
Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products



Abstract
1. Introduction
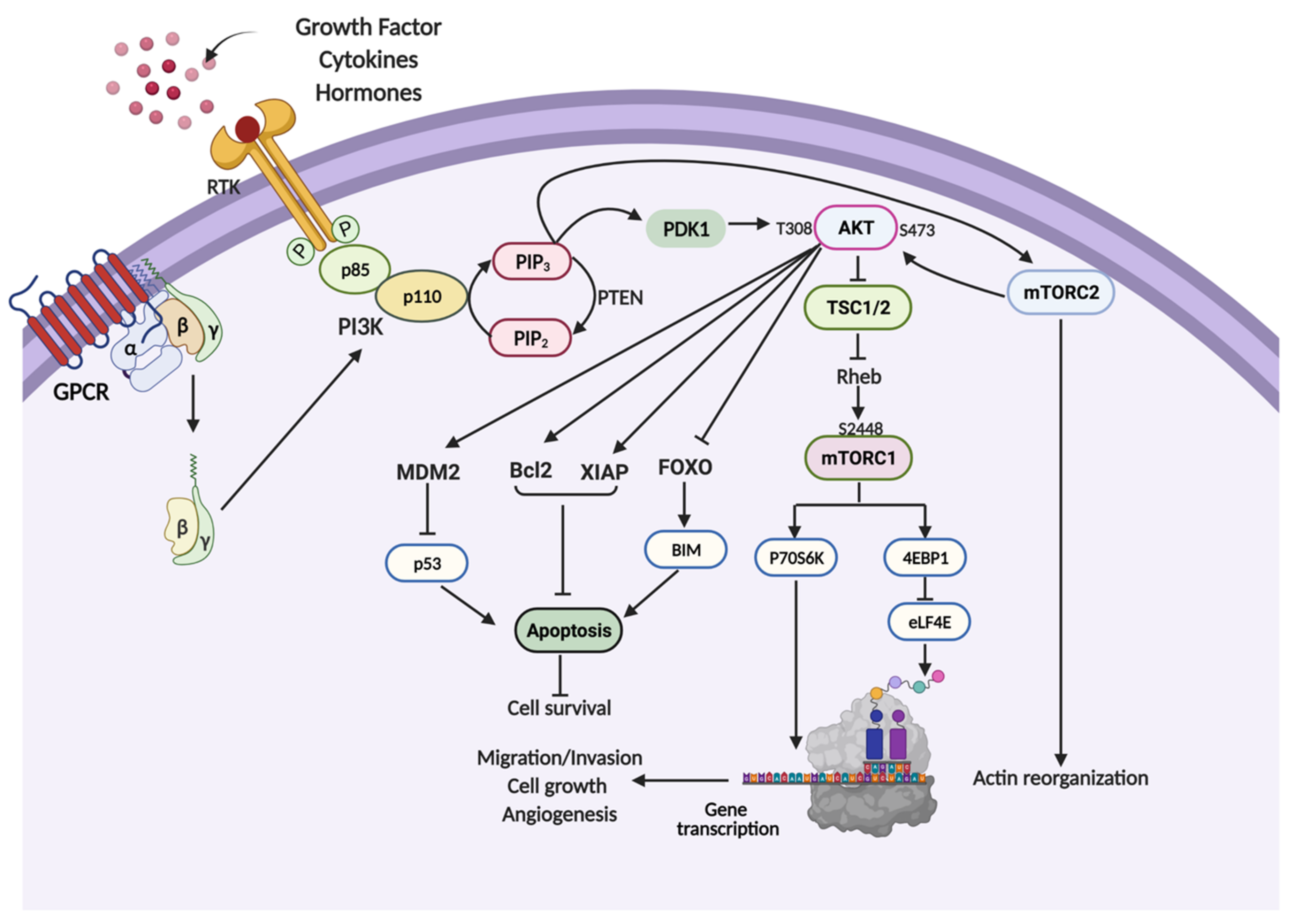
2. PI3K/AKT/mTOR Signaling Pathway
3. PI3K/AKT/mTOR Pathway in Cell Survival and Chemotherapeutic Resistance
4. PI3K/AKT/mTOR Pathway in Cell Proliferation
5. PI3K/AKT/mTOR Pathway in Cancer Cell Metastasis
6. PI3K/AKT/mTOR Pathway in Cancer Angiogenesis
7. Current Research on PI3K/AKT/mTOR Inhibitors in Lung Cancer
7.1. PI3K Inhibitors
7.2. AKT Inhibitors
7.3. mTOR Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Mechanism of Actions | Combination with | Phase | Refs. |
|---|---|---|---|---|
| Buparlisib (BKM120) | Class I Pan-PI3K inhibitor | Carboplatin and pemetrexed disodium | Phase I | [119] |
| Gefitinib | Phase I | [120] | ||
| Docetaxel | Phase I | [21] | ||
| Cisplatin and etoposide | Phase I | [121] | ||
| Pictilisib (GDC-0941) | PI3Kα/δ inhibitor | Paclitaxel Carboplatin (with or without bevacizumab) or pemetrexed, cisplatin, and bevacizumab | Phase I | [125] |
| Idelalisib | PI3Kδ inhibitor | Pembrolizumab | Phase IB/II | [129] |
| Alpelisib (BYL719) | PI3Kα inhibitor | MEK162 | Phase I | [134] |
| Serabelisib | PI3K inhibitor | Canagliflozin | Phase IB/II | [137] |
| Taselisib (GDC-0032) | PI3Kα, δ, and γ inhibitor | - | Phase I | [139] |
| - | Phase II | [140] | ||
| Gedatolisib (PF05212384) | Dual PI3K/mTOR inhibitor | Paclitaxel Carboplatin | Phase I/II | [143] |
| Palbociclib | Phase I | [144] | ||
| Voxtalisib (SAR245409/XL765) | Dual PI3K/mTOR inhibitor | MSC1936369B (Pimasertib) | Phase I | [147] |
| MK2206 | AKT inhibitor | Erlotinib | Phase II | [162] |
| Erlotinib | Phase II | [22,156] | ||
| Standard chemotherapy and erlotinib | Phase I | [163] | ||
| Gefitinib | Phase I | [161] | ||
| Capivarsetib (AZD5363) | AKT inhibitor | - | Phase I | [166] |
| Perifosine | Dual PI3K/AKT inhibitor | - | Phase I/II | [24] |
| Uprosertib (GSK-2141795) | AKT inhibitor | Trametinib dimethyl sulfoxide | Phase I/II | [169] |
| Aspirin | Decrease AKT phosphorylation | Osimertinib | Phase I | [175] |
| Rapamycin | mTORC1 inhibitor | Sunitinib | Phase I | [184] |
| Afatinib (BIBW2992) | Phase I | [23,185] | ||
| Temsirolimus | mTORC1 inhibitor | Neratinib | Phase II | [187] |
| - | Phase II | [188] | ||
| Pemetrexed | Phase I | [189] | ||
| Radiation | Phase I/II | [192] | ||
| Metformin | mTOR inhibitor | - | Phase II | [197] |
| Sintilimab | Phase II | [198] | ||
| Onatasertib (CC223) | Dual mTOR inhibitor | Erlotinib Azacytidine | Phase I | [200] |
| Sapanisertib | Dual mTOR inhibitor | - | Phase II | [201] |
| Vistusertib (AZD2014) | Dual mTOR inhibitor | - | Phase II | [202] |
8. Natural Compounds Targeting the PI3K/AKT/mTOR Pathway in Lung Cancer
8.1. Bibenzyl
8.2. Phenanthrene
8.3. Phenolic and Flavonoids
8.4. Quinoline
| Groups | Compound | Sources | Cell Lines | Mechanism of Actions | Refs. |
|---|---|---|---|---|---|
| Bibenzyl | 4,5,4′-trihydroxy-3,3′-dimethoxybibenzyl (TDB) | Dendrobium ellipsophyllum | H292, H460 and H23 | Induce apoptosis by downregulating AKT and upregulating p53 and proapoptotic proteins | [208] |
| H292 | Inhibit migration and invasion by downregulating AKT, FAK, CDC42 and integrins | [209] | |||
| H292 | Suppress metastasis by downregulating AKT and EMT signaling | [210] | |||
| Gigantol | Dendrobium draconis | H460 and H292 | Inhibit migration by downregulating AKT, CDC42 and Cav-1 | [212] | |
| H460 | Decrease the cancer stemness properties by inhibiting PI3K/AKT and JAK/STAT signaling | [213] | |||
| H460 | Downregulate active AKT, EMT markers and induce slug degradation | [214] | |||
| H460 | Sensitize cells to anoikis by suppressing the expression of AKT, ERK, Cav-1 and EMT markers | [215] | |||
| Phenanthrene | Ephemeranthol A | Dendrobium infundibulum | H460 | Downregulate AKT, FAK and EMT markers | [218] |
| Cypripedin | Dendrobium densiflorum | H460 and H23 | Downregulate AKT and EMT markers | [220] | |
| Erianthridin | Dendrobium formosum | A549 and H460 | Downregulate AKT/mTOR/p70S6K signaling | [96] | |
| Phenolic and Flavonoid Compounds | Phoyunnanin E | Dendrobium venustum | H460 | Inhibit migration by downregulating AKT and FAK signaling together with their downstream targets | [224] |
| Curcumin | Curcuma longa | A549 | Induce apoptosis and inhibit cell proliferation through the suppression of PI3K/AKT signaling and upregulation of miR-192-5p | [226] | |
| A549 | Inhibit cell migration and invasion by decreasing PI3K/AKT/mTOR signaling and increasing miR-206 | [227] | |||
| Sotetsuflavone | Cycas revolute | A549 | Induce autophagy by downregulating PI3K/AKT/mTOR signaling | [230] | |
| A549 | Suppress the expression of HIF1α and its downstream targets, such as VEGF and MMPs, by downregulating PI3K/AKT and TNF-α/NF-κB | [25] | |||
| Luteoloside | Chrysanthemum morifolium | A549 and H292 | Induce cell cycle arrest and autophagy by inhibiting PI3K/AKT/mTOR/p70S6K signaling | [231] | |
| Cardamonin | Boesenbergia rotunda | H460, H1975, A549, H292, H1299 and HCC827 | Inhibit proliferation and metastasis by downregulating the PI3K/Akt/mTOR pathway and its downstream targets | [233] | |
