
Synthesis, Molecular Docking, and Biofilm Formation Inhibitory Activity of Bis(Indolyl)Pyridines Analogues of the Marine Alkaloid Nortopsentin

 ,
,
Abstract
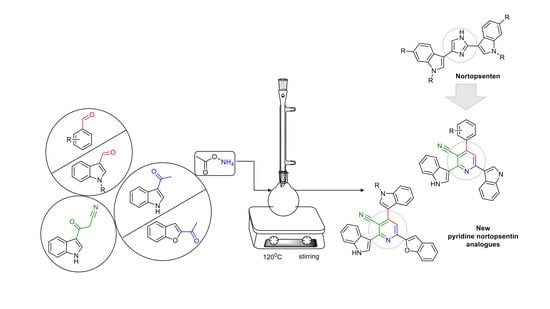
:
1. Introduction
2. Results and Discussion
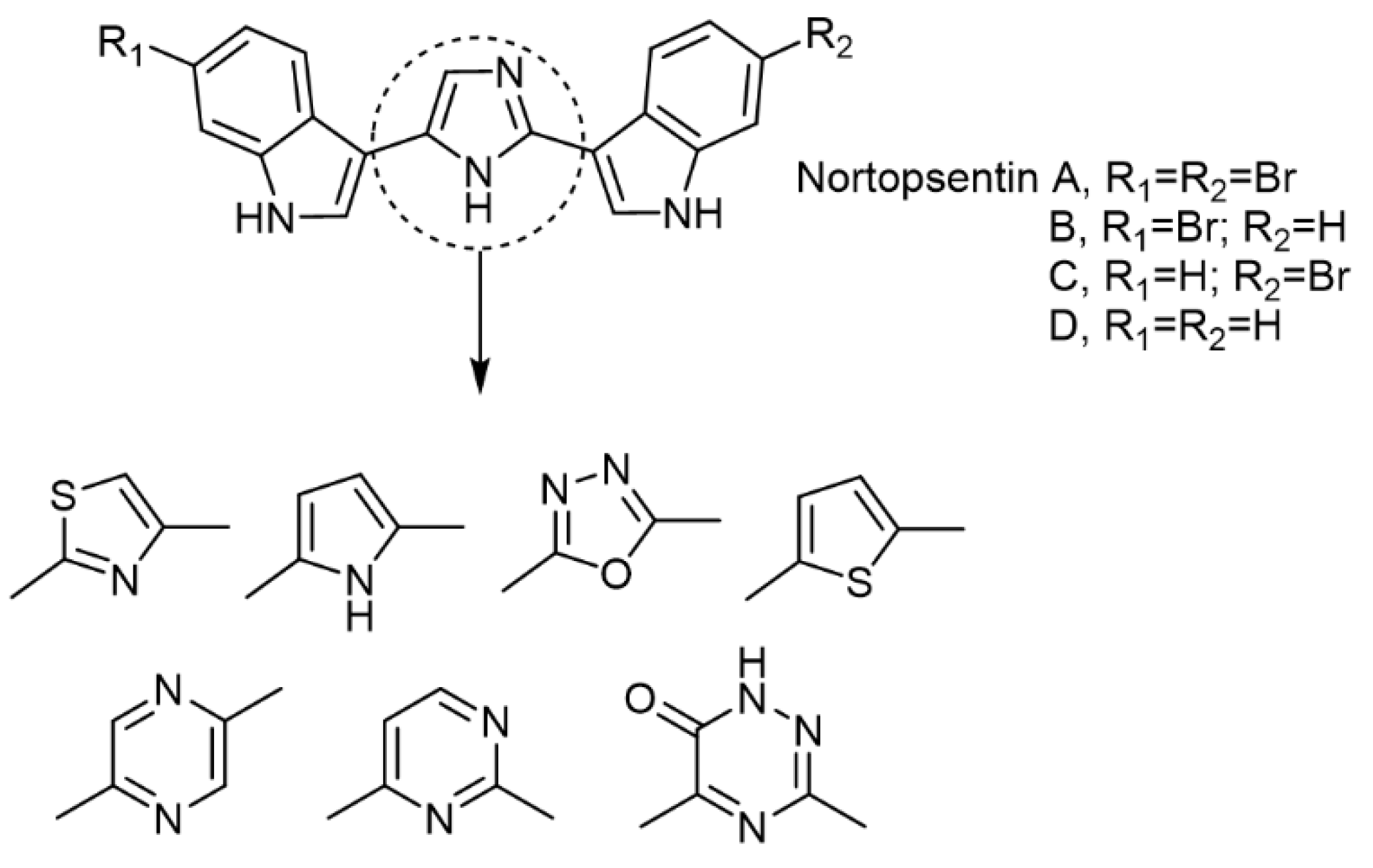
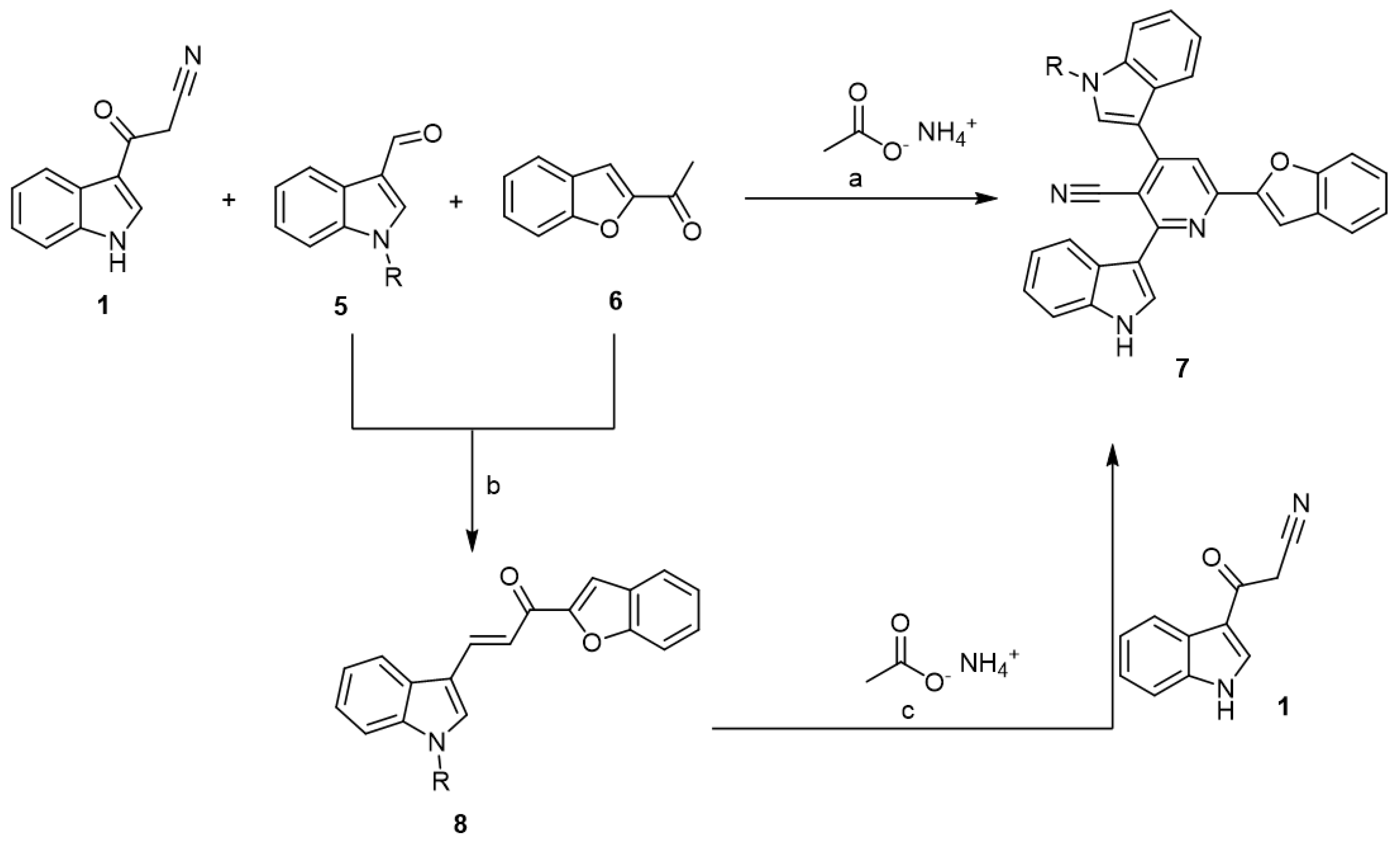
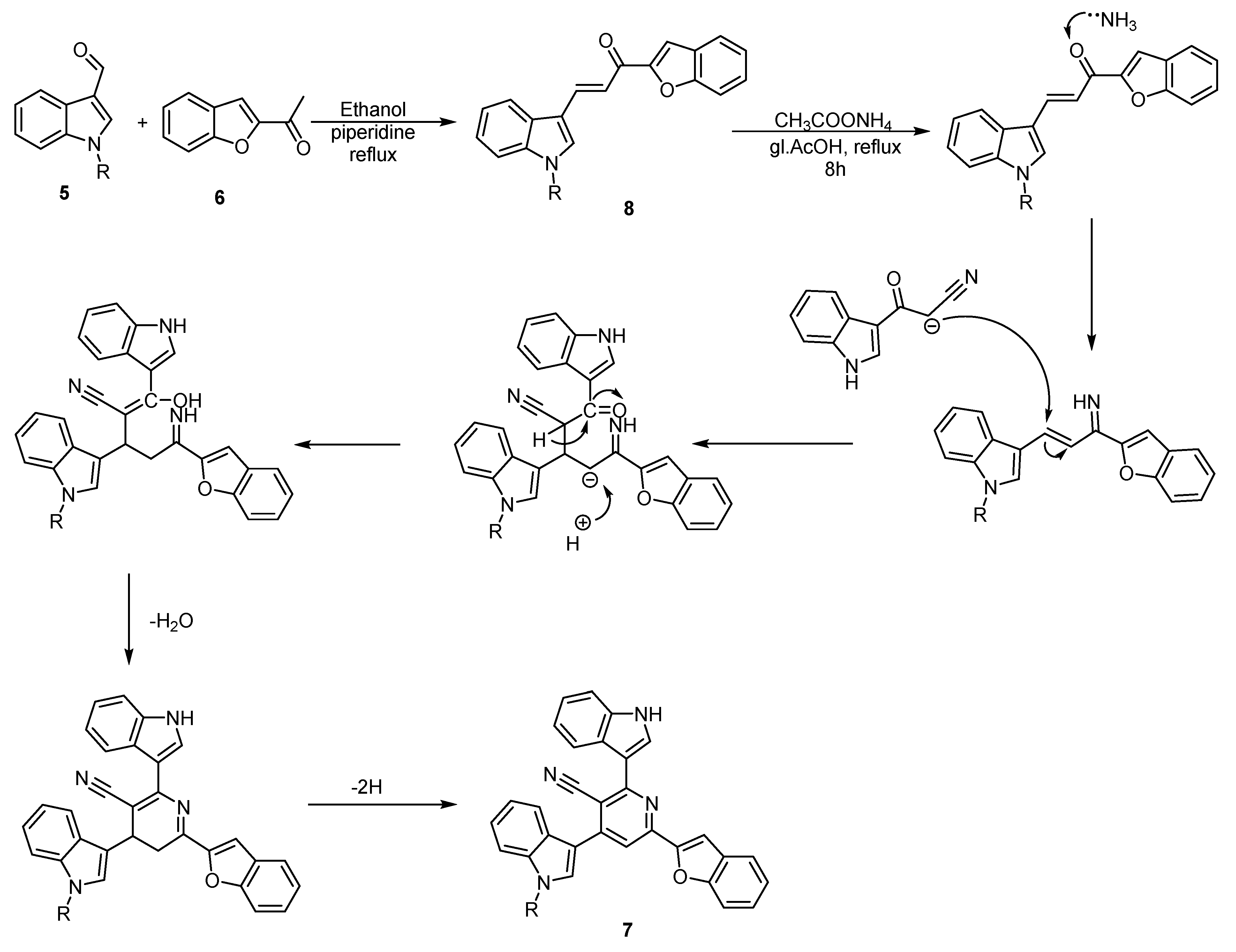
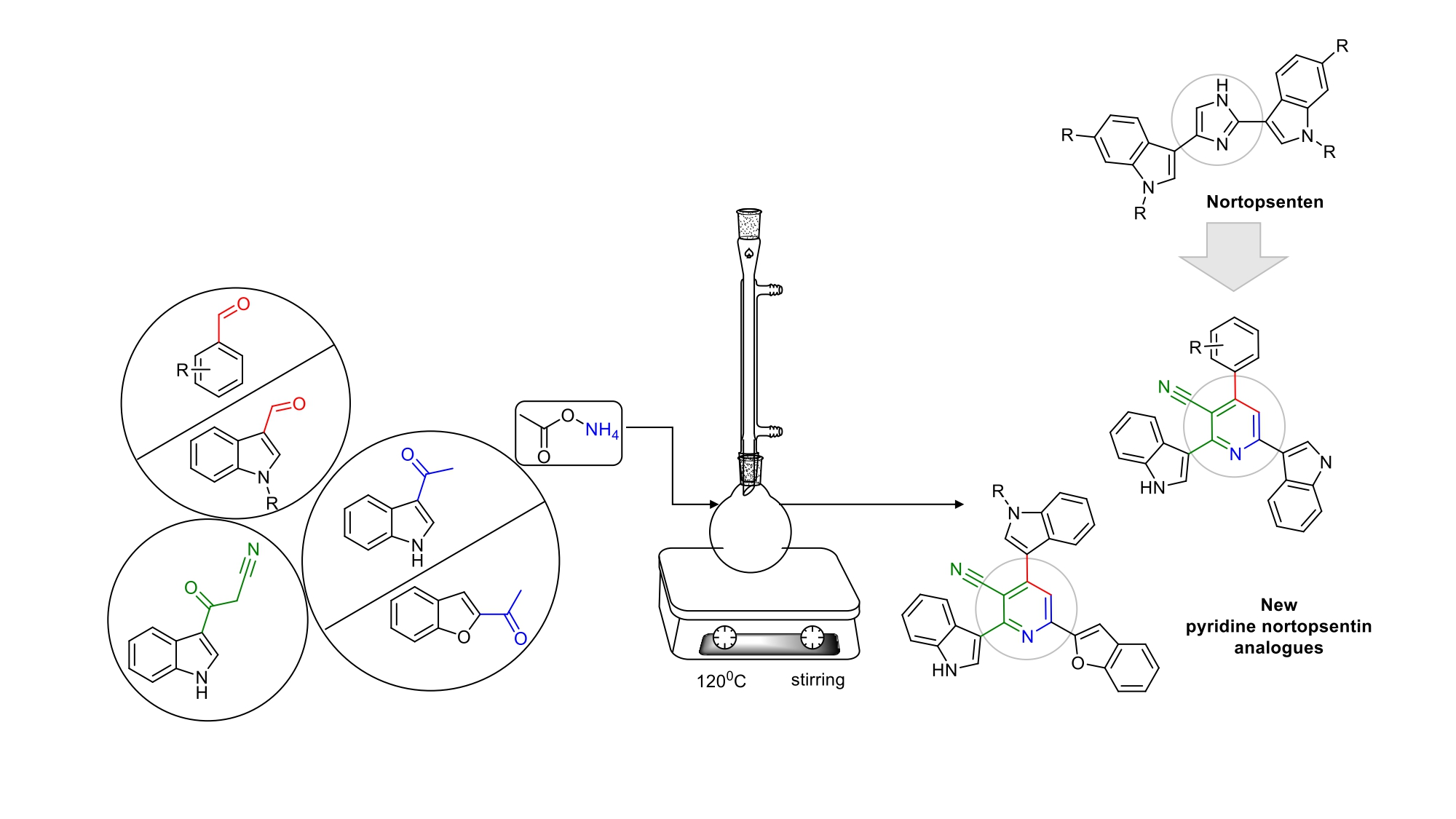
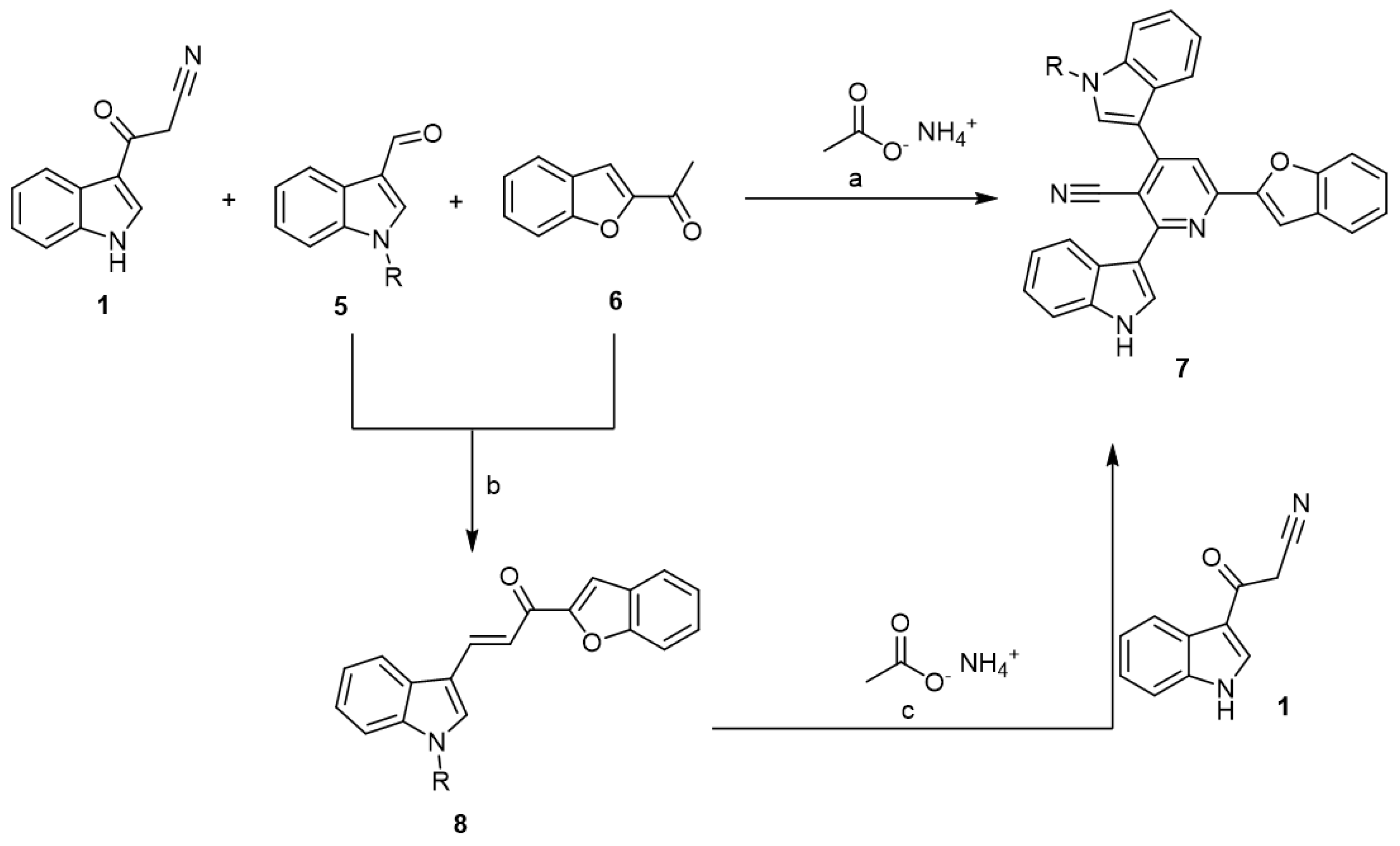
2.1. Chemistry
2.2. Biological Studies
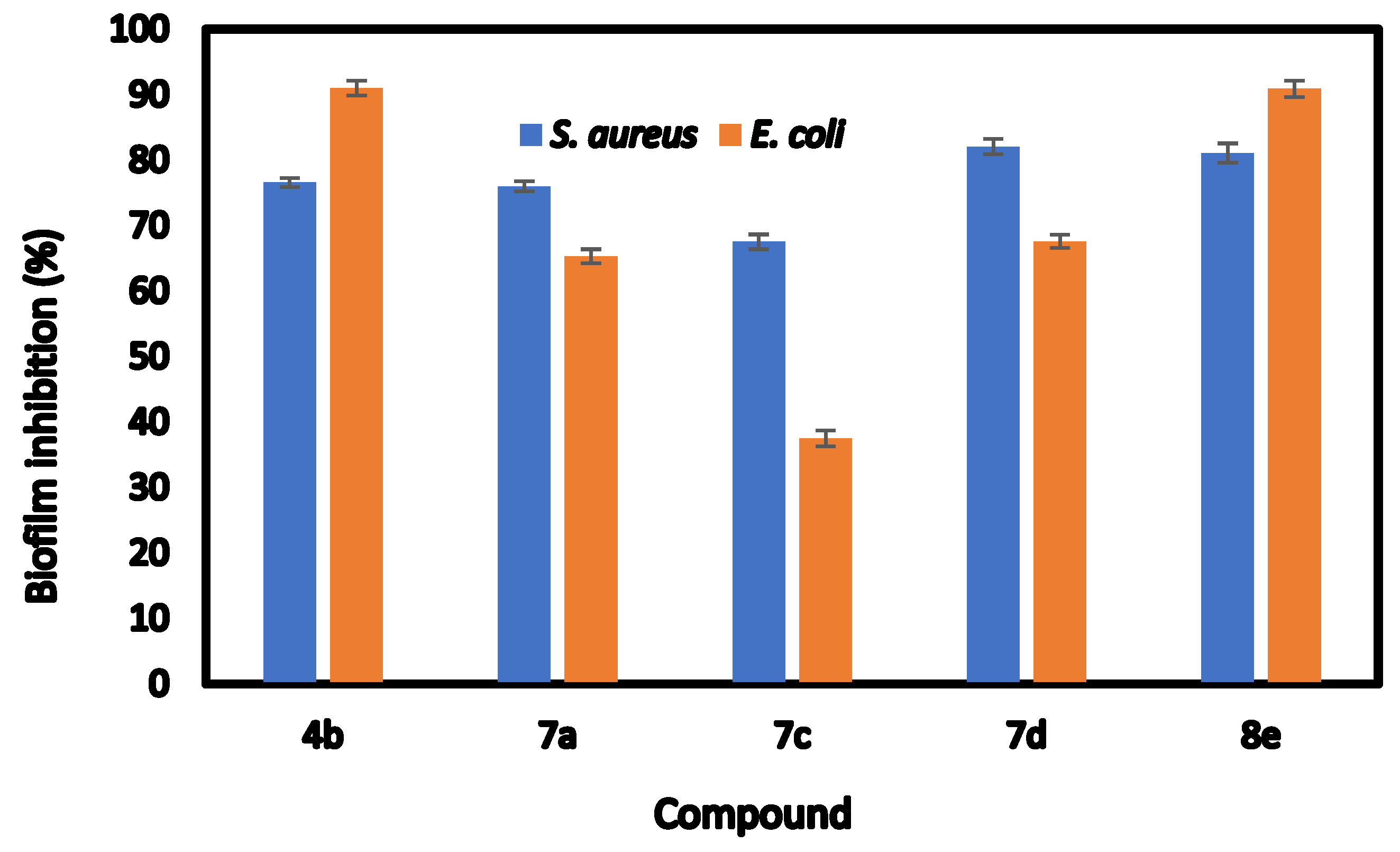
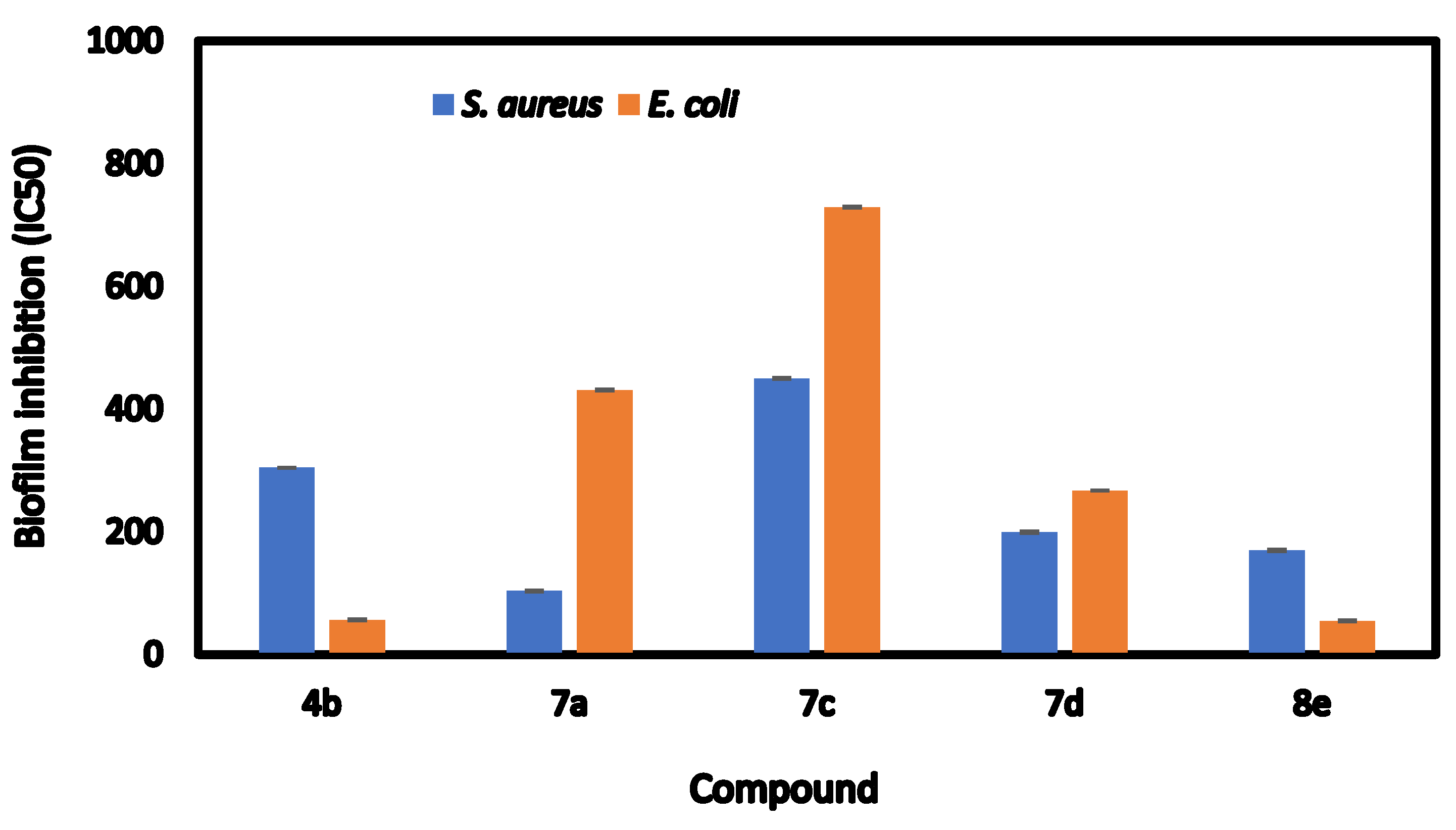
2.3. Inhibition of Biofilm Formation
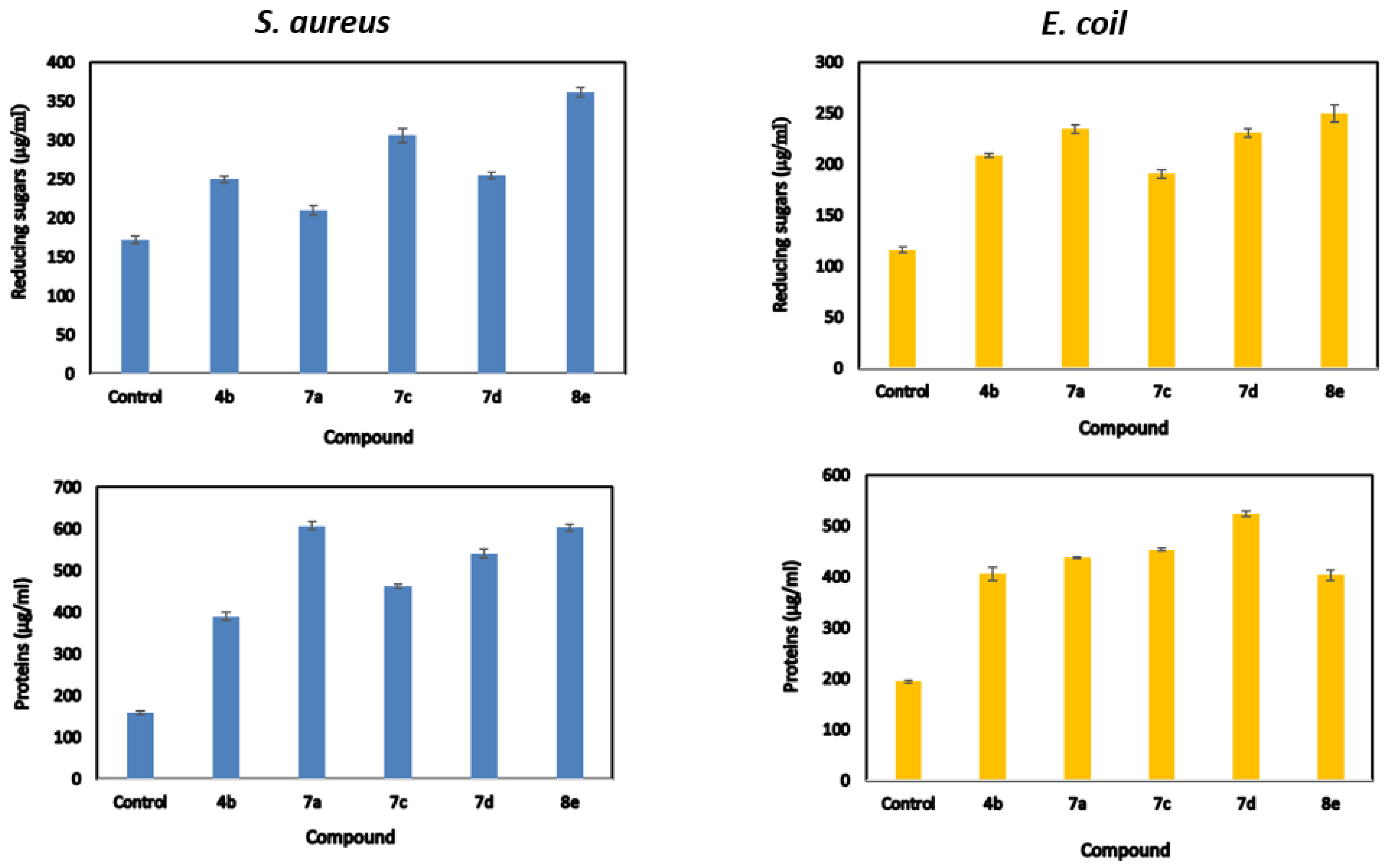
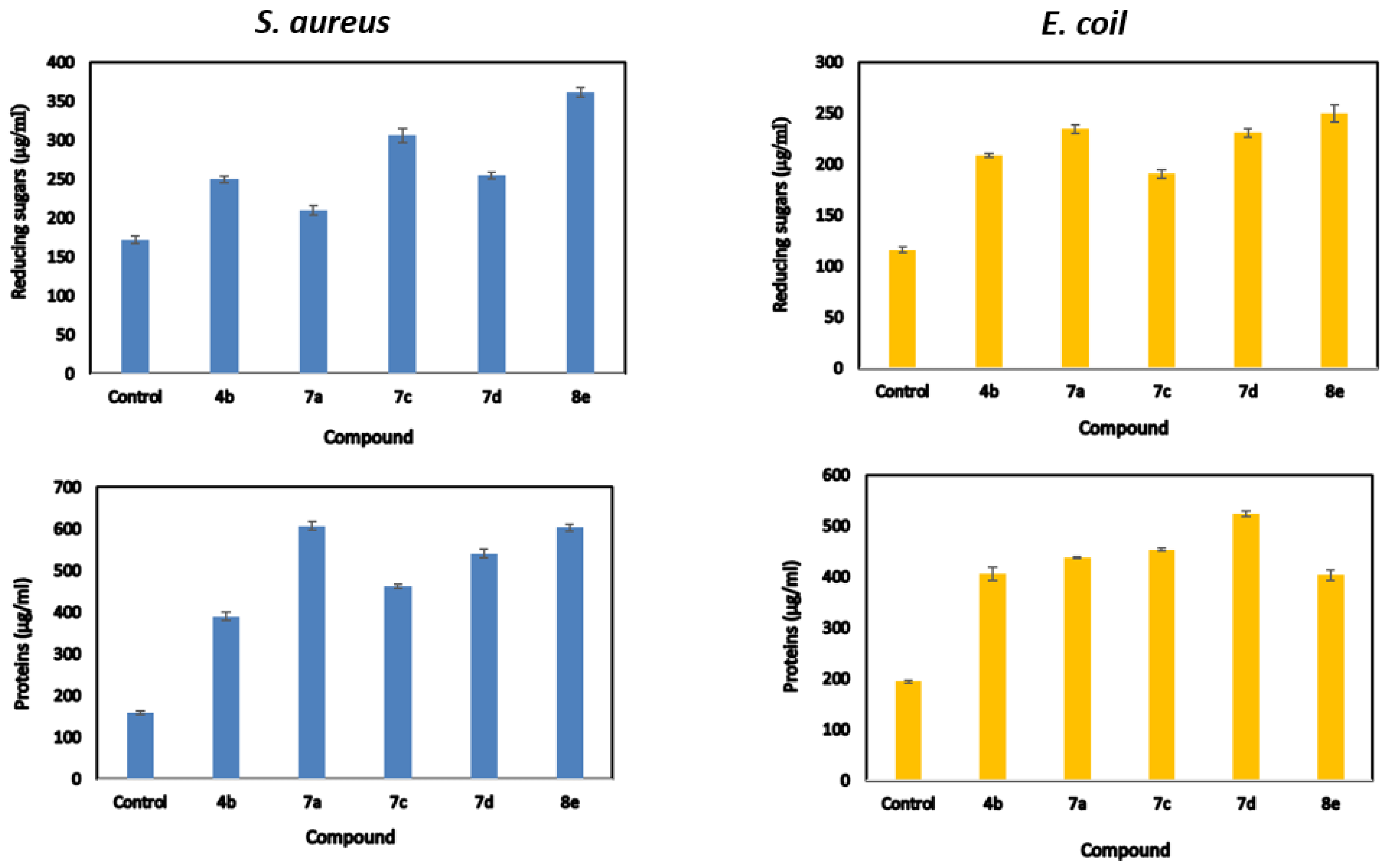
2.4. Release of Reducing Sugars and Proteins
2.5. Molecular Docking Results
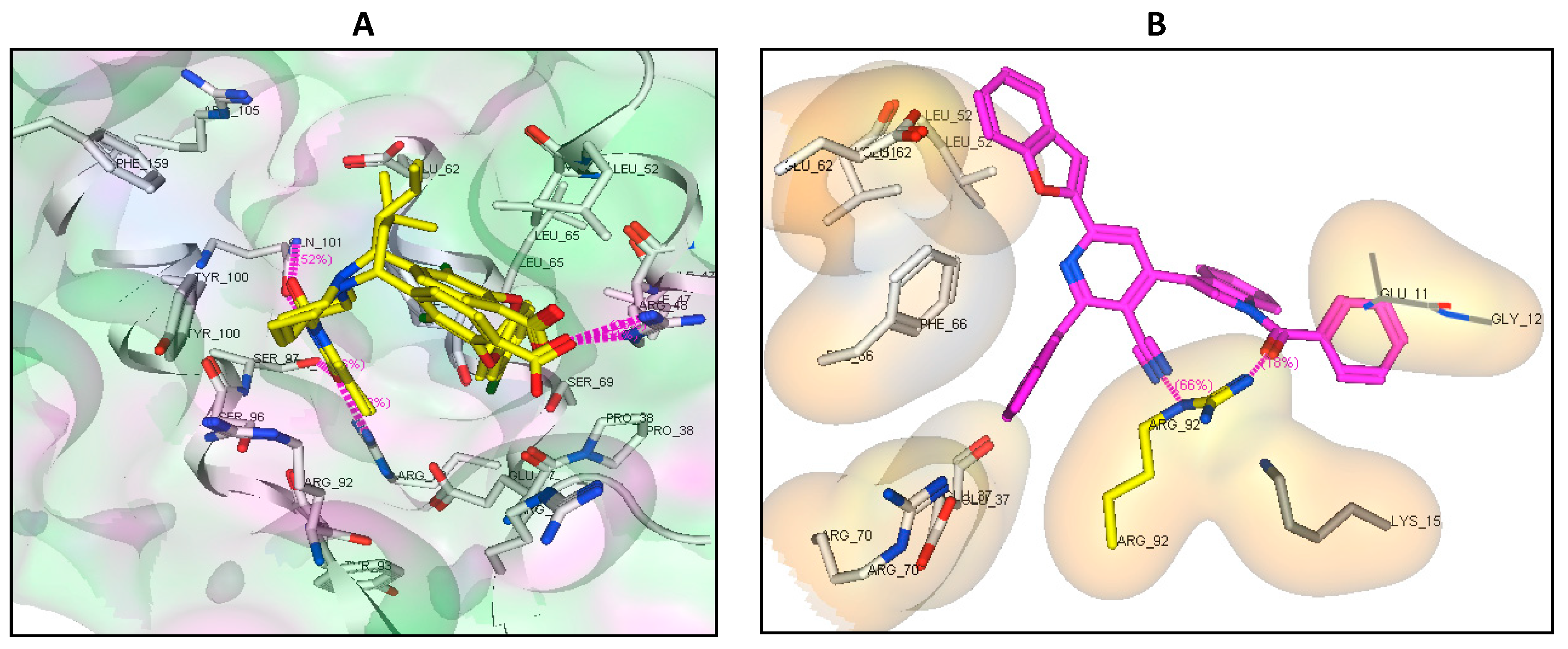
2.6. Binding Mode of the Studied Compounds with Thymidylate Kinase (ID: 4QGG)
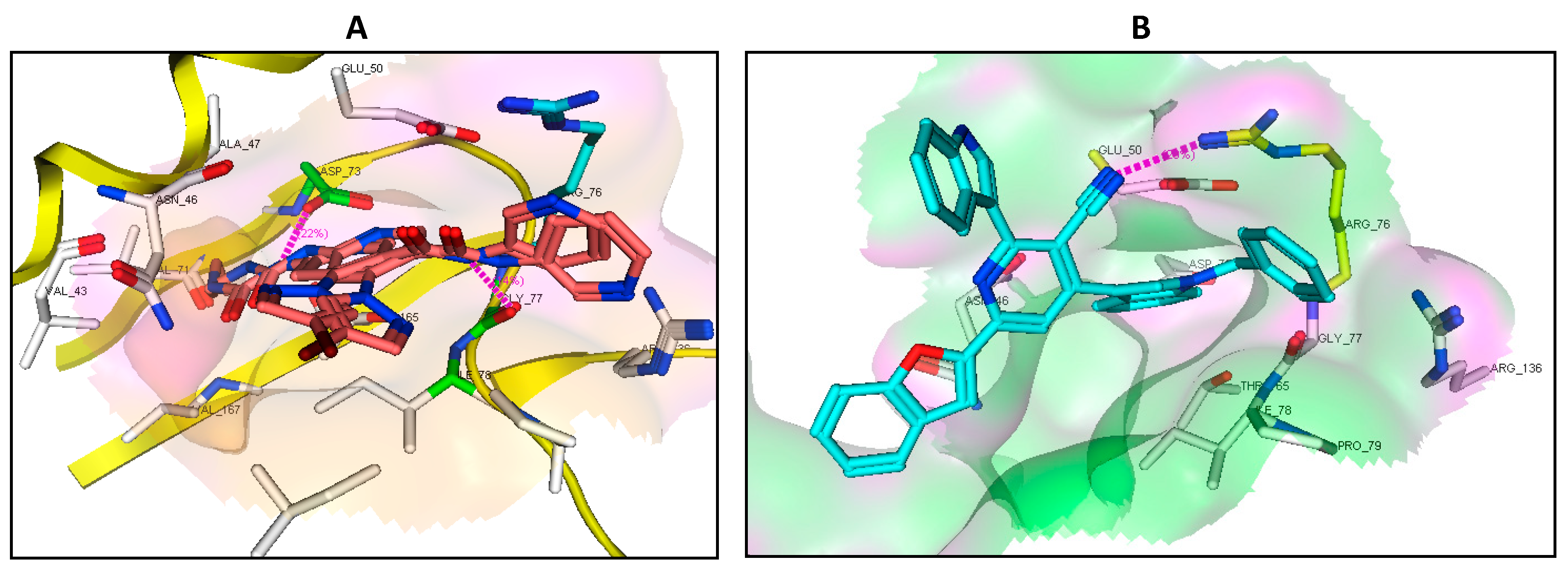
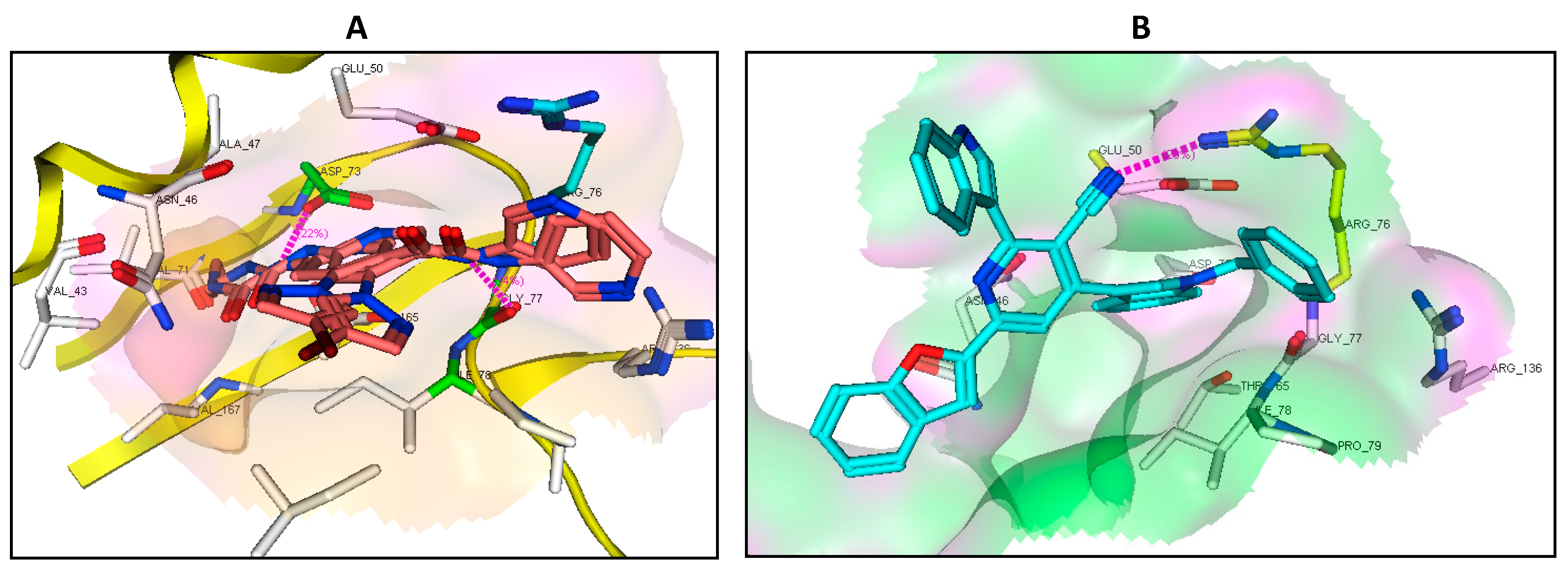
2.7. Binding Mode of the Studied Compounds with DNA Gyrase B (ID: 6F86)
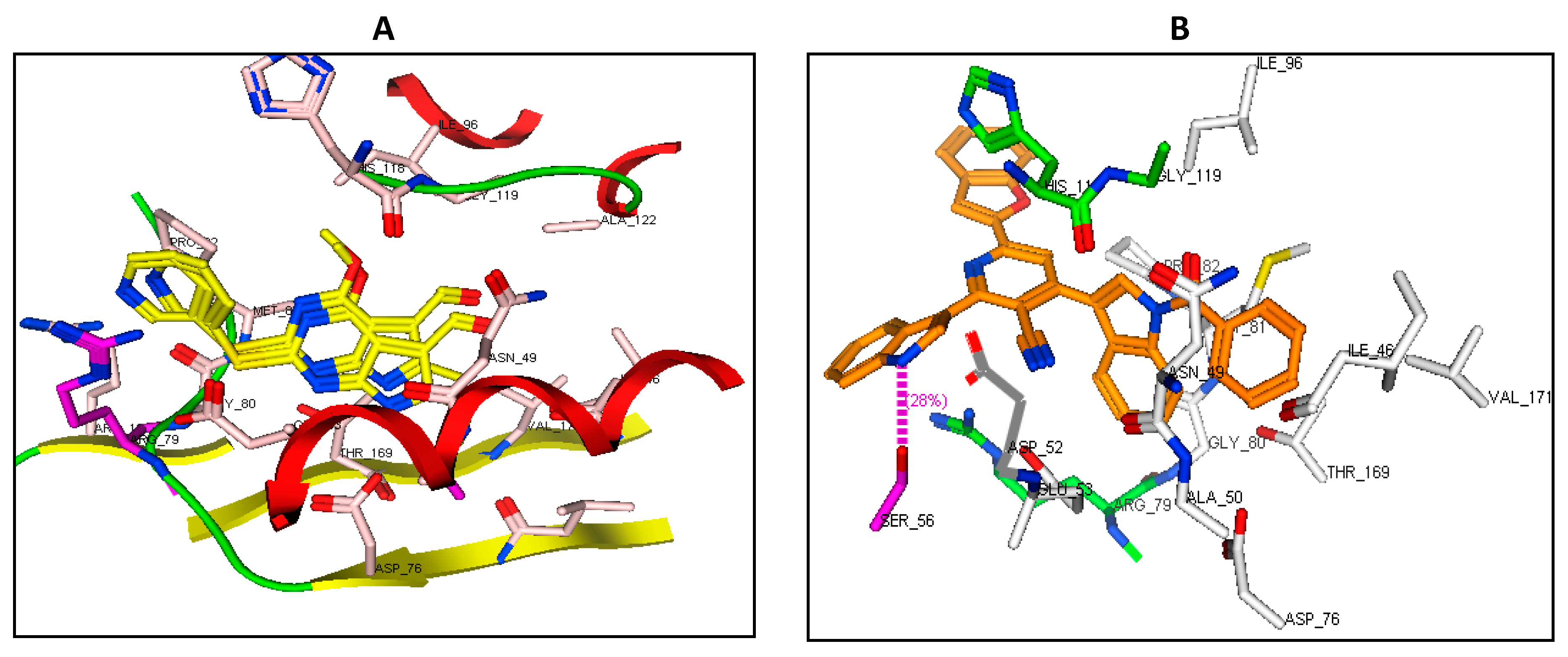
2.8. Binding Mode of the Studied Compounds with DNA Topoisomerase IV Subunit B (ID: 4HZ5)
