
Argon Adsorption on Cationic Gold Clusters Aun+ (n ≤ 20)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Experimental
2.2. Computational
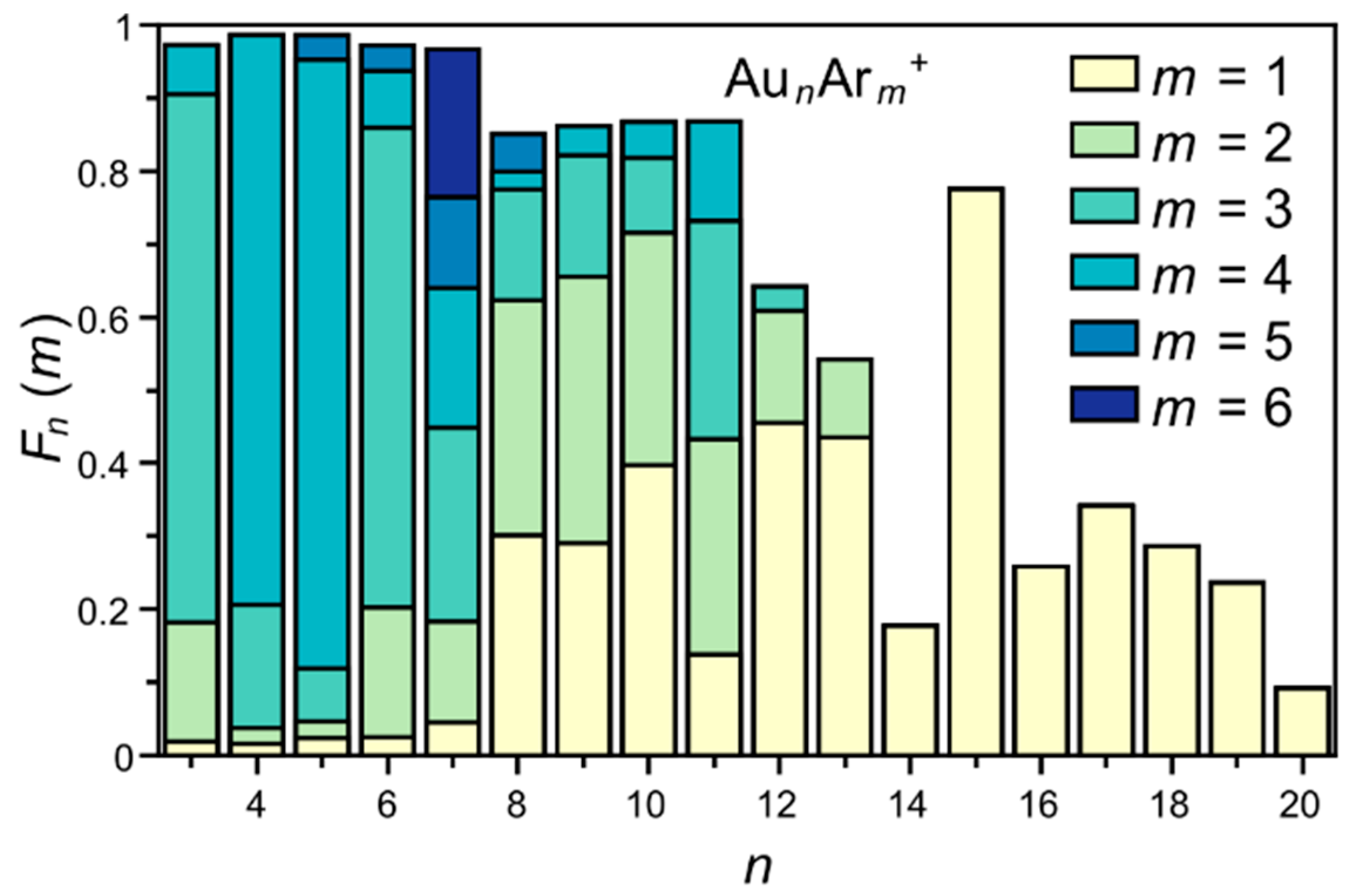
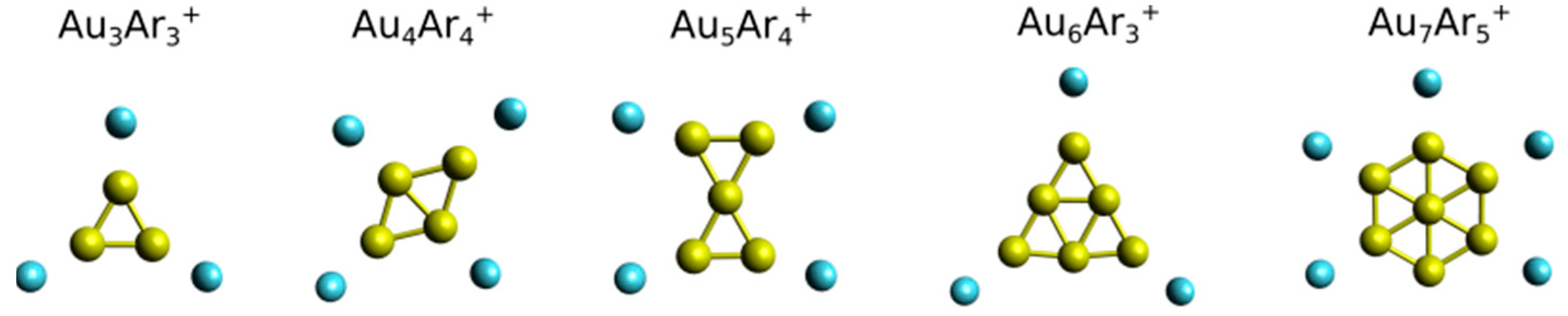