| Quinoline | Jorunnamycin A | Xestospongia sp. | H460 | Inhibition of AKT and EMT markers | [236] |
| Renieramycin M | Xestospongia sp. | H460 | Sensitize cells to anoikis by suppressing the expression of AKT and ERK, and downregulating Mcl-1 and Bcl-2 | [239] |
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Liu, Y.; Mao, C.; Wang, M.; Liu, N.; Ouyang, L.; Liu, S.; Tang, H.; Cao, Y.; Liu, S.; Wang, X.; et al. Cancer Progression Is Mediated by Proline Catabolism in Non-Small Cell Lung Cancer. Oncogene 2020, 38, 2358–2376. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Riely, G.J. Systemic Therapy for Locally Advanced and Metastatic Non-Small Cell Lung Cancer: A Review. Jama J. Am. Med. Assoc. 2019, 322, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hu, Q.; Pang, Z.; Xu, X. LncRNA MAGI2-AS3 Upregulates Cytokine Signaling 1 by Sponging MiR-155 in Non-Small Cell Lung Cancer. Cancer Biother. Radiopharm. 2020, 35, 72–76. [Google Scholar] [CrossRef]
- Cersosimo, R.J. Lung Cancer: A Review. Am. J. Health Pharm. 2002, 59, 611–642. [Google Scholar] [CrossRef]
- Liao, R.G.; Watanabe, H.; Meyerson, M.; Hammerman, P.S. Targeted Therapy for Squamous Cell Lung Cancer. Lung Cancer Manag. 2012, 1, 293–300. [Google Scholar] [CrossRef]
- Goldstraw, P.; Crowley, J.; Chansky, K.; Giroux, D.J.; Groome, P.A.; Rami-Porta, R.; Postmus, P.E.; Rusch, V.; Sobin, L. The IASLC Lung Cancer Staging Project: Proposals for the Revision of the TNM Stage Groupings in the Forthcoming (Seventh) Edition of the TNM Classification of Malignant Tumours. J. Thorac. Oncol. 2007, 2, 706–714. [Google Scholar] [CrossRef]
- Badawy, A.A.; Khedr, G.; Omar, A.; Bae, S.; Arafat, W.; Grant, S. Site of Metastases as Prognostic Factors in Unselected Population of Stage IV Non-Small Cell Lung Cancer. Asian Pac. J. Cancer Prev. 2018, 19, 1907–1910. [Google Scholar]
- Socinski, M.A. The Significant Impact of Chemotherapy in Lung Cancer. Clin. Adv. Hematol. Oncol. 2014, 12, 767–768. [Google Scholar]
- Yu, J.L.; Simmons, C.; Victor, J.C.; Han, D.; Hogeveen, S.; Leighl, N.; Verma, S. Impact of New Chemotherapeutic and Targeted Agents on Survival in Stage IV Non-Small Cell Lung Cancer. Oncologist 2011, 16, 1307–1315. [Google Scholar] [CrossRef][Green Version]
- Ke, B.; Wei, T.; Huang, Y.; Gong, Y.; Wu, G.; Liu, J.; Chen, X.; Shi, L. Interleukin-7 Resensitizes Non-Small-Cell Lung Cancer to Cisplatin via Inhibition of ABCG2. Mediat. Inflamm. 2019, 2019, 7241418. [Google Scholar] [CrossRef] [PubMed]
- Naylor, E.C.; Desani, J.K.; Chung, P.K. Targeted Therapy and Immunotherapy for Lung Cancer. Surg. Oncol. Clin. N. Am. 2016, 25, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Lemjabbar-Alaoui, H.; Hassan, O.U.I.; Yang, Y.W.; Buchanan, P. Lung Cancer: Biology and Treatment Options. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, V.S.; Giuranno, L.; Dubois, L.J.; Theys, J.; Vooijs, M. Drug Resistance in Non-Small Cell Lung Cancer: A Potential for NOTCH Targeting? Front. Oncol. 2018, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Chemotherapy for Lung Cancer in the Era of Personalized Medicine. Tuberc. Respir. Dis. 2019, 82, 179–189. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Chemotherapy Resistance in Lung Cancer. Adv. Exp. Med. Biol. 2016, 893, 189–209. [Google Scholar]
- Camidge, D.R. Targeted therapy vs chemotherapy: which has had more impact on survival in lung cancer? Does targeted therapy make patients live longer? Hard to prove, but impossible to ignore. Clin. Adv. Hematol. Oncol. H&Q 2014, 12, 763–766. [Google Scholar]
- Suda, K.; Mitsudomi, T. Successes and Limitations of Targeted Cancer Therapy in Lung Cancer. Prog. Tumor Res. 2014, 41, 62–77. [Google Scholar]
- Schettino, C.; Bareschino, M.; Sacco, P.; Maione, P.; Rossi, A.; Casaluce, F.; Sgambato, A.; Gridelli, C. New Molecular Targets in the Treatment of NSCLC. Curr. Pharm. Des. 2013, 19, 5333–5343. [Google Scholar] [CrossRef]
- Phase II Study of Buparlisib + Docetaxel in Advanced or Metastatic Squamous Non-Small Cell Lung Cancer (NSCLC) Patients—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01911325?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=5 (accessed on 18 February 2021).
- MK2206 and Erlotinib Hydrochloride in Treating Patients with Advanced Non-Small Cell Lung Cancer Who Have Progressed After Previous Response to Erlotinib Hydrochloride Therapy—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01294306?term=AKT+inhibitor&cond=Lung+Cancer&draw=2&rank=1 (accessed on 18 February 2021).
- Moran, T.; Palmero, R.; Provencio, M.; Insa, A.; Majem, M.; Reguart, N.; Bosch-Barrera, J.; Isla, D.; Costa, E.C.; Lee, C.; et al. A Phase Ib Trial of Continuous Once-Daily Oral Afatinib plus Sirolimus in Patients with Epidermal Growth Factor Receptor Mutation-Positive Non-Small Cell Lung Cancer and/or Disease Progression Following Prior Erlotinib or Gefitinib. Lung Cancer 2017, 108, 154–160. [Google Scholar] [CrossRef]
- A Phase 1/2 Trial of Perifosine in the Treatment of Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00399789?term=Perifosine&cond=Cancer&draw=2&rank=8 (accessed on 10 January 2021).