3. Materials and Methods
3.1. Chemistry
3.2. Biological Studies
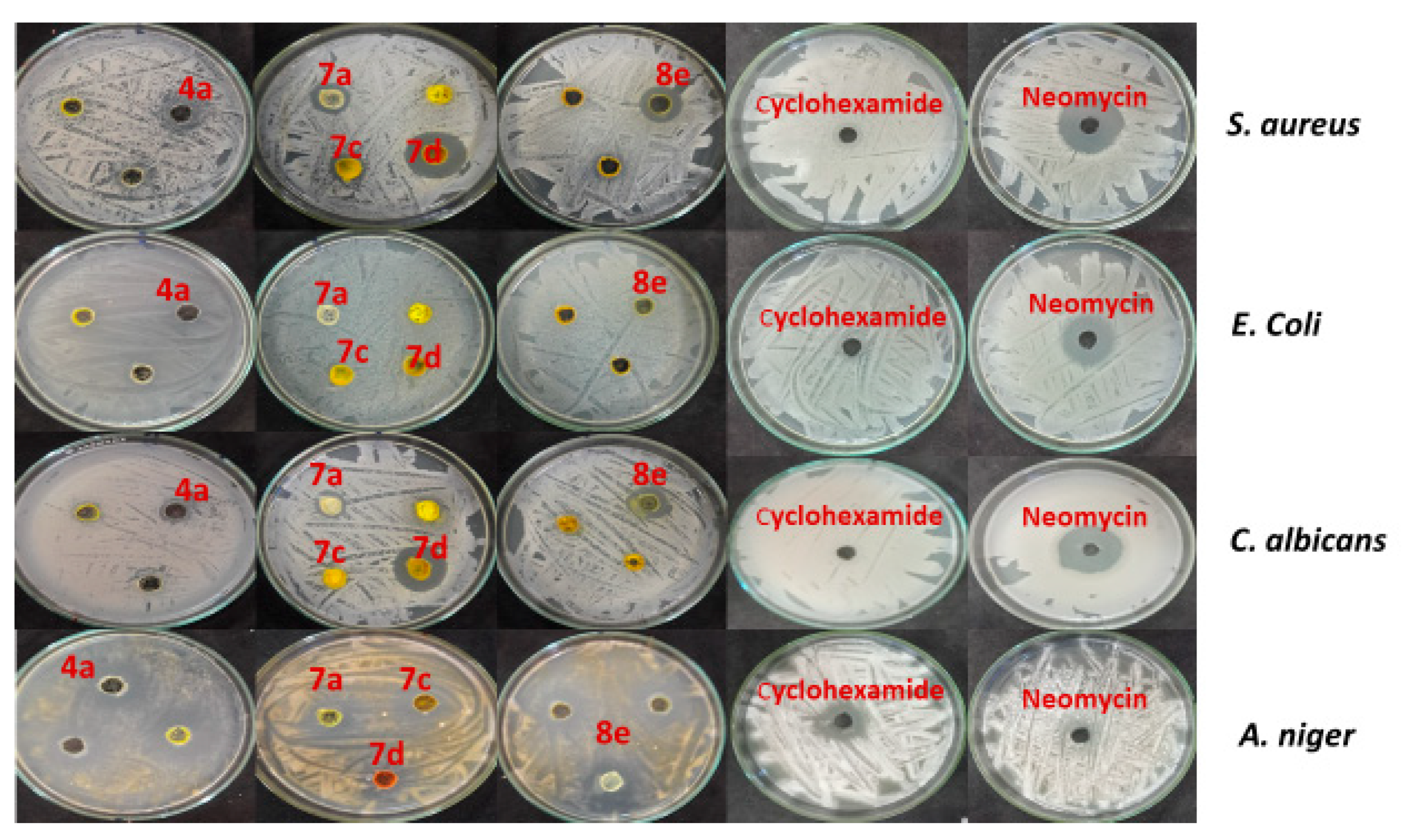
Antimicrobial Activity
3.3. MICs and MBCs Evaluation
3.4. Inhibition of Biofilm Formation (Crystal Violet Method)
3.5. Release of Cellular Sugars and Proteins
3.6. Molecular Docking
3.6.1. Target Preparation
3.6.2. Ligand Preparation
3.6.3. Docking Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb. Drug Resist. 2019, 25, 890–908. [Google Scholar] [CrossRef]
- Sun, Y.; Ye, J.; Hou, Y.; Chen, H.; Cao, J.; Zhou, T. Predation Efficacy of Bdellovibrio Bacteriovorus on Multidrug-Resistant Clinical Pathogens and Their Corresponding Biofilms. Jpn. J. Infect. Dis. 2017, 70, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Hiramatsu, K.; Cui, L.; Kuroda, M.; Ito, T. The Emergence and Evolution of Methicillin-Resistant Staphylococcus Aureus. Trends Microbiol. 2001, 9, 486–493. [Google Scholar] [CrossRef]
- Mishra, R.; Panda, A.K.; De Mandal, S.; Shakeel, M.; Bisht, S.S.; Khan, J. Natural Anti-Biofilm Agents: Strategies to Control Biofilm-Forming Pathogens. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus Biofilm: An Emerging Battleground in Microbial Communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef]