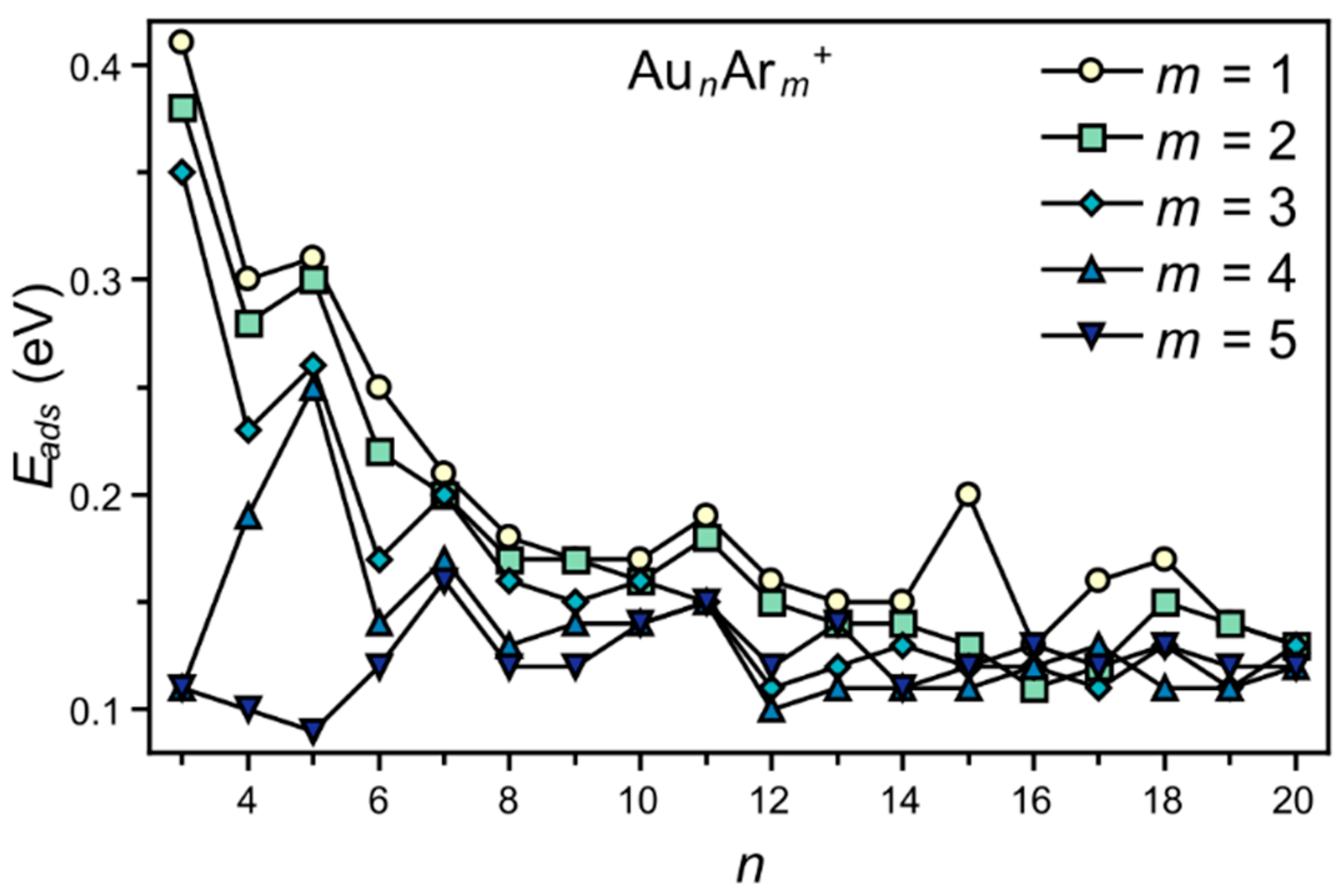
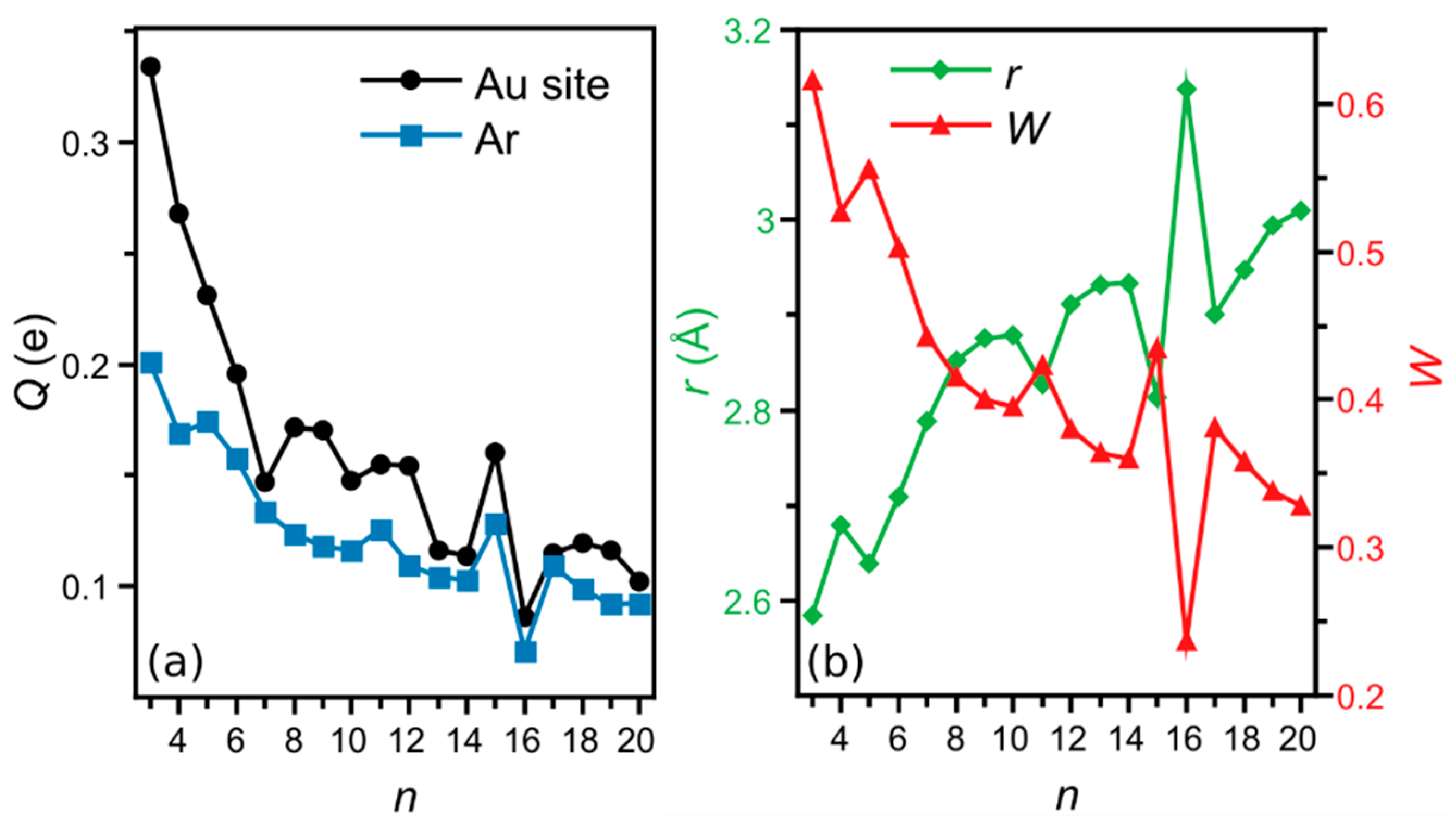