- Wang, S.; Yan, Y.; Cheng, Z.; Hu, Y.; Liu, T. Sotetsuflavone Suppresses Invasion and Metastasis in Non-Small-Cell Lung Cancer A549 Cells by Reversing EMT via the TNF-α/NF-ΚB and PI3K/AKT Signaling Pathway. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef]
- Chen, L.M.; Song, T.J.; Xiao, J.H.; Huang, Z.H.; Li, Y.; Lin, T.Y. Tripchlorolide Induces Autophagy in Lung Cancer Cells by Inhibiting the PI3K/AKT/MTOR Pathway and Improves Cisplatin Sensitivity in A549/DDP Cells. Oncotarget 2017, 8, 63911–63922. [Google Scholar] [CrossRef]
- Liu, F.; Gao, S.; Yang, Y.; Zhao, X.; Fan, Y.; Ma, W.; Yang, D.; Yang, A.; Yu, Y. Antitumor Activity of Curcumin by Modulation of Apoptosis and Autophagy in Human Lung Cancer A549 Cells through Inhibiting PI3K/Akt/MTOR Pathway. Oncol. Rep. 2018, 39, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P. The Diverse Mechanisms and Anticancer Potential of Naphthoquinones. Cancer Cell Int. 2019, 19, 1–20. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The Evolution of Phosphatidylinositol 3-Kinases as Regulators of Growth and Metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Arcaro, A.; Guerreiro, A. The Phosphoinositide 3-Kinase Pathway in Human Cancer: Genetic Alterations and Therapeutic Implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef]
- Maruyama, I. Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef]
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T. Regulation and Targets of Receptor Tyrosine Kinases. Eur. J. Cancer 2002, 38 (Suppl. S5), S3–S10. [Google Scholar] [CrossRef]
- Denley, A.; Gymnopoulos, M.; Kang, S.; Mitchell, C.; Vogt, P.K. Requirement of Phosphatidylinositol(3,4,5)Trisphosphate in Phosphatidylinositol 3-Kinase-Induced Oncogenic Transformation. Mol. Cancer Res. 2009, 7, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Kunter, I.; Erdal, E.; Nart, D.; Yilmaz, F.; Karademir, S.; Sagol, O.; Atabey, N. Active Form of AKT Controls Cell Proliferation and Response to Apoptosis in Hepatocellular Carcinoma. Oncol. Rep. 2014, 31, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Qiao, M.; Sheng, S.; Pardee, A.B. Metastasis and AKT Activation. Cell Cycle 2008, 7, 2991–2996. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/MTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 1–8. [Google Scholar] [CrossRef]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 Tumor Suppressor Negatively Regulates MTOR Signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Tee, A.R.; Anjum, R.; Blenis, J. Inactivation of the Tuberous Sclerosis Complex-1 and -2 Gene Products Occurs by Phosphoinositide 3-Kinase/Akt-Dependent and -Independent Phosphorylation of Tuberin. J. Biol. Chem. 2003, 278, 37288–37296. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. MTOR Signaling in Cancer and Mtor Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. MTOR Signaling Pathway and MTOR Inhibitors in Cancer: Progress and Challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Cook, R.S.; Chen, J. MTORC1 and MTORC2 in Cancer and the Tumor Microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Xie, Z.; Case, N.; Thompson, W.R.; Uzer, G.; Styner, M.; Rubin, J. MTORC2 Regulates Mechanically Induced Cytoskeletal Reorganization and Lineage Selection in Marrow-Derived Mesenchymal Stem Cells. J. Bone Miner. Res. 2014, 29, 78–89. [Google Scholar] [CrossRef]
- Yang, X.; Yang, C.; Farberman, A.; Rideout, T.C.; de Lange, C.F.; France, J.; Fan, M.Z. The Mammalian Target of Rapamycin-Signaling Pathway in Regulating Metabolism and Growth. J. Anim. Sci. 2008, 86, 36–50. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and Downstream of MTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Hanrahan, J.; Blenis, J. Rheb Activation of MTOR and S6K1 Signaling. Methods Enzymol. 2006, 407, 542–555. [Google Scholar] [PubMed]
- Yuan, T.L.; Cantley, L.C. PI3K Pathway Alterations in Cancer: Variations on a Theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef] [PubMed]
- Shimoi, T.; Hamada, A.; Yamagishi, M.; Hirai, M.; Yoshida, M.; Nishikawa, T.; Sudo, K.; Shimomura, A.; Noguchi, E.; Yunokawa, M.; et al. PIK3CA Mutation Profiling in Patients with Breast Cancer, Using a Highly Sensitive Detection System. Cancer Sci. 2018, 109, 2558–2566. [Google Scholar] [CrossRef]
- Gasparri, M.L.; Bardhi, E.; Ruscito, I.; Papadia, A.; Farooqi, A.A.; Marchetti, C.; Bogani, G.; Ceccacci, I.; Mueller, M.D.; Benedetti Panici, P. PI3K/AKT/MTOR Pathway in Ovarian Cancer Treatment: Are We on the Right Track? Geburtshilfe und Frauenheilkunde 2017, 77, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Li, J.; Li, J.; Che, G. Clinical Significance of PIK3CA Gene in Non-Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Biomed. Res. Int. 2020, 2020, 3608241. [Google Scholar] [CrossRef]
- Carson, J.D.; Van Aller, G.; Lehr, R.; Sinnamon, R.H.; Kirkpatrick, R.B.; Auger, K.R.; Dhanak, D.; Copeland, R.A.; Gontarek, R.R.; Tummino, P.J.; et al. Effects of Oncogenic P110α Subunit Mutations on the Lipid Kinase Activity of Phosphoinositide 3-Kinase. Biochem. J. 2007, 409, 519–524. [Google Scholar] [CrossRef]
- Miller, M.S.; Maheshwari, S.; McRobb, F.M.; Kinzler, K.W.; Amzel, L.M.; Vogelstein, B.; Gabelli, S.B. Identification of Allosteric Binding Sites for PI3Kα Oncogenic Mutant Specific Inhibitor Design. Bioorg. Med. Chem. 2017, 25, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Galdadas, I.; Athanasiou, C.; Cournia, Z. Insights into the Mechanism of the PIK3CA E545K Activating Mutation Using MD Simulations. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Guha, S.; Wang, H.; Fok, J.Y.; Koul, D.; Abbruzzese, J.; Mehta, K. Tissue Transglutaminase Regulates Focal Adhesion Kinase/AKT Activation by Modulating PTEN Expression in Pancreatic Cancer Cells. Clin. Cancer Res. 2008, 14, 1997–2005. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials 06 Biological Sciences 0601 Biochemistry and Cell Biology. Mol. Cancer 2019, 18, 1–28. [Google Scholar]
- Milella, M.; Falcone, I.; Conciatori, F.; Incani, U.C.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.A.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, L.; Dong, Y.; Tao, C.; Zhang, R.; Shao, H.; Shen, H. Effect of AKT1 (p. E17K) Hotspot Mutation on Malignant Tumorigenesis and Prognosis. Front. Cell Dev. Biol. 2020, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dodhia, S.; Su, G.H. Dysregulations in the PI3K Pathway and Targeted Therapies for Head and Neck Squamous Cell Carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef]
- Dan, H.C.; Sun, M.; Kaneko, S.; Feldman, R.I.; Nicosia, S.V.; Wang, H.G.; Tsang, B.K.; Cheng, J.Q. Akt Phosphorylation and Stabilization of X-Linked Inhibitor of Apoptosis Protein (XIAP). J. Biol. Chem. 2004, 279, 5405–5412. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A Phosphatidylinositol 3-Kinase/Akt Pathway Promotes Translocation of Mdm2 from the Cytoplasm to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, T.M.; Martinez Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-Talk between Akt, P53 and Mdm2: Possible Implications for the Regulation of Apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.M.; Bhattacharya, S.; Johnson, L.R. Mdm2 Inhibition Induces Apoptosis in P53 Deficient Human Colon Cancer Cells by Activating P73- and E2F1-Mediated Expression of PUMA and Siva-1. Apoptosis 2011, 16, 35–44. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1965–1970. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The BCL-2 Arbiters of Apoptosis and Their Growing Role as Cancer Targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein Kinase B Inhibits Cell Death by Preventing the Release of Cytochrome c from Mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Xu, T.; Masters, S.; Haian, F.; Gotoh, Y.; Greenberg, M.E. Akt Phosphorylation of BAD Couples Survival Signals to the Cell- Intrinsic Death Machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Prabhu, K.S.; Siveen, K.S.; Kuttikrishnan, S.; Iskandarani, A.; Tsakou, M.; Achkar, I.W.; Therachiyil, L.; Krishnankutty, R.; Parray, A.; Kulinski, M.; et al. Targeting of X-Linked Inhibitor of Apoptosis Protein and PI3-Kinase/AKT Signaling by Embelin Suppresses Growth of Leukemic Cells. PLoS ONE 2017, 12, e0180895. [Google Scholar] [CrossRef]