- Carbone, A.; Parrino, B.; Cusimano, M.G.; Spanò, V.; Montalbano, A.; Barraja, P.; Schillaci, D.; Cirrincione, G.; Diana, P.; Cascioferro, S. New Thiazole Nortopsentin Analogues Inhibit Bacterial Biofilm Formation. Mar. Drugs 2018, 16, 274. [Google Scholar] [CrossRef] [Green Version]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and Antifungal Imidazolediylbis[Indoles] from the Sponge Spongosorites Ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Kawasaki, I.; Yamashita, M.; Ohta, S. Total Synthesis of Nortopsentins A-D, Marine Alkaloids. Chem. Pharm. Bull. 1996, 44, 1831–1839. [Google Scholar] [CrossRef] [Green Version]
- Kamel, M.M.; Abdel-hameid, M.K.; El-Nassan, H.B.; El-Khouly, E.A. Recent Advances in the Synthesis and Biological Applications of Nortopsentin Analogs. Chem. Heterocycl. Comp. 2020, 56, 499–502. [Google Scholar] [CrossRef]
- Alvarado, S.; Roberts, B.F.; Wright, A.E.; Chakrabarti, D. The Bis(Indolyl)Imidazole Alkaloid Nortopsentin A Exhibits Antiplasmodial Activity. Antimicrob. Agents Chemother. 2013, 57, 2362–2364. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Cascioferro, S.; Parrino, B.; Carbone, D.; Pecoraro, C.; Schillaci, D.; Cusimano, M.G.; Cirrincione, G.; Diana, P. Thiazole Analogues of the Marine Alkaloid Nortopsentin as Inhibitors of Bacterial Biofilm Formation. Molecules 2021, 26, 81. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and Antitumor Activity of 2,5-Bis(3′-Indolyl)-Furans and 3,5-Bis(3′-Indolyl)-Isoxazoles, Nortopsentin Analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Martorana, A.; Dattolo, G.; Gia, O.; Via, L.D.; Cirrincione, G. Synthesis and Antitumor Properties of 2,5-Bis(3′-Indolyl)Thiophenes: Analogues of Marine Alkaloid Nortopsentin. Bioorganic Med. Chem. Lett. 2007, 17, 2342–2346. [Google Scholar] [CrossRef]
- Diana, P.; Carbone, A.; Barraja, P.; Martorana, A.; Gia, O.; DallaVia, L.; Cirrincione, G. 3,5-Bis(3′-Indolyl)Pyrazoles, Analogues of Marine Alkaloid Nortopsentin: Synthesis and Antitumor Properties. Bioorganic Med. Chem. Lett. 2007, 17, 6134–6137. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.-H. Synthesis and Antiproliferative Activity of 2,5-Bis(3′-Indolyl)Pyrroles, Analogues of the Marine Alkaloid Nortopsentin. Mar. Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Gu, X.-H. Syntheses and Cytotoxicity Evaluation of Bis(Indolyl)Thiazole, Bis(Indolyl)Pyrazinone and Bis(Indolyl)Pyrazine: Analogues of Cytotoxic Marine Bis(Indole) Alkaloid. Bioorg. Med. Chem. 2000, 8, 363–371. [Google Scholar] [CrossRef]
- Sreenivasulu, R.; Durgesh, R.; Jadav, S.S.; Sujitha, P.; Ganesh Kumar, C.; Raju, R.R. Synthesis, Anticancer Evaluation and Molecular Docking Studies of Bis(Indolyl) Triazinones, Nortopsentin Analogs. Chem. Pap. 2018, 72, 1369–1378. [Google Scholar] [CrossRef]
- Kumar, D.; Arun, V.; MaruthiKumar, N.; Acosta, G.; Noel, B.; Shah, K. A Facile Synthesis of Novel Bis-(Indolyl)-1,3,4-Oxadiazoles as Potent Cytotoxic Agents. ChemMedChem 2012, 7, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spanò, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and Antiproliferative Activity of Substituted 3[2-(1H-Indol-3-Yl)-1,3-Thiazol-4-Yl]-1H-Pyrrolo[3,2-b]Pyridines, Marine Alkaloid Nortopsentin Analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef]
