [Google Scholar] [CrossRef] [PubMed]
- Kubas, A.; Hoffmann, F.; Heck, A.; Oberhofer, H.; Elstner, M.; Blumberger, J. Electronic couplings for molecular charge transfer: Benchmarking CDFT, FODFT, and FODFTB against high-level ab initio calculations. J. Chem. Phys. 2014, 140, 104105. [Google Scholar] [CrossRef] [Green Version]
- Kubas, A.; Gajdos, F.; Heck, A.; Oberhofer, H.; Elstner, M.; Blumberger, J. Electronic couplings for molecular charge transfer: Benchmarking CDFT, FODFT and FODFTB against high-level ab initio calculations. II. Phys. Chem. Chem. Phys. 2015, 17, 14342–14354. [Google Scholar] [CrossRef] [PubMed]
- Canola, S.; Pecoraro, C.; Negri, F. Dimer and cluster approach for the evaluation of electronic couplings governing charge transport: Application to two pentacene polymorphs. Chem. Phys. 2016, 478, 130–138. [Google Scholar] [CrossRef]
- Troisi, A.; Orlandi, G. The hole transfer in DNA: Calculation of electron coupling between close bases. Chem. Phys. Lett. 2001, 344, 509–518. [Google Scholar] [CrossRef]
- Senthilkumar, K.; Grozema, F.C.; Bickelhaupt, F.M.; Siebbeles, L.D.A. Charge transport in columnar stacked triphenylenes: Effects of conformational fluctuations on charge transfer integrals and site energies. J. Chem. Phys. 2003, 119, 9809–9817. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.E.; Brédas, J.-L. Theoretical characterization of titanyl phthalocyanine as a p-type organic semiconductor: Short intermolecular π–π interactions yield large electronic couplings and hole transport bandwidths. J. Chem. Phys. 2008, 128, 034701. [Google Scholar] [CrossRef] [Green Version]
- Valeev, E.F.; Coropceanu, V.; da Silva Filho, D.; Salman, S.; Brédas, J.-L. Effect of electronic polarization on charge-transport parameters in molecular organic semiconductors. J. Am. Chem. Soc. 2006, 128, 9882–9886. [Google Scholar] [CrossRef] [PubMed]
- Schober, C.; Reuter, K.; Oberhofer, H. Critical analysis of fragment-orbital DFT schemes for the calculation of electronic coupling values. J. Chem. Phys. 2016, 144, 054103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.E.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Barbara, P.F.; Meyer, T.J.; Ratner, M.A. Contemporary Issues in Electron Transfer Research. J. Phys. Chem. 1996, 100, 13148–13168. [Google Scholar] [CrossRef]
- Olivier, Y.; Lemaur, V.; Brédas, J.L.; Cornil, J. Charge hopping in organic semiconductors: Influence of molecular parameters on macroscopic mobilities in model one-dimensional stacks. J. Phys. Chem. A 2006, 110, 6356–6364. [Google Scholar] [CrossRef]
- Schrader, M.; Körner, C.; Elschner, C.; Andrienko, D. Charge transport in amorphous and smectic mesophases of dicyanovinyl-substituted oligothiophenes. J. Mater. Chem. 2012, 22, 22258–22264. [Google Scholar] [CrossRef]
- Baumeier, B.; Stenzel, O.; Poelking, C.; Andrienko, D.; Schmidt, V. Stochastic modeling of molecular charge transport networks. Phys. Rev. B 2012, 86, 184202. [Google Scholar] [CrossRef] [Green Version]
- Marsh, R.A.; Groves, C.; Greenham, N.C. A microscopic model for the behavior of nanostructured organic photovoltaic devices. J. Appl. Phys. 2007, 101, 083509. [Google Scholar] [CrossRef] [Green Version]
- Baumeier, B.; May, F.; Lennartz, C.; Andrienko, D. Challenges for in silico design of organic semiconductors. J. Mater. Chem. 2012, 22, 10971. [Google Scholar] [CrossRef]