- Miyamoto, M.; Takano, M.; Iwaya, K.; Shinomiya, N.; Kato, M.; Aoyama, T.; Sasaki, N.; Goto, T.; Suzuki, A.; Hitrata, J.; et al. X-Chromosome-Linked Inhibitor of Apoptosis as a Key Factor for Chemoresistance in Clear Cell Carcinoma of the Ovary. Br. J. Cancer 2014, 110, 2881–2886. [Google Scholar] [CrossRef]
- Marine, J.C.; Lozano, G. Mdm2-Mediated Ubiquitylation: P53 and Beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef]
- Shinozaki, T.; Nota, A.; Taya, Y.; Okamoto, K. Functional Role of Mdm2 Phosphorylation by ATR in Attenuation of P53 Nuclear Export. Oncogene 2003, 22, 8870–8880. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Maurer, U.; Green, D.R.; Schuler, M. Pharmacologic Activation of P53 Elicits Bax-Dependent Apoptosis in the Absence of Transcription. Cancer Cell 2003, 4, 371–381. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Das, T.P.; Suman, S.; Alatassi, H.; Ankem, M.K.; Damodaran, C. Inhibition of AKT Promotes FOXO3a-Dependent Apoptosis in Prostate Cancer. Cell Death Dis. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO Transcription Factors; Regulation by AKT and 14-3-3 Proteins. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1938–1945. [Google Scholar] [CrossRef]
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-MTOR Singnaling Network in Cancer. Chin. J. Cancer 2013, 32, 253–265. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Abukhdeir, A.M.; Park, B.H. P21 and P27: Roles in Carcinogenesis and Drug Resistance. Expert Rev. Mol. Med. 2008, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K. MTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhou, H. Role of MTOR Signaling in Tumor Cell Motility, Invasion and Metastasis. Curr. Protein Pept. Sci. 2012, 12, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and MTOR Signaling Pathways in the Induction of Epithelial-Mesenchymal Transition (EMT) Process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Attar-Schneider, O.; Drucker, L.; Gottfried, M. Migration and Epithelial-to-Mesenchymal Transition of Lung Cancer Can Be Targeted via Translation Initiation Factors EIF4E and EIF4GI. Lab. Investig. 2016, 96, 1004–1015. [Google Scholar] [CrossRef][Green Version]
- Robichaud, N.; Del Rincon, S.V.; Huor, B.; Alain, T.; Petruccelli, L.A.; Hearnden, J.; Goncalves, C.; Grotegut, S.; Spruck, C.H.; Furic, L.; et al. Phosphorylation of EIF4E Promotes EMT and Metastasis via Translational Control of SNAIL and MMP-3. Oncogene 2014, 34, 2032–2042. [Google Scholar] [CrossRef]
- Qian, Y.; Corum, L.; Meng, Q.; Blenis, J.; Zheng, J.Z.; Shi, X.; Flynn, D.C.; Jiang, B.H. PI3K Induced Actin Filament Remodeling through Akt and P70S6K1: Implication of Essential Role in Cell Migration. Am. J. Physiol. Cell Physiol. 2004, 286, 153–163. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. The Actin-Bundling Protein Palladin Is an Akt1-Specific Substrate That Regulates Breast Cancer Cell Migration. Mol. Cell 2010, 38, 333–344. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Wong, A.S.T. P70 S6 Kinase and Actin Dynamics. Spermatogenesis 2012, 2, 44–52. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Cheung, A.N.Y.; Ngan, H.Y.S.; Wong, A.S.T. P70 S6 Kinase in the Control of Actin Cytoskeleton Dynamics and Directed Migration of Ovarian Cancer Cells. Oncogene 2011, 30, 2420–2432. [Google Scholar] [CrossRef]
- Jeong, Y.-J.; Hwang, S.-K.; Magae, J.; Chang, Y.-C. Ascofuranone Suppresses Invasion and F-Actin Cytoskeleton Organization in Cancer Cells by Inhibiting the MTOR Complex 1 Signaling Pathway. Cell. Oncol. 2020, 397, 473–805. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Wong, A.S.T. Activation of P70S6K Induces Expression of Matrix Metalloproteinase 9 Associated with Hepatocyte Growth Factor-Mediated Invasion in Human Ovarian Cancer Cells. Endocrinology 2006, 147, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Pothongsrisit, S.; Arunrungvichian, K.; Hayakawa, Y.; Sritularak, B.; Mangmool, S.; Pongrakhananon, V. Erianthridin Suppresses Non-Small-Cell Lung Cancer Cell Metastasis through Inhibition of Akt/MTOR/P70S6K Signaling Pathway. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Furic, L.; Rong, L.; Larsson, O.; Koumakpayi, I.H.; Yoshida, K.; Brueschke, A.; Petroulakis, E.; Robichaud, N.; Pollak, M.; Gabouryd, L.A.; et al. EIF4E Phosphorylation Promotes Tumorigenesis and Is Associated with Prostate Cancer Progression. Proc. Natl. Acad. Sci. USA 2010, 107, 14134–14139. [Google Scholar] [CrossRef]
- Adair, T.H.; Montani, J.-P. Overview of Angiogenesis. In Angiogenesis; Morgan & Claypool Life Sciences: Williston, VT, USA, 2010; ISBN 9781615043316. [Google Scholar]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.L.; Dufour, E.; Dassa, E.P.; Rustin, P.; Jacobs, H.T.; Taylor, C.T.; Hagen, T. Stabilization of Hypoxia-Inducible Factor-1α Protein in Hypoxia Occurs Independently of Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2010, 285, 31277–31284. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/Pseudohypoxia-Mediated Activation of Hypoxia-Inducible Factor-1α in Cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef]
- Wan, R.; Mo, Y.; Chien, S.; Li, Y.; Li, Y.; Tollerud, D.J.; Zhang, Q. The Role of Hypoxia Inducible Factor-1α in the Increased MMP-2 and MMP-9 Production by Human Monocytes Exposed to Nickel Nanoparticles. Nanotoxicology 2011, 5, 568–582. [Google Scholar] [CrossRef]
- Stetler-Stevenson, W.G. Matrix Metalloproteinases in Angiogenesis: A Moving Target for Therapeutic Intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Kalluri, R.; Cho, Y.J. The VEGF Pathway in Cancer and Disease: Responses, Resistance, and the Path Forward. Cold Spring Harb. Perspect. Med. 2012, 2, 1–17. [Google Scholar] [CrossRef]
- Saraswati, S.; Kumar, S.; Alhaider, A. α-Santalol Inhibits the Angiogenesis and Growth of Human Prostate Tumor Growth by Targeting Vascular Endothelial Growth Factor Receptor 2-Mediated AKT/mTOR/P70S6K Signaling Pathway. Mol. Cancer 2013, 12, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xiu, Z.; Zhou, Z.; Huang, B.; Liu, J.; Wu, X.; Li, S.; Tang, X. Cytochalasin H Inhibits Angiogenesis via the Suppression of HIF-1α Protein Accumulation and VEGF Expression through PI3K/AKt/P70S6K and ERK1/2 Signaling Pathways in Non-Small Cell Lung Cancer Cells. J. Cancer 2019, 10, 1997–2005. [Google Scholar] [CrossRef]
- Cui, W.; Cai, Y.; Zhou, X. Advances in Subunits of PI3K Class I in Cancer. Pathology 2014, 46, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Amzel, L.M.; Huang, C.H.; Mandelker, D.; Lengauer, C.; Gabelli, S.B.; Vogelstein, B. Structural Comparisons of Class I Phosphoinositide 3-Kinases. Nat. Rev. Cancer 2008, 8, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.S.; Thompson, P.E.; Gabelli, S.B. Structural Determinants of Isoform Selectivity in Pi3k Inhibitors. Biomolecules 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Knapp, S.; Ahmed, A.A. The Structural Basis of PI3K Cancer Mutations: From Mechanism to Therapy. Cancer Res. 2014, 74, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Vadas, O.; Dbouk, H.A.; Shymanets, A.; Perisic, O.; Burke, J.E.; Saab, W.F.A.; Khalil, B.D.; Harteneck, C.; Bresnick, A.R.; Nürnberg, B.; et al. Molecular Determinants of PI3Kγ-Mediated Activation Downstream of G-Protein-Coupled Receptors (GPCRs). Proc. Natl. Acad. Sci. USA 2013, 110, 18862–18867. [Google Scholar] [CrossRef]
- Staben, S.T. Isoform Selective PI3K Inhibitors for Treating Cancer. In Cancer II; Topics in Medicinal Chemistry, Waring, M.J., Eds.; Springer: Chem, Switzerland, 2018; Volume 28, ISBN 9783319759241. [Google Scholar]
- Zhang, M.; Jang, H.; Nussinov, R.; Nussinov, R. PI3K Inhibitors: Review and New Strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K Pathway in Cancer: Are We Making Headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class i PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Qiu, Y.; Kong, D. Class I Phosphatidylinositol 3-Kinase Inhibitors for Cancer Therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- PI3K Inhibitor BKM120, Carboplatin, and Pemetrexed Disodium in Treating Patients with Stage IV Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01723800?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=1 (accessed on 18 February 2021).