- Parrino, B.; Attanzio, A.; Spanò, V.; Cascioferro, S.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Diana, P.; Cirrincione, G.; Carbone, A. Synthesis, Antitumor Activity and CDK1 Inhibiton of New Thiazole Nortopsentin Analogues. Eur. J. Med. Chem. 2017, 138, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Spanò, V.; Attanzio, A.; Cascioferro, S.; Carbone, A.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. Synthesis and Antitumor Activity of New Thiazole Nortopsentin Analogs. Mar. Drugs 2016, 14, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Hao, Y.; Ji, X.; Wang, Z.; Liu, Y.; Ma, D.; Li, Y.; Pang, H.; Ni, J.; Wang, Q. Optimization, Structure-Activity Relationship, and Mode of Action of Nortopsentin Analogues Containing Thiazole and Oxazole Moieties. J. Agric. Food Chem. 2019, 67, 10018–10031. [Google Scholar] [CrossRef] [PubMed]
- França, P.H.B.; Barbosa, D.P.; da Silva, D.L.; Ribeiro, Ê.A.N.; Santana, A.E.G.; Santos, B.V.O.; Barbosa-Filho, J.M.; Quintans, J.S.S.; Barreto, R.S.S.; Quintans-Júnior, L.J.; et al. Indole Alkaloids from Marine Sources as Potential Leads against Infectious Diseases. Available online: https://www.hindawi.com/journals/bmri/2014/375423/ (accessed on 2 January 2021).