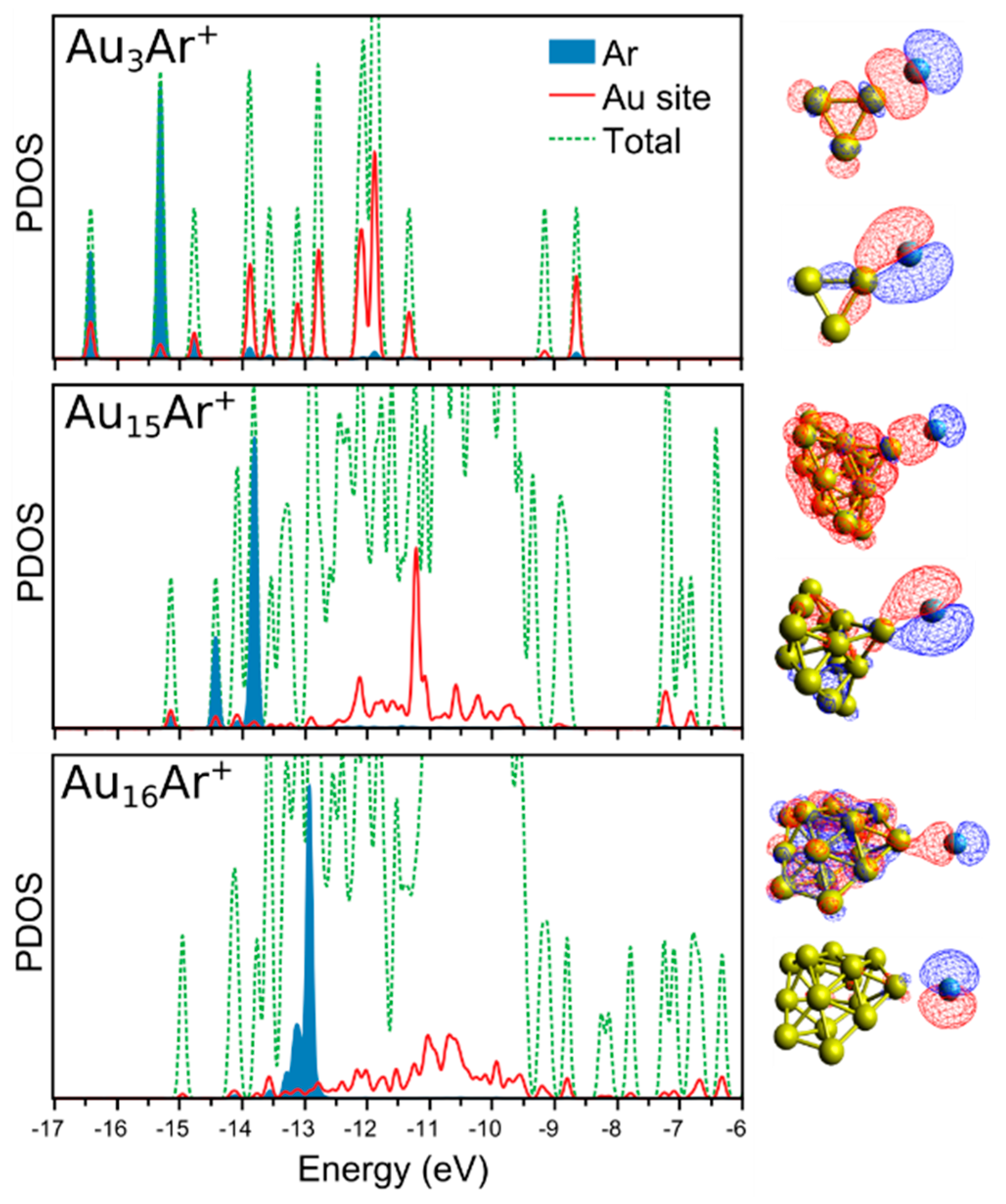