- A Trial of Gefitinib in Combination with BKM120 in Patients with Advanced Non-Small Cell Lung Cancer, with Enrichment for Patients Whose Tumours Harbour Molecular Alterations of PI3K Pathway and Known to Overexpress EGFR—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01570296?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=3 (accessed on 18 February 2021).
- Cisplatin, Etoposide and PI3K Inhibitor BKM120 in Treating Patients with Advanced Solid Tumors or Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02194049 (accessed on 22 February 2021).
- Rodon, J.; Braña, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I Dose-Escalation and -Expansion Study of Buparlisib (BKM120), an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. Invest. N. Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Folkes, A.J.; Ahmadi, K.; Alderton, W.K.; Alix, S.; Baker, S.J.; Box, G.; Chuckowree, I.S.; Clarke, P.A.; Depledge, P.; Eccles, S.A.; et al. The Identification of 2-(1H-Indazol-4-Yl)-6-(4-Methanesulfonyl-Piperazin-1- Ylmethyl)-4-Morpholin-4-Yl-Thieno[3,2-d]Pyrimidine (GDC-0941) as a Potent, Selective, Orally Bioavailable Inhibitor of Class I PI3 Kinase for the Treatment of Cancer. J. Med. Chem. 2008, 51, 5522–5532. [Google Scholar] [CrossRef]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan-Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef]
- A Study of the Safety and Pharmacology of PI3-Kinase Inhibitor GDC-0941 In Combination with Either Paclitaxel and Carboplatin (with or without Bevacizumab) or Pemetrexed, Cisplatin, And Bevacizumab in Patients with Advanced Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00974584?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=2 (accessed on 18 February 2021).
- Soria, J.C.; Adjei, A.A.; Bahleda, R.; Besse, B.; Ferte, C.; Planchard, D.; Zhou, J.; Ware, J.; Morrissey, K.; Shankar, G.; et al. A Phase IB Dose-Escalation Study of the Safety and Pharmacokinetics of Pictilisib in Combination with Either Paclitaxel and Carboplatin (with or without Bevacizumab) or Pemetrexed and Cisplatin (with or without Bevacizumab) in Patients with Advanced Non–Small Cell Lung Cancer. Eur. J. Cancer 2017, 86, 186–196. [Google Scholar]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.M.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a P110δ Selective Phosphatidylinositol-3-Kinase Inhibitor for the Treatment of B-Cell Malignancies, Inhibits PI3K Signaling and Cellular Viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef]
- Zirlik, K.; Veelken, H. Idelalisib. In Recent Results in Cancer Research; Springer: New York, NY, USA, 2018; Volume 212, pp. 243–264. [Google Scholar]
- Pembrolizumab + Idelalisib for Lung Cancer Study—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03257722 (accessed on 18 February 2021).
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a Potent and Selective Phosphatidylinositol-3 Kinase Alpha Inhibitor Selected for Clinical Evaluation. Bioorganic Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the Novel and Specific PI3Ka Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef]
- Study of Alpelisib (BYL719) in Combination with Trastuzumab and Pertuzumab as Maintenance Therapy in Patients with HER2-Positive Advanced Breast Cancer with a PIK3CA Mutation—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04208178 (accessed on 23 February 2021).
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus Fulvestrant for PIK3CA-Mutated, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor-2–Negative Advanced Breast Cancer: Final Overall Survival Results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef]
- A Phase Ib Study of MEK162 Plus BYL719 in Adult Patients with Selected Advanced Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01449058 (accessed on 18 February 2021).
- Juric, D.; Soria, J.-C.; Sharma, S.; Banerji, U.; Azaro, A.; Desai, J.; Ringeisen, F.P.; Kaag, A.; Radhakrishnan, R.; Hourcade-Potelleret, F.; et al. A Phase 1b Dose-Escalation Study of BYL719 plus Binimetinib (MEK162) in Patients with Selected Advanced Solid Tumors. J. Clin. Oncol. 2014, 32, 9051. [Google Scholar] [CrossRef]
- Juric, D.; De Bono, J.S.; LoRusso, P.M.; Nemunaitis, J.; Heath, E.I.; Kwak, E.L.; Mercade, T.M.; Geuna, E.; De Miguel-Luken, M.J.; Patel, C.; et al. A First-in-Human, Phase I, Dose-Escalation Study of TAK-117, A Selective PI3Ka Isoform Inhibitor, in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 5015–5023. [Google Scholar] [CrossRef] [PubMed]
- A Phase 1b/2 Study of Serabelisib in Combination with Canagliflozin in Patients with Advanced Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04073680?term=PI3K+inhibitor&cond=Lung+Cancer&draw=2&rank=15 (accessed on 18 February 2021).
- Ndubaku, C.O.; Heffron, T.P.; Staben, S.T.; Baumgardner, M.; Blaquiere, N.; Bradley, E.; Bull, R.; Do, S.; Dotson, J.; Dudley, D.; et al. Discovery of 2-{3-[2-(1-Isopropyl-3-Methyl-1H-1,2-4-Triazol-5-Yl)-5,6- Dihydrobenzo[f]Imidazo[1,2- d ][1,4]Oxazepin-9-Yl]-1H-Pyrazol-1-Yl}-2- Methylpropanamide (GDC-0032): A β-Sparing Phosphoinositide 3-Kinase Inhibitor with High Unbound Exposure and Robust in Vivo Antitumor Activity. J. Med. Chem. 2013, 56, 4597–4610. [Google Scholar] [PubMed]
- Langer, C.J.; Redman, M.W.; Wade, J.L.; Aggarwal, C.; Bradley, J.D.; Crawford, J.; Stella, P.J.; Knapp, M.H.; Miao, J.; Minichiello, K.; et al. SWOG S1400B (NCT02785913), a Phase II Study of GDC-0032 (Taselisib) for Previously Treated PI3K-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Sub-Study). J. Thorac. Oncol. 2019, 14, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Lung-MAP: Taselisib as Therapy in Treating Patients with Stage IV Squamous Cell Lung Cancer and Positive Biomarker Matches—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02785913?term=taselisib&cond=lung+cancer&draw=2&rank=1 (accessed on 24 February 2021).
- Venkatesan, A.M.; Dehnhardt, C.M.; Delos Santos, E.D.; Chen, Z.; Dos Santos, O.D.; Ayral-Kaloustian, S.; Khafizova, G.; Brooijmans, N.; Mallon, R.; Hollander, I.; et al. Bis(Morpholino-l,3,5-Triazine) Derivatives: Potent Adenosine 5′-Triphosphate Competitive Phosphatidylinositol-3-Kinase/Mammalian Target of Rapamycin Inhibitors: Discovery of Compound 26 (PKI-587), a Highly Efficacious Dual Inhibitor. J. Med. Chem. 2010, 53, 2636–2645. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.M.; Chen, Z.; Dos Santos, O.; Dehnhardt, C.; Santos, E.D.; Ayral-Kaloustian, S.; Mallon, R.; Hollander, I.; Feldberg, L.; Lucas, J.; et al. PKI-179: An Orally Efficacious Dual Phosphatidylinositol-3-Kinase (PI3K)/Mammalian Target of Rapamycin (MTOR) Inhibitor. Bioorganic Med. Chem. Lett. 2010, 20, 5869–5873. [Google Scholar] [CrossRef]
- Study of Paclitaxel, Carboplatin, and PF-05212384 in Advanced or Metastatic NSCLC (UF-STO-LUNG-002)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02920450?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=6 (accessed on 18 February 2021).
- Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination with the PI3K/MTOR Inhibitor Gedatolisib (PF-05212384) for Patients with Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03065062?term=mTOR+inhibitor&cond=Lung+Cancer&draw=3&rank=11 (accessed on 18 February 2021).
- Garcia-Echeverria, C.; Sellers, W.R. Drug Discovery Approaches Targeting the PI3K/Akt Pathway in Cancer. Oncogene 2008, 27, 5511–5526. [Google Scholar] [CrossRef]
- Inaba, K.; Oda, K.; Aoki, K.; Sone, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Fukuda, T.; Makii, C.; Arimoto, T.; et al. Synergistic Antitumor Effects of Combination PI3K/MTOR and MEK Inhibition (SAR245409 and Pimasertib) in Mucinous Ovarian Carcinoma Cells by Fluorescence Resonance Energy Transfer Imaging. Oncotarget 2016, 7, 29577–29591. [Google Scholar] [CrossRef]
- Trial of MEK Inhibitor and PI3K/MTOR Inhibitor in Subjects with Locally Advanced or Metastatic Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01390818?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=8 (accessed on 18 February 2021).