- Ahmad, G.; Rasool, N.; Ikram, H.M.; Gul Khan, S.; Mahmood, T.; Ayub, K.; Zubair, M.; Al-Zahrani, E.; Ali Rana, U.; Akhtar, M.N.; et al. Efficient Synthesis of Novel Pyridine-Based Derivatives via Suzuki Cross-Coupling Reaction of Commercially Available 5-Bromo-2-Methylpyridin-3-Amine: Quantum Mechanical Investigations and Biological Activities. Molecules 2017, 22, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.N.; Mallareddy, G.; Nagender, P.; Rao, P.S.; Poornachandra, Y.; Ranjithreddy, P.; Kumar, C.G.; Narsaiah, B. Synthesis of Novel Triazole Functionalized Pyridine Derivatives as Potential Antimicrobial and Anti-Biofilm Agents. Indian J. Chem. Sect. B Org. Med. Chem. 2016, 55, 1361–1375. [Google Scholar]
- Xiong, W.-N.; Yang, C.-G.; Jiang, B. Synthesis of Novel Analogues of Marine Indole Alkaloids: Mono(Indolyl)-4-Trifluoromethylpyridines and Bis(Indolyl)-4-Trifluoromethylpyridines as Potential Anticancer Agents. Bioorg. Med. Chem. 2001, 9, 1773–1780. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Wang, Y.-L.; Zhou, R.-R.; Wu, G.-C.; Yin, S.-Y.; Yan, X.; Wang, B. N-Bromosuccinimide (NBS)-Catalyzed C–H Bond Functionalization: An Annulation of Alkynes with Electron Withdrawing Group (EWG)-Substituted Acetyl Indoles for the Synthesis of Carbazoles. Org. Lett. 2017, 19, 6140–6143. [Google Scholar] [CrossRef]
- Abdel-Aziem, A. Utility of 2-Acetyl Benzofuran for the Synthesis of New Heterocycles as Potential Anticancer Agents. J. Heterocycl. Chem. 2017, 54, 2985–2995. [Google Scholar] [CrossRef]
- Hiremathad, A.; Patil, M.R.; Chethana, K.R.; Chand, K.; Santos, M.A.; Keri, R.S. Benzofuran: An Emerging Scaffold for Antimicrobial Agents. RSC Adv. 2015, 5, 96809–96828. [Google Scholar] [CrossRef]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, M.E. Design, Synthesis, Antimicrobial Evaluation, and Laccase Catalysis Effect of Novel Benzofuran–Oxadiazole and Benzofuran–Triazole Hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Rajesh, S.; Dharanishanthi, V.; Kanna, A.V. Antibacterial Mechanism of Biogenic Silver Nanoparticles of Lactobacillus Acidophilus. J. Exp. Nanosci. 2015, 10, 1143–1152. [Google Scholar] [CrossRef]
- Mohi El-Deen, E.M.; Abd El-Meguid, E.A.; Karam, E.A.; Nossier, E.S.; Ahmed, M.F. Synthesis and Biological Evaluation of New Pyridothienopyrimidine Derivatives as Antibacterial Agents and Escherichia Coli Topoisomerase II Inhibitors. Antibiotics 2020, 9, 695. [Google Scholar] [CrossRef] [PubMed]
- Döhren, H.V. Antibiotics: Actions, Origins, Resistance, by C. Walsh. 2003. Washington, DC: ASM Press. 345 Pp. $99.95 (Hardcover). Protein Sci. 2004, 13, 3059–3060. [Google Scholar] [CrossRef] [Green Version]
- Silver, L.L. Multi-Targeting by Monotherapeutic Antibacterials. Nat. Rev. Drug Discov 2007, 6, 41–55. [Google Scholar] [CrossRef]
- Tari, L.W.; Trzoss, M.; Bensen, D.C.; Li, X.; Chen, Z.; Lam, T.; Zhang, J.; Creighton, C.J.; Cunningham, M.L.; Kwan, B.; et al. Pyrrolopyrimidine Inhibitors of DNA Gyrase B (GyrB) and Topoisomerase IV (ParE). Part I: Structure Guided Discovery and Optimization of Dual Targeting Agents with Potent, Broad-Spectrum Enzymatic Activity. Bioorganic Med. Chem. Lett. 2013, 23, 1529–1536. [Google Scholar] [CrossRef]
- Kawatkar, S.P.; Keating, T.A.; Olivier, N.B.; Breen, J.N.; Green, O.M.; Guler, S.Y.; Hentemann, M.F.; Loch, J.T.; McKenzie, A.R.; Newman, J.V.; et al. Antibacterial Inhibitors of Gram-Positive Thymidylate Kinase: Structure-Activity Relationships and Chiral Preference of a New Hydrophobic Binding Region. J. Med. Chem. 2014, 57, 4584–4597. [Google Scholar] [CrossRef]
- Collins, C.H.; Lyne, P.M.; Grange, J.M. Collins and Lyne’s Microbiological Methods, 7th ed.; Hodder Education Publishers: London, UK; New York, NY, USA, 1995; ISBN 978-0-7506-0653-0. [Google Scholar]
- Maigali, S.S.; Abd-El-Maksoud, M.A.; El-Hussieny, M.; Soliman, F.M.; Abdel-Aziz, M.S.; Shalaby, E.-S.M. Chemistry of Phosphorus Ylides: Part 41 Synthesis of Antimicrobial Agents from the Reaction of Aminoantipyrine, Coumarin- and Quinoline-Carbaldehyde with Phosphacumulene and Phosphaallene Ylides. J. Chem. Res. 2014, 38, 754–761. [Google Scholar] [CrossRef]
- Sarker, S.D.; Nahar, L.; Kumarasamy, Y. Microtitre Plate-Based Antibacterial Assay Incorporating Resazurin as an Indicator of Cell Growth, and Its Application in the in Vitro Antibacterial Screening of Phytochemicals. Methods 2007, 42, 321–324. [Google Scholar] [CrossRef]
- Miller, G.L. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
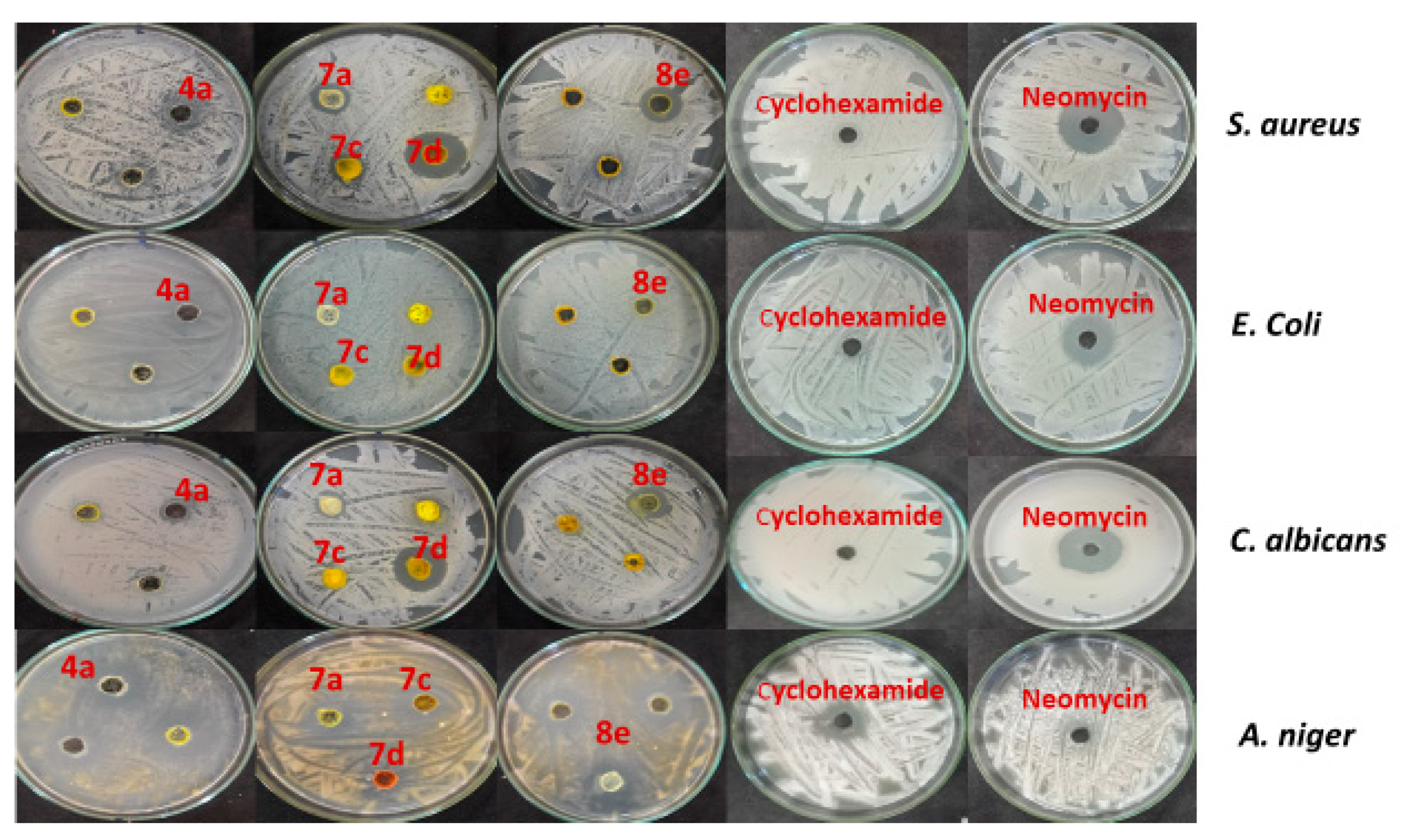
| Compd. No b | Inhibition Zone (ф mm) | |||
|---|---|---|---|---|
| S. aureus ATCC 6538 | E. coli ATCC 25922 | C. albicans ATCC 10231 | A. niger NRRL A-326 | |
| 4a | 0 | 0 | 13 | 29 |
| 4b | 20 | 15 | 16 | 26 |
| 4c | 15 | 13 | 13 | 20 |
| 4d | 16 | 0 | 15 | 18 |
| 4e | 14 | 16 | 14 | 16 |
| 4f | 14 | 0 | 12 | 19 |
| 4g | 15 | 0 | 12 | 18 |
| 4h | 16 | 14 | 0 | 16 |
| 4i | 14 | 0 | 12 | 17 |
| 4j | 0 | 0 | 0 | 0 |
| 7a | 18 | 0 | 17 | 13 |
| 7b | 0 | 12 | 13 | 14 |
| 7c | 17 | 13 | 12 | 0 |
| 7d | 36 | 12 | 27 | 14 |
| 7e | 12 | 14 | 0 | 13 |
| 8a | 12 | 13 | 14 | 12 |
| 8b | 0 | 0 | 14 | 0 |