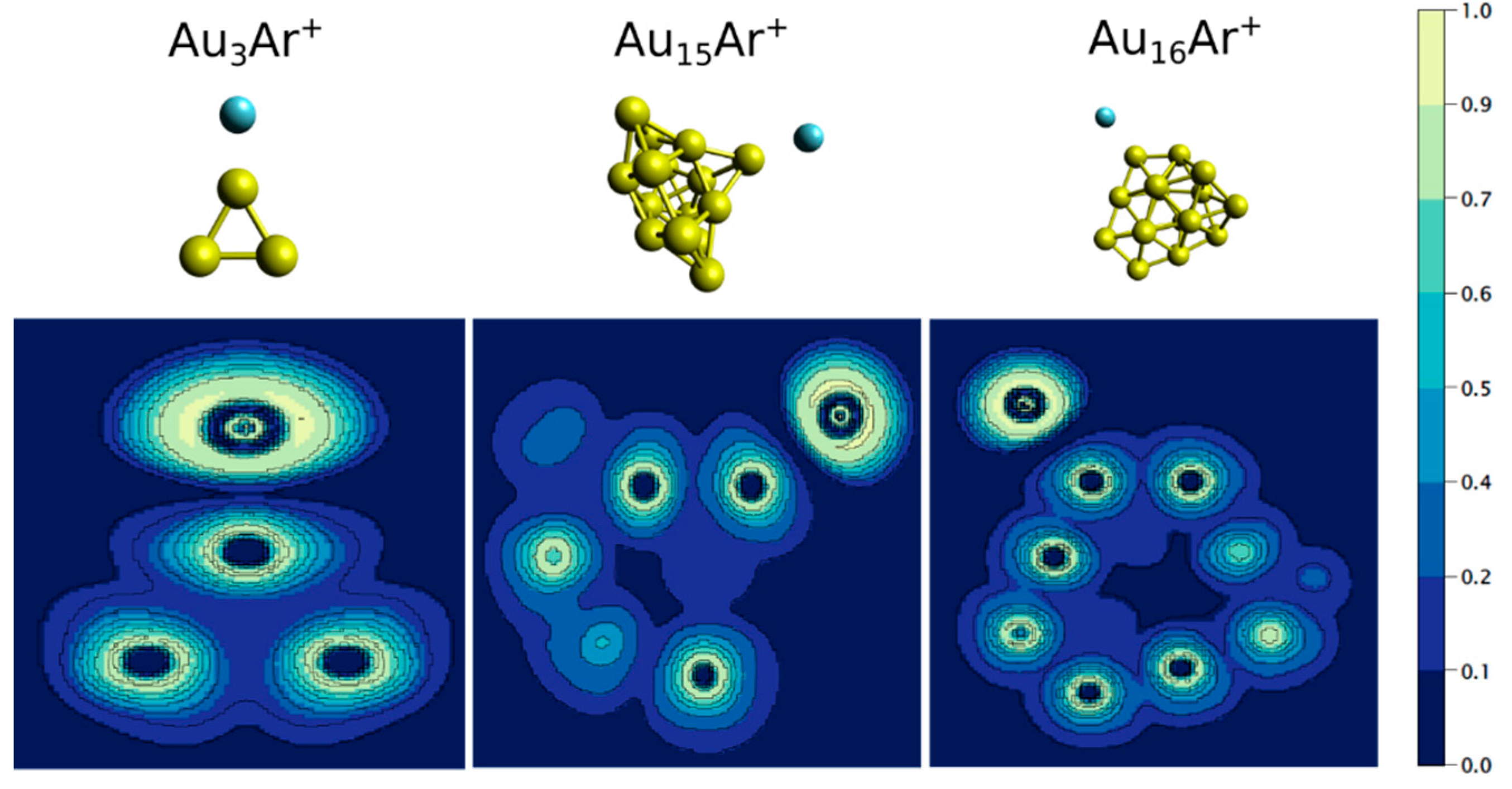
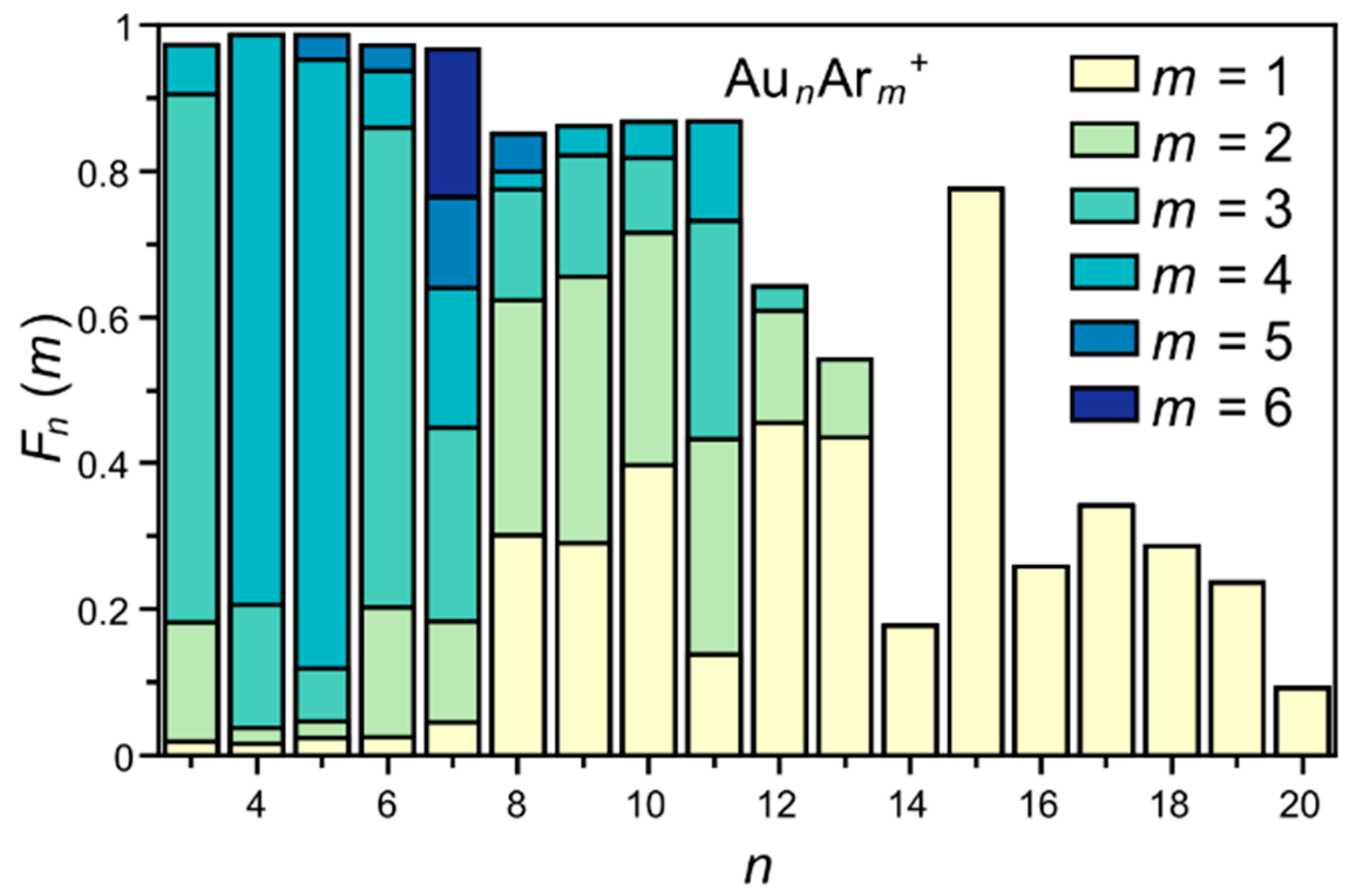