- Kumar, C.C.; Madison, V. AKT Crystal Structure and AKT-Specific Inhibitors. Oncogene 2005, 24, 7493–7501. [Google Scholar] [CrossRef]
- Yudushkin, I. Control of Akt Activity and Substrate Phosphorylation in Cells. IUBMB Life 2020, 72, 1115–1125. [Google Scholar] [CrossRef]
- Chu, N.; Viennet, T.; Bae, H.; Salguero, A.; Boeszoermenyi, A.; Arthanari, H.; Cole, P.A. The Structural Determinants of Ph Domain-Mediated Regulation of Akt Revealed by Segmental Labeling. eLife 2020, 9, 1–23. [Google Scholar] [CrossRef]
- Lučic, I.; Rathinaswamy, M.K.; Truebestein, L.; Hamelin, D.J.; Burke, J.E.; Leonard, T.A. Conformational Sampling of Membranes by Akt Controls Its Activation and Inactivation. Proc. Natl. Acad. Sci. USA 2018, 115, E3940–E3949. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.O.; Pascal, J.M.; Armen, R.S.; Rodeck, U. Autoregulation of Kinase Dephosphorylation by ATP Binding to AGC Protein Kinases. Cell Cycle 2012, 11, 475–478. [Google Scholar] [CrossRef][Green Version]
- Lazaro, G.; Kostaras, E.; Vivanco, I. Inhibitors in AKTion: ATP-Competitive vs Allosteric. Biochem. Soc. Trans. 2020, 48, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lin, J.; Wu, W.I.; Ballard, J.; Lee, B.B.; Gloor, S.L.; Vigers, G.P.A.; Morales, T.H.; Friedman, L.S.; Skelton, N.; et al. An ATP-Site on-off Switch That Restricts Phosphatase Accessibility of Akt. Sci. Signal. 2012, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brown, T. Harnessing Allostery: A Novel Approach to Drug Discovery. Harv. Bus. Rev. 2008, 86, 84–92. [Google Scholar]
- Lara, P.N.; Longmate, J.; Mack, P.C.; Kelly, K.; Socinski, M.A.; Salgia, R.; Gitlitz, B.; Li, T.; Koczywas, M.; Reckamp, K.L.; et al. Phase II Study of the AKT Inhibitor MK-2206 plus Erlotinib in Patients with Advanced Non-Small Cell Lung Cancer Who Previously Progressed on Erlotinib. Clin. Cancer Res. 2015, 21, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Yan, L. Abstract #DDT01-1: MK-2206: A Potent Oral Allosteric AKT Inhibitor. Cancer Res. 2009, 69, DDT01-1. Available online: https://cancerres.aacrjournals.org/content/69/9_Supplement/DDT01-1 (accessed on 24 February 2021).
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an Allosteric Akt Inhibitor, Enhances Antitumor Efficacy by Standard Chemotherapeutic Agents or Molecular Targeted Drugs in Vitro and in Vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-Man Clinical Trial of the Oral Pan-AKT Inhibitor MK-206 in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Yan, L.; Vitfell-Rasmussen, J.; Zernhelt, A.M.; Sullivan, D.M.; Cassier, P.A.; Chen, E.; Biondo, A.; Tetteh, E.; Siu, L.L.; et al. Phase 1 Trial of the Oral AKT Inhibitor MK-2206 plus Carboplatin/ Paclitaxel, Docetaxel, or Erlotinib in Patients with Advanced Solid Tumors. J. Hematol. Oncol. 2014, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Study of MK-2206 in Combination with Standard Chemotherapy in Participants with Locally Advanced or Metastatic Solid Tumors (MK-2206-003)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00848718 (accessed on 25 February 2021).
- Wu, Y.L.; Zhou, C.; Cheng, Y.; Lu, S.; Chen, G.Y.; Huang, C.; Huang, Y.S.; Yan, H.H.; Ren, S.; Liu, Y.; et al. Erlotinib as Second-Line Treatment in Patients with Advanced Non-Small-Cell Lung Cancer and Asymptomatic Brain Metastases: A Phase II Study (CTONG-0803). Ann. Oncol. 2013, 24, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Dose Defining Study For MK-2206 Combined with Gefitinib in Non-Small Cell Lung Cancer (NSCLC)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01147211?term=AKT+inhibitor&cond=Lung+Cancer&draw=2&rank=2 (accessed on 18 February 2021).
- Addie, M.; Ballard, P.; Buttar, D.; Crafter, C.; Currie, G.; Davies, B.R.; Debreczeni, J.; Dry, H.; Dudley, P.; Greenwood, R.; et al. Discovery of 4-Amino-N-[(1S)-1-(4-Chlorophenyl)-3-Hydroxypropyl]-1-(7H- Pyrrolo[2,3-d]Pyrimidin-4-Yl)Piperidine-4-Carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. J. Med. Chem. 2013, 56, 2059–2073. [Google Scholar] [CrossRef]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A Phase I Dose-Escalation Study of Enzalutamide in Combination with the AKT Inhibitor AZD5363 (Capivasertib) in Patients with Metastatic Castration-Resistant Prostate Cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [PubMed]
- National Lung Matrix Trial: Multi-Drug Phase II Trial in Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02664935?term=AKT+inhibitor&cond=Lung+Cancer&draw=2&rank=3 (accessed on 22 December 2020).
- Cheraghchi-Bashi, A.; Parker, C.A.; Curry, E.; Salazar, J.F.; Gungor, H.; Saleem, A.; Cunnea, P.; Rama, N.; Salinas, C.; Mills, G.B.; et al. A Putative Biomarker Signature for Clinically Effective AKT Inhibition: Correlation of in Vitro, in Vivo and Clinical Data Identifies the Importance of Modulation of the MTORC1 Pathway. Oncotarget 2015, 6, 41736–41749. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Kurzrock, R.; Valero, V.; Gonzalez, R.; Heist, R.S.; Tan, A.R.; Means-Powell, J.; Werner, T.L.; Becerra, C.; Wang, C.; et al. Phase I Dose-Escalation Trial of the Oral AKT Inhibitor Uprosertib in Combination with the Oral MEK1/MEK2 Inhibitor Trametinib in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2020, 85, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Uprosertib, Dabrafenib, and Trametinib in Treating Patients with Stage IIIC-IV Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01902173 (accessed on 18 February 2021).
- Patel, V.; Lahusen, T.; Sy, T.; Sausville, E.A.; Gutkind, J.S.; Senderowicz, A.M. Perifosine, a Novel Alkylphospholipid, Induces P21WAF1 Expression in Squamous Carcinoma Cells through a P53-Independent Pathway, Leading to Loss in Cyclin-Dependent Kinase Activity and Cell Cycle Arrest. Cancer Res. 2002, 62, 1401–1409. [Google Scholar]
- Bendell, J.C.; Ervin, T.J.; Senzer, N.N.; Richards, D.A.; Firdaus, I.; Lockhart, A.C.; Cohn, A.L.; Saleh, M.N.; Gardner, L.R.; Sportelli, P.; et al. Results of the X-PECT Study: A Phase III Randomized Double-Blind, Placebo-Controlled Study of Perifosine plus Capecitabine (P-CAP) versus Placebo plus Capecitabine (CAP) in Patients (Pts) with Refractory Metastatic Colorectal Cancer (MCRC). J. Clin. Oncol. 2012, 30, LBA3501. [Google Scholar]
- Han, R.; Hao, S.; Lu, C.; Zhang, C.; Lin, C.; Li, L.; Wang, Y.; Hu, C.; He, Y. Aspirin Sensitizes Osimertinib-Resistant NSCLC Cells in Vitro and in Vivo via Bim-Dependent Apoptosis Induction. Mol. Oncol. 2020, 14, 1152–1169. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, C.; Dong, H.; Wang, X.; Gao, F.; Zhang, S.; Zhang, X. Aspirin Has a Better Effect on PIK3CA Mutant Colorectal Cancer Cells by PI3K/Akt/Raptor Pathway. Mol. Med. 2020, 26, 1–13. [Google Scholar] [CrossRef]
- Din, F.V.N.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin Inhibits MTOR Signaling, Activates AMP-Activated Protein Kinase, and Induces Autophagy in Colorectal Cancer Cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar] [CrossRef]
- Combination of Osimertinib and Aspirin to Treat Osimertinib Resistance Non-Small Cell Lung Cancer ( NSCLC)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03532698?term=PI3K+inhibitor&cond=Non+Small+Cell+Lung+Cancer&draw=2&rank=7 (accessed on 18 February 2021).