| 8c | 0 | 0 | 0 | 0 |
| 8d | 0 | 0 | 13 | 14 |
| 8e | 21 | 14 | 22 | 14 |
| neomycin | 27 | 25 | 28 | 0 |
| cyclohexamide | 0 | 0 | 0 | 22 |
| Compounds | Pathogenic Microorganisms | |||||
|---|---|---|---|---|---|---|
| S. aureus ATCC 6538 | E. coli ATCC 25922 | C. albicans ATCC 10231 | ||||
| MIC (μg/mL) | MBC (μg/mL) | MIC (μg/mL) | MBC (μg/mL) | MIC (μg/mL) | MBC (μg/mL) | |
| 4b | 78.125 | 312.5 | 312.5 | 1250 | 78.13 | 312.5 |
| 7a | 39.063 | 625 | 312.5 | 625 | 39.06 | 156.25 |
| 7c | 312.5 | 625 | 625 | 1250 | 39.06 | 78.13 |
| 7d | 9.766 | 19.53 | 312.5 | 1250 | 19.53 | 19.53 |
| 8e | 19.53 | 19.53 | 312.5 | 625 | 19.53 | 39.06 |
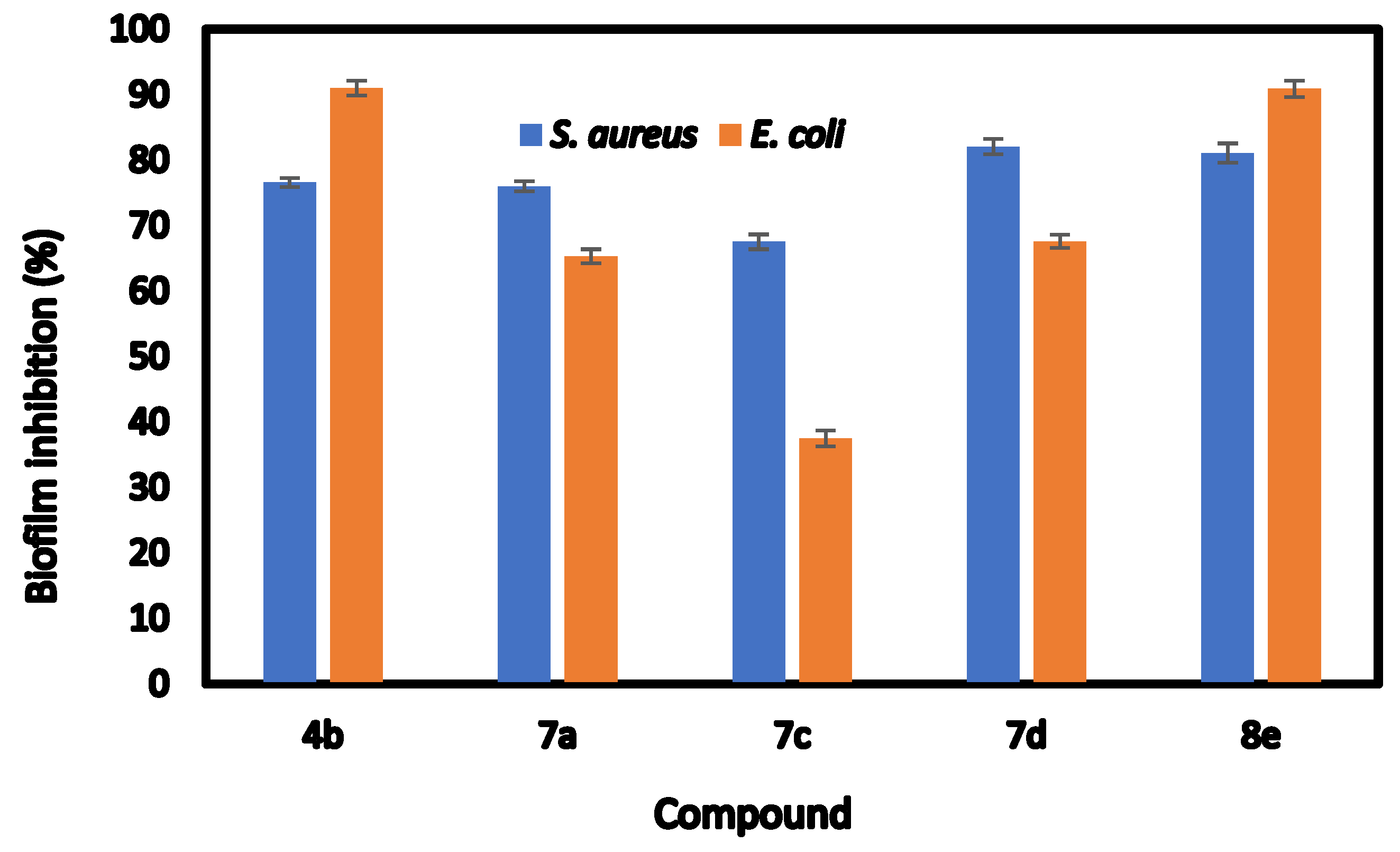
| Compds | Biofilm Inhibition (IC50) | Biofilm Inhibition (%) | ||
|---|---|---|---|---|
| Staphylococcus aureus | Escherichia coli | Staphylococcus aureus | Escherichia coli | |
| 4b | 304.42 ± 0.32 b | 56.52 ± 0.76 d | 76.59 ± 0.34 c | 91.02 ± 0.65 a |
| 7a | 103.52 ± 0.76 e | 431.21 ± 0.68 b | 76.21 ± 0.45 c | 65.33 ± 0.61 c |
| 7c | 449.81 ± 0.66 a | 729.32 ± 0.76 a | 67.52 ± 0.65 d | 37.51 ± 0.69 d |
| 7d | 199.32 ± 0.71 c | 266.91 ± 0.17 c | 82.06 ± 0.66 a | 67.61 ± 0.57 b |
| 8e | 169.63 ± 0.81 d | 54.65 ± 0.62 e | 81.08 ± 0.84 b | 90.88 ± 0.72 a |
| Compds | Reducing Sugars (μg/mL) | Proteins Release (μg/mL) | ||
|---|---|---|---|---|
| Staphylococcus aureus | Escherichia coli | Staphylococcus aureus | Escherichia coli | |
| Control | 172.365 ± 2.81 f | 116.198 ± 1.69 f | 158.9 ± 2.09 f | 194.5 ± 1.68 f |
| 4b | 249.533 ± 2.45 d | 208.533 ± 1.23 d | 389.95 ± 5.71 e | 405.9 ± 7.71 e |
| 7a | 327.2 ± 3.88 a | 227.318 ± 3.11 c | 480.8 ± 5.25 c | 562.9 ± 4.59 a |
| 7c | 209.818 ± 3.19 e | 234.368 ± 2.33 a | 607.4 ± 2.46 a | 437.8 ± 1.46 d |
| 7d | 306.203 ± 5.36 b | 190.368 ± 2.47 e | 462.5 ± 16.14 d | 453.4 ± 1.54 c |
| 8e | 254.533 ± 2.54 c | 230.7 ± 2.37 b | 540.9 ± 5.76 b | 523.8 ± 3.64 b |
| Comp. No. | Moieties from the Compound | Amino Acid Residues | Type of Interaction Distance | Score Kcal/mol |
|---|---|---|---|---|
| Thymidylate kinase (ID: 4QGG) | ||||
| Native ligand | NH | Gln101 | H-don (1.68, 2.85) | −24.34 RMSD: 1.42 |
| CO | Arg48, Arg70, Ser97 | H-acc (2.24, 2.79, 2.75) | ||
| 4b | Indole moiety | Lys15, Arg92 | π-Cation | −22.43 |
| Indole moiety and Pyridine ring | Phe66, Tyr100 | π-π | ||
| 7a | Indole moiety and benzofuran | Arg48 | π-Cation | −15.52 |
| Indole moiety | Arg36 | π-Cation | ||
| 7c | Indole moiety and benzofuran | Arg92 | π-Cation | −23.01 |
| Benzofuran | Lys15 | π-Cation | ||
| 7d | CN and CO | Arg92 | H-acc (2.88, 2.51) | −25.36 |
| Indole moiety | Arg92 | π-Cation | ||
| 8e | CO | Arg48 | H-acc (2.51) | −20.61 |
| benzofuran | Arg36 | π-Cation | ||
| BNA gyrase B (ID: 6F86) | ||||
| Native ligand | NH | Asp73 | H-don (1.85) | −22.84 RMSD: 1.65 |
| NH | Gly77 | H-don (2.10) | ||
| Pyridine ring | Arg76 | π-Cation | ||
| 4b | Pyridine ring | Arg76 | π-Cation | −14.64 |
| 7a | - | - | - | −19.35 |
| 7c | CN | Arg76 | H-acc (2.93) | −22.31 |
| 7d | Indole moiety | Arg76 | π-Cation | −16.90 |
| 8e | Indole moiety | Arg76 | π-Cation | −7.77 |
| DNA topoisomerase IV subunit B (ID: 4HZ5) | ||||
| Native ligand | Pyridine | Arg79 | π-Cation | −14.90 RMSD: 0.84 |
| 4b | Pyridine | Arg79 | π-Cation | −20.85 |
| Indole moiety | Arg130 | π-Cation | ||
| 7a | CN | Arg138 | H-acc (3.21) | −19.09 |
| Indole moiety | Arg79 | π-Cation | ||
| Benzofuran | His118 | π-Cation | ||
| 7c | Indole moiety | Arg79 | π-Cation | −24.90 |
| Benzofuran | His118 | π-Cation | ||
| NH | Ser56 | H-don (2.23) | ||
| 7d | NH | Ser56 | H-don (2.08) | −26.53 |
| Benzofuran | His118 | π-Cation | ||
| Indole moiety | Arg79 | π-Cation | ||
| 8e | CO | GLY 80 | H-acc (2.87) | −23.80 |
| SO2 | ARG 138 | H-acc (2.47, 2.90) | ||
| Indole moiety | Arg79 | π-Cation | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abo-Salem, H.M.; Abd El Salam, H.A.; Abdel-Aziem, A.; Abdel-Aziz, M.S.; El-Sawy, E.R. Synthesis, Molecular Docking, and Biofilm Formation Inhibitory Activity of Bis(Indolyl)Pyridines Analogues of the Marine Alkaloid Nortopsentin. Molecules 2021, 26, 4112. https://doi.org/10.3390/molecules26144112
Abo-Salem HM, Abd El Salam HA, Abdel-Aziem A, Abdel-Aziz MS, El-Sawy ER. Synthesis, Molecular Docking, and Biofilm Formation Inhibitory Activity of Bis(Indolyl)Pyridines Analogues of the Marine Alkaloid Nortopsentin. Molecules. 2021; 26(14):4112. https://doi.org/10.3390/molecules26144112
Chicago/Turabian StyleAbo-Salem, Heba M., Hayam A. Abd El Salam, Anhar Abdel-Aziem, Mohamed S. Abdel-Aziz, and Eslam Reda El-Sawy. 2021. "Synthesis, Molecular Docking, and Biofilm Formation Inhibitory Activity of Bis(Indolyl)Pyridines Analogues of the Marine Alkaloid Nortopsentin" Molecules 26, no. 14: 4112. https://doi.org/10.3390/molecules26144112
APA StyleAbo-Salem, H. M., Abd El Salam, H. A., Abdel-Aziem, A., Abdel-Aziz, M. S., & El-Sawy, E. R. (2021). Synthesis, Molecular Docking, and Biofilm Formation Inhibitory Activity of Bis(Indolyl)Pyridines Analogues of the Marine Alkaloid Nortopsentin. Molecules, 26(14), 4112. https://doi.org/10.3390/molecules26144112