- Troisi, A.; Orlandi, G. Charge-Transport Regime of Crystalline organic semiconductors: Diffusion limited by thermal off-diagonal electronic disorder. Phys. Rev. Lett. 2006, 96, 086601. [Google Scholar] [CrossRef]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software tools for molecular design. J. Chem. Theory Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q.; Shuai, Z.; Chen, L.; Shi, Q. Multiscale study of charge mobility of organic semiconductor with dynamic disorders. Phys. Chem. Chem. Phys. 2010, 12, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Carrera, R.S.; Paramonov, P.; Day, G.M.; Coropceanu, V.; Brédas, J.-L. Interaction of charge carriers with lattice vibrations in oligoacene crystals from naphthalene to pentacene. J. Am. Chem. Soc. 2010, 132, 14437–14446. [Google Scholar] [CrossRef]
- Vener, M.V.; Parashchuk, O.D.; Kharlanov, O.G.; Maslennikov, D.R.; Dominskiy, D.I.; Yu. Chernyshov, I.; Yu. Paraschuk, D.; Yu. Sosorev, A. Non-local electron-phonon interaction in naphthalene diimide derivatives, its experimental probe and impact on charge-carrier mobility. Adv. Electron. Mater. 2021, 7, 2001281. [Google Scholar] [CrossRef]
- Beratan, D.N.; Skourtis, S.S.; Balabin, I.A.; Balaeff, A.; Keinan, S.; Venkatramani, R.; Xiao, D. Steering electrons on moving pathways. Acc. Chem. Res. 2009, 42, 1669–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dimer or Charge Transfer Path | Molecules Forming the Dimer | Intermolecular Distance (Å) | Interplanar Distance (Å) | Vij (LUMO) (meV) | keT a (s−1) |
|---|---|---|---|---|---|
| α-NDTI | |||||
| P1 | ANTI-1 | 6.654 | 3.646 | 36 | 5.62 × 1012 |
| ANTI-2 | 6.654 | 3.646 | 40 | 6.88 × 1012 | |
| mix | 6.654 | 3.646 | 38 | ||
| N-NDTI | |||||
| P1 | ANTI-1 | 4.197 | 3.623 | 35 | 6.87 × 1012 |
| ANTI-2 | 4.197 | 3.623 | 50 | 1.36 × 1013 | |
| mix | 4.193–4.202 | 3.700–3.744 | 46–38 | 1.15 × 1013–7.91 × 1012 | |
| P2 | ANTI-1 | 11.002 | 0.144 | 10 | 5.26 × 1011 |
| ANTI-2 | 11.002 | 0.144 | 8 | 3.05 × 1011 | |
| mix | 11.002–11.002 | 0.244–0.278 | 9–8 | 4.91 × 1011–3.89 × 1011 | |
| P3 | ANTI-1 | 11.238 | 3.467 | 2 | 2.59 × 1010 |
| ANTI-2 | 11.238 | 3.467 | 1 | 5.12 × 109 | |
| P4 | ANTI-1 | 12.290 | 3.033 | 0 | 4.40 × 108 |
| ANTI-2 | 12.290 | 3.033 | 0 | 1.56 × 108 | |
(eV) | ωeff a (cm−1) | Seff a | (eV) | (eV) | |
|---|---|---|---|---|---|
| α-NDTI ANTI-1 | 0.286 | 920 | 2.51 | 0.032 | 0.01 |
| α-NDTIANTI-2 | 0.286 | 925 | 2.49 | 0.033 | 0.01 |
| N-NDTId | 0.294 | 849 | 2.79 | 0.011 | 0.01 |
| Zero Field (Brownian) | TOF | TOF | Exp | |
|---|---|---|---|---|
| μ (cm2 V−1 s−1) | μmax (cm2 V−1 s−1) | μmin (cm2 V−1 s−1) | μ a (cm2 V−1 s−1) | |
| α-NDTI ANTI-1 | 0.32 | 1.00 b | 3 × 10−6 b | 0.037 |
| α-NDTI ANTI-2 | 0.39 | 1.23 b | 2 × 10−6 b | |
| N-NDTI ANTI-1 | 0.24 | 0.46 c | 0.25 c | 1.27 |
| N-NDTI ANTI-2 | 0.34 | 0.92 c | 0.14 c | |
| N-NDTI mix | 0.28 | 0.63 c | 0.21 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricci, G.; Canola, S.; Dai, Y.; Fazzi, D.; Negri, F. Impact of Fluoroalkylation on the n-Type Charge Transport of Two Naphthodithiophene Diimide Derivatives. Molecules 2021, 26, 4119. https://doi.org/10.3390/molecules26144119
Ricci G, Canola S, Dai Y, Fazzi D, Negri F. Impact of Fluoroalkylation on the n-Type Charge Transport of Two Naphthodithiophene Diimide Derivatives. Molecules. 2021; 26(14):4119. https://doi.org/10.3390/molecules26144119
Chicago/Turabian StyleRicci, Gaetano, Sofia Canola, Yasi Dai, Daniele Fazzi, and Fabrizia Negri. 2021. "Impact of Fluoroalkylation on the n-Type Charge Transport of Two Naphthodithiophene Diimide Derivatives" Molecules 26, no. 14: 4119. https://doi.org/10.3390/molecules26144119
APA StyleRicci, G., Canola, S., Dai, Y., Fazzi, D., & Negri, F. (2021). Impact of Fluoroalkylation on the n-Type Charge Transport of Two Naphthodithiophene Diimide Derivatives. Molecules, 26(14), 4119. https://doi.org/10.3390/molecules26144119