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting MTOR for Cancer Therapy. J. Hematol. Oncol. 2019, 12, 71–89. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Liu, M.; Chen, X.; Huang, M.; Tan, D.; Dong, M.Q.; Wong, C.C.L.; Wang, J.; Xu, Y.; et al. 4.4 Å Resolution Cryo-EM Structure of Human MTOR Complex 1. Protein Cell 2016, 7, 878–887. [Google Scholar] [CrossRef]
- Melick, C.H.; Jewell, J.L. Regulation of Mtorc1 by Upstream Stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef]
- Yuan, H.X.; Guan, K.L. Structural Insights of MTOR Complex 1. Cell Res. 2016, 26, 267–268. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Arriola Apelo, S.I.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A Novel Rapamycin Analog Is Highly Selective for MTORC1 in Vivo. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-Competitive Inhibitors of MTOR: An Update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Xie, G.; Zhou, G.; Cheng, Y.; Zhang, G.; Yao, G.; Chen, Y.; Li, Y.; Zhao, G. NVP-BEZ235, a Novel Dual PI3K-MTOR Inhibitor Displays Anti-Glioma Activity and Reduces Chemoresistance to Temozolomide in Human Glioma Cells. Cancer Lett. 2015, 367, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Lin, X.; Zhang, C.; Liu, Z.; Chen, Z.; Li, Z.; Wang, J.; Li, B.; Hu, Y.; Dong, B.; et al. Dual PI3K/MTOR Inhibitor BEZ235 as a Promising Therapeutic Strategy against Paclitaxel-Resistant Gastric Cancer via Targeting PI3K/Akt/MTOR Pathway Article. Cell Death Dis. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Study of SUNITINIB and Rapamycin in Advanced Non-Small Cell Lung Cancer (NSCLC)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00555256?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=1 (accessed on 18 February 2021).
- Trial of Continuous Once Daily Oral Treatment Using BIBW 2992 (Afatinib) Plus Sirolimus (Rapamune®) in Patients with Non-Small Cell Lung Cancer Harbouring an EGFR Mutation and/or Disease Progression Following Prior Erlotinib or Gefitinib—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00993499?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=10 (accessed on 18 February 2021).
- Ohara, T.; Takaoka, M.; Toyooka, S.; Tomono, Y.; Nishikawa, T.; Shirakawa, Y.; Yamatsuji, T.; Tanaka, N.; Fujiwara, T.; Naomoto, Y. Inhibition of MTOR by Temsirolimus Contributes to Prolonged Survival of Mice with Pleural Dissemination of Non-Small-Cell Lung Cancer Cells. Cancer Sci. 2011, 102, 1344–1349. [Google Scholar] [CrossRef]
- Neratinib with and without Temsirolimus for Patients with HER2 Activating Mutations in Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01827267?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=2 (accessed on 18 February 2021).
- CCI-779 in Treating Patients with Stage IIIB (with Pleural Effusion) or Stage IV Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00079235?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=3 (accessed on 18 February 2021).
- CCI-779 in Treating Patients with Extensive-Stage Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00028028?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=4 (accessed on 18 February 2021).
- Temsirolimus and Pemetrexed for Recurrent or Refractory Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00921310?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=6 (accessed on 18 February 2021).
- Waqar, S.N.; Baggstrom, M.Q.; Morgensztern, D.; Williams, K.; Rigden, C.; Govindan, R. A Phase i Trial of Temsirolimus and Pemetrexed in Patients with Advanced Non-Small Cell Lung Cancer. Chemotherapy 2016, 61, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Temsirolimus and Radiation for Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00796796?term=mTOR+inhibitor&cond=Lung+Cancer&draw=2&rank=5 (accessed on 18 February 2021).
- Waqar, S.N.; Robinson, C.; Bradley, J.; Goodgame, B.; Rooney, M.; Williams, K.; Gao, F.; Govindan, R. A Phase i Study of Temsirolimus and Thoracic Radiation in Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2014, 15, 119–123. [Google Scholar] [CrossRef]
- Amin, S.; Lux, A.; O’Callaghan, F. The Journey of Metformin from Glycaemic Control to MTOR Inhibition and the Suppression of Tumour Growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46. [Google Scholar] [CrossRef]
- Lin, J.; Gill, A.; Zahm, S.H.; Carter, C.A.; Shriver, C.D.; Nations, J.A.; Anderson, W.F.; McGlynn, K.A.; Zhu, K. Metformin Use and Survival after Non-Small Cell Lung Cancer: A Cohort Study in the US Military Health System. Int. J. Cancer 2017, 141, 254–263. [Google Scholar] [CrossRef]
- Chuang, M.C.; Yang, Y.H.; Tsai, Y.H.; Hsieh, M.J.; Lin, Y.C.; Lin, C.K.; Chen, P.C.; Yang, T.M. Survival Benefit Associated with Metformin Use in Inoperable Non-Small Cell Lung Cancer Patients with Diabetes: A Population-Based Retrospective Cohort Study. PLoS ONE 2018, 13, 1–11. [Google Scholar]
- Phase II Lung Metcore—Preoperative Metformin for Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03086733?term=metformin&cond=Lung+Cancer&draw=2&rank=4 (accessed on 26 February 2021).
- Sintilimab Combined with Metformin in First-Line Chemotherapy Refractory Advanced NSCLC Patients—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03874000 (accessed on 3 June 2021).
- Mortensen, D.S.; Fultz, K.E.; Xu, S.; Xu, W.; Packard, G.; Khambatta, G.; Gamez, J.C.; Leisten, J.; Zhao, J.; Apuy, J.; et al. CC-223, a Potent and Selective Inhibitor of MTOR Kinase: In Vitro and in Vivo Characterization. Mol. Cancer Ther. 2015, 14, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Study Assessing Safety, Pharmacokinetics and Efficacy of CC-223 with Either Erlotinib or Oral Azacitidine in Advanced Non-Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT01545947 (accessed on 18 February 2021).
- Sapanisertib in Treating Patients with Stage IV or Recurrent Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02417701 (accessed on 18 February 2021).
- Vistusertib (AZD2014) Monotherapy in Relapsed Small Cell Lung Cancer Patients Harboring RICTOR Amplification—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03106155 (accessed on 18 February 2021).