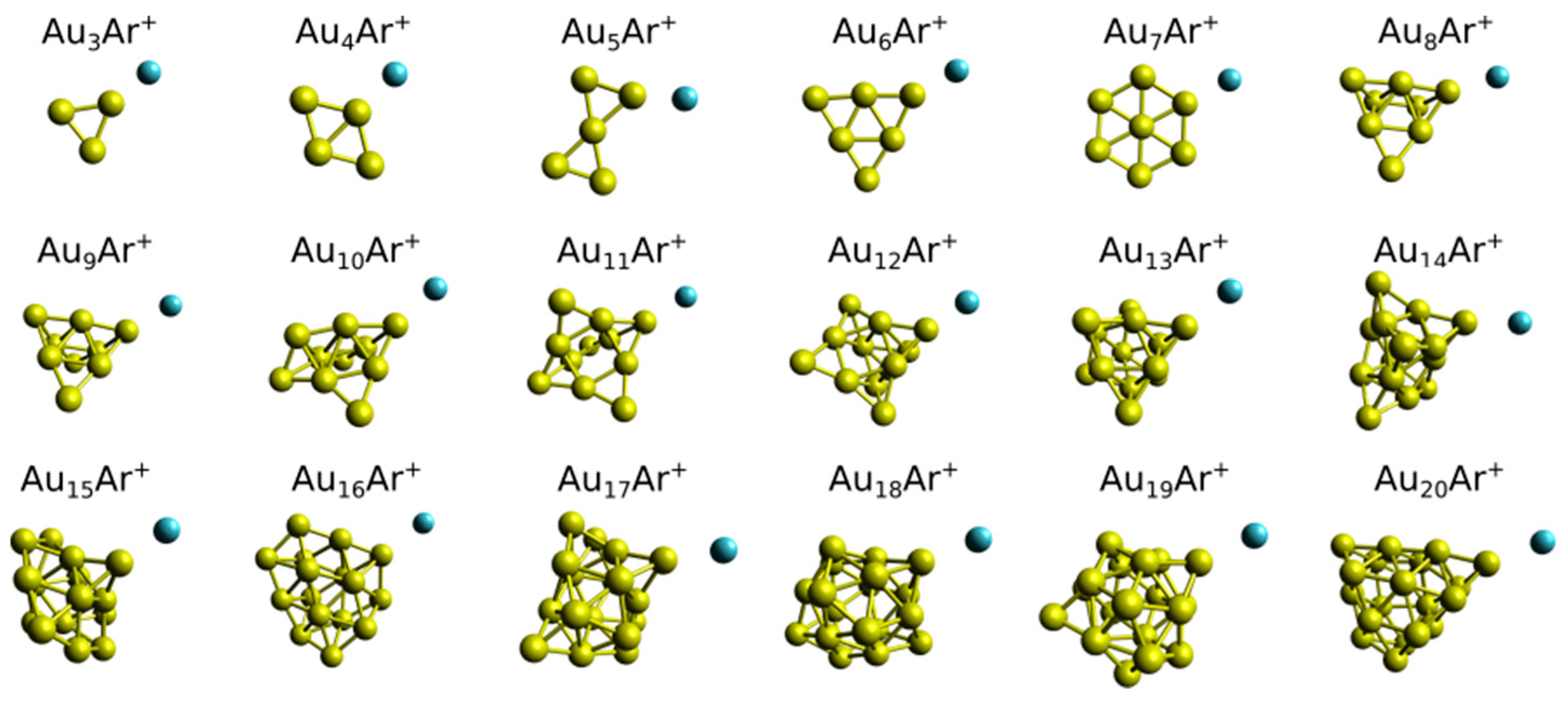
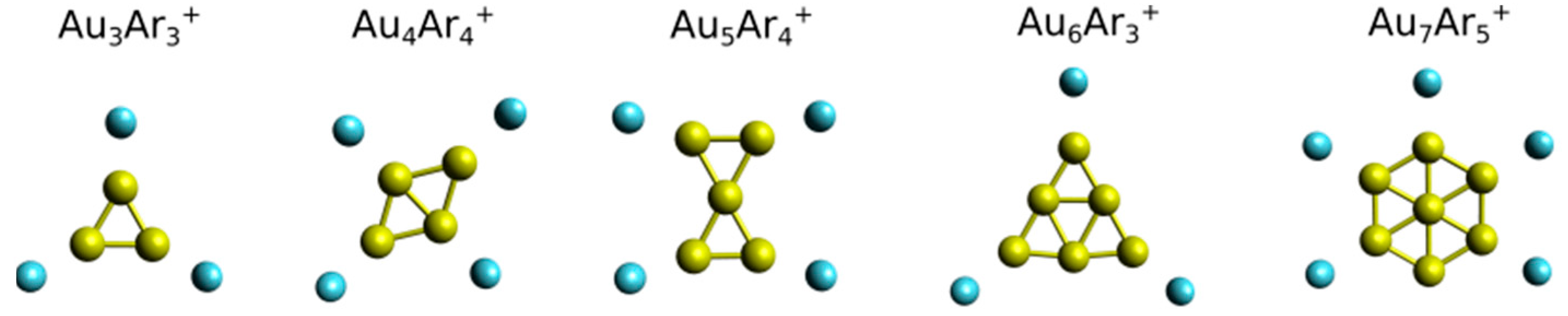