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Mushtaq, S.; Abbasi, B.H.; Uzair, B.; Abbasi, R. Natural Products as Reservoirs of Novel Therapeutic Agents. EXCLI J. 2018, 17, 420–451. [Google Scholar] [PubMed]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Sritularak, B.; Duangrak, N.; Likhitwitayawuid, K. A New Bibenzyl from Dendrobium Secundum. Zeitschrift für Naturforschung C 2011, 66, 205–208. [Google Scholar] [CrossRef]
- He, L.; Su, Q.; Bai, L.; Li, M.; Liu, J.; Liu, X.; Zhang, C.; Jiang, Z.; He, J.; Shi, J.; et al. Recent Research Progress on Natural Small Molecule Bibenzyls and Its Derivatives in Dendrobium Species. Eur. J. Med. Chem. 2020, 204, 1–17. [Google Scholar] [CrossRef]
- Hlosrichok, A.; Sumkhemthong, S.; Sritularak, B.; Chanvorachote, P.; Chaotham, C. A Bibenzyl from Dendrobium Ellipsophyllum Induces Apoptosis in Human Lung Cancer Cells. J. Nat. Med. 2018, 72, 615–625. [Google Scholar] [CrossRef]
- Chaotham, C.; Chanvorachote, P. A Bibenzyl from Dendrobium Ellipsophyllum Inhibits Migration in Lung Cancer Cells. J. Nat. Med. 2015, 69, 565–574. [Google Scholar] [CrossRef]
- Chaotham, C.; Pongrakhananon, V.; Sritularak, B.; Chanvorachote, P. A Bibenzyl from Dendrobium Ellipsophyllum Inhibits Epithelial-to-Mesenchymal Transition and Sensitizes Lung Cancer Cells to Anoikis. Anticancer Res. 2014, 34, 1931–1938. [Google Scholar]
- Tanagornmeatar, K.; Chaotham, C.; Sritularak, B.; Likhitwitayawuid, K.; Chanvorachote, P. Cytotoxic and Anti-Metastatic Activities of Phenolic Compounds from Dendrobium Ellipsophyllum. Anticancer Res. 2014, 34, 6573–6579. [Google Scholar] [PubMed]
- Charoenrungruang, S.; Chanvorachote, P.; Sritularak, B.; Pongrakhananon, V. Gigantol, a Bibenzyl from Dendrobium Draconis, Inhibits the Migratory Behavior of Non-Small Cell Lung Cancer Cells. J. Nat. Prod. 2014, 77, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Losuwannarak, N.; Maiuthed, A.; Kitkumthorn, N.; Leelahavanichkul, A.; Roytrakul, S.; Chanvorachote, P. Gigantol Targets Cancer Stem Cells and Destabilizes Tumors via the Suppression of the PI3K/AKT and JAK/STAT Pathways in Ectopic Lung Cancer Xenografts. Cancers 2019, 11, 2032. [Google Scholar] [CrossRef] [PubMed]
- Unahabhokha, T.; Chanvorachote, P.; Sritularak, B.; Kitsongsermthon, J.; Pongrakhananon, V. Gigantol Inhibits Epithelial to Mesenchymal Process in Human Lung Cancer Cells. Evid. Based Complement. Altern. Med. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Unahabhokha, T.; Chanvorachote, P.; Pongrakhananon, V. The Attenuation of Epithelial to Mesenchymal Transition and Induction of Anoikis by Gigantol in Human Lung Cancer H460 Cells. Tumor Biol. 2016, 37, 8633–8641. [Google Scholar] [CrossRef] [PubMed]
- Kovács, A.; Vasas, A.; Hohmann, J. Natural Phenanthrenes and Their Biological Activity. Phytochemistry 2008, 69, 1084–1110. [Google Scholar] [CrossRef] [PubMed]
- Tóth, B.; Hohmann, J.; Vasas, A. Phenanthrenes: A Promising Group of Plant Secondary Metabolites. J. Nat. Prod. 2018, 81, 661–678. [Google Scholar] [CrossRef] [PubMed]
- Nonpanya, N.; Prakhongcheep, O.; Petsri, K.; Jitjaicham, C.; Tungsukruthai, S.; Sritularak, B.; Chanvorachote, P. Ephemeranthol a Suppresses Epithelial to Mesenchymal Transition and FAK-Akt Signaling in Lung Cancer Cells. Anticancer Res. 2020, 40, 4989–4999. [Google Scholar] [CrossRef] [PubMed]
- Wattanathamsan, O.; Treesuwan, S.; Sritularak, B.; Pongrakhananon, V. Cypripedin, a Phenanthrenequinone from Dendrobium Densiflorum, Sensitizes Non-Small Cell Lung Cancer H460 Cells to Cisplatin-Mediated Apoptosis. J. Nat. Med. 2018, 72, 503–513. [Google Scholar] [CrossRef]
- Treesuwan, S.; Sritularak, B.; Chanvorachote, P.; Pongrakhananon, V. Cypripedin Diminishes an Epithelial-to-Mesenchymal Transition in Non-Small Cell Lung Cancer Cells through Suppression of Akt/GSK-3β Signalling. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Boonjing, S.; Pothongsrisit, S.; Wattanathamsan, O.; Sritularak, B.; Pongrakhananon, V. Erianthridin Induces Non-Small Cell Lung Cancer Cell Apoptosis through the Suppression of Extracellular Signal-Regulated Kinase Activity. Planta Med. 2021, 87, 283–293. [Google Scholar] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Sci. World J. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Phiboonchaiyanan, P.P.; Petpiroon, N.; Sritularak, B.; Chanvorachote, P. Phoyunnanin e Induces Apoptosis of Non-Small Cell Lung Cancer Cells via P53 Activation and down-Regulation of Survivin. Anticancer Res. 2018, 38, 6281–6290. [Google Scholar] [CrossRef]
- Petpiroon, N.; Sritularak, B.; Chanvorachote, P. Phoyunnanin E Inhibits Migration of Non-Small Cell Lung Cancer Cells via Suppression of Epithelial-to-Mesenchymal Transition and Integrin Av and Integrin Β3. BMC Complement. Altern. Med. 2017, 17, 1–16. [Google Scholar] [CrossRef]
- Hewlings, S.; Kalman, D. Curcumin: A Review of Its Effects on Human Health. Foods 2017, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Qiao, F.; Wang, Y.; Xu, Y.; Shang, Y. Curcumin Inhibits Cell Proliferation and Induces Apoptosis of Human Non-Small Cell Lung Cancer Cells through the Upregulation of MiR-192-5p and Suppression of PI3K/Akt Signaling Pathway. Oncol. Rep. 2015, 34, 2782–2789. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Feng, T.; Liu, X.; Liu, Q. Curcumin Inhibits Migration and Invasion of Non-Small Cell Lung Cancer Cells through up-Regulation of MiR-206 and Suppression of PI3K/AKT/MTOR Signaling Pathway. Acta Pharm. 2020, 70, 399–409. [Google Scholar] [CrossRef]
- Gontijo, V.S.; dos Santos, M.H.; Viegas, C., Jr. Biological and Chemical Aspects of Natural Biflavonoids from Plants: A Brief Review. Mini Rev. Med. Chem. 2016, 17, 834–862. [Google Scholar] [CrossRef]
- Wang, S.; Hu, Y.; Yan, Y.; Cheng, Z.; Liu, T. Sotetsuflavone Inhibits Proliferation and Induces Apoptosis of A549 Cells through ROS-Mediated Mitochondrial-Dependent Pathway. Bmc Complement. Altern. Med. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, X.; Hu, Y.; Lei, T.; Liu, T. Sotetsuflavone Induces Autophagy in Non-Small Cell Lung Cancer through Blocking PI3K/Akt/MTOR Signaling Pathway in Vivo and in Vitro. Front. Pharmacol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Zhou, M.; Shen, S.; Zhao, X.; Gong, X. Luteoloside Induces G0/G1 Arrest and pro-Death Autophagy through the ROS-Mediated AKT/MTOR/P70S6K Signalling Pathway in Human Non-Small Cell Lung Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2017, 494, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.M.; Valente, I.M.; Rodrigues, J.A. An Overview on Cardamonin. J. Med. Food 2014, 17, 633–640. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, R.; Li, Q.; Jie, X.; Hong, J.; Zong, Y.; Dong, X.; Zhang, S.; Li, Z.; Wu, G. Cardamonin Inhibits the Proliferation and Metastasis of Non-Small-Cell Lung Cancer Cells by Suppressing the PI3K/Akt/MTOR Pathway. Anticancer. Drugs 2019, 30, 241–250. [Google Scholar] [CrossRef]
- Barone, A.; Chi, D.C.; Theoret, M.R.; Chen, H.; He, K.; Kufrin, D.; Helms, W.S.; Subramaniam, S.; Zhao, H.; Patel, A.; et al. FDA Approval Summary: Trabectedin for Unresectable or Metastatic Liposarcoma or Leiomyosarcoma Following an Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 7448–7453. [Google Scholar] [CrossRef][Green Version]
- Sirimangkalakitti, N.; Chamni, S.; Charupant, K.; Chanvorachote, P.; Mori, N.; Saito, N.; Suwanborirux, K. Chemistry of Renieramycins. 15. Synthesis of 22- O -Ester Derivatives of Jorunnamycin A and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cells. J. Nat. Prod. 2016, 79, 2089–2093. [Google Scholar] [CrossRef]
- Ecoy, G.A.U.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P.; Chaotham, C. Jorunnamycin A from Xestospongia Sp. Suppresses Epithelial to Mesenchymal Transition and Sensitizes Anoikis in Human Lung Cancer Cells. J. Nat. Prod. 2019, 82, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Suwanborirux, K.; Amnuoypol, S.; Plubrukarn, A.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of Renieramycins. Part 3. Isolation and Structure of Stabilized Renieramycin Type Derivatives Possessing Antitumor Activity from Thai Sponge Xestospongia Species, Pretreated with Potassium Cyanide. J. Nat. Prod. 2003, 66, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M Attenuates Cancer Stem Cell-like Phenotypes in H460 Lung Cancer Cells. Anticancer Res. 2017, 37, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M Sensitizes Anoikis-Resistant H460 Lung Cancer Cells to Anoikis. Anticancer Res. 2016, 36, 1665–1671. [Google Scholar] [PubMed]
- Halim, H.; Chunhacha, P.; Suwanborirux, K.; Chanvorachote, P. Anticancer and Antimetastatic Activities of Renieramyein M, a Marine Tetrahydroisoquinoline Alkaloid, in Human Non-Small Cell Lung Cancer Cells. Anticancer Res. 2011, 31, 193–201. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iksen; Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100. https://doi.org/10.3390/molecules26134100
Iksen, Pothongsrisit S, Pongrakhananon V. Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules. 2021; 26(13):4100. https://doi.org/10.3390/molecules26134100
Chicago/Turabian StyleIksen, Sutthaorn Pothongsrisit, and Varisa Pongrakhananon. 2021. "Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products" Molecules 26, no. 13: 4100. https://doi.org/10.3390/molecules26134100
APA StyleIksen, Pothongsrisit, S., & Pongrakhananon, V. (2021). Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules, 26(13), 4100. https://doi.org/10.3390/molecules26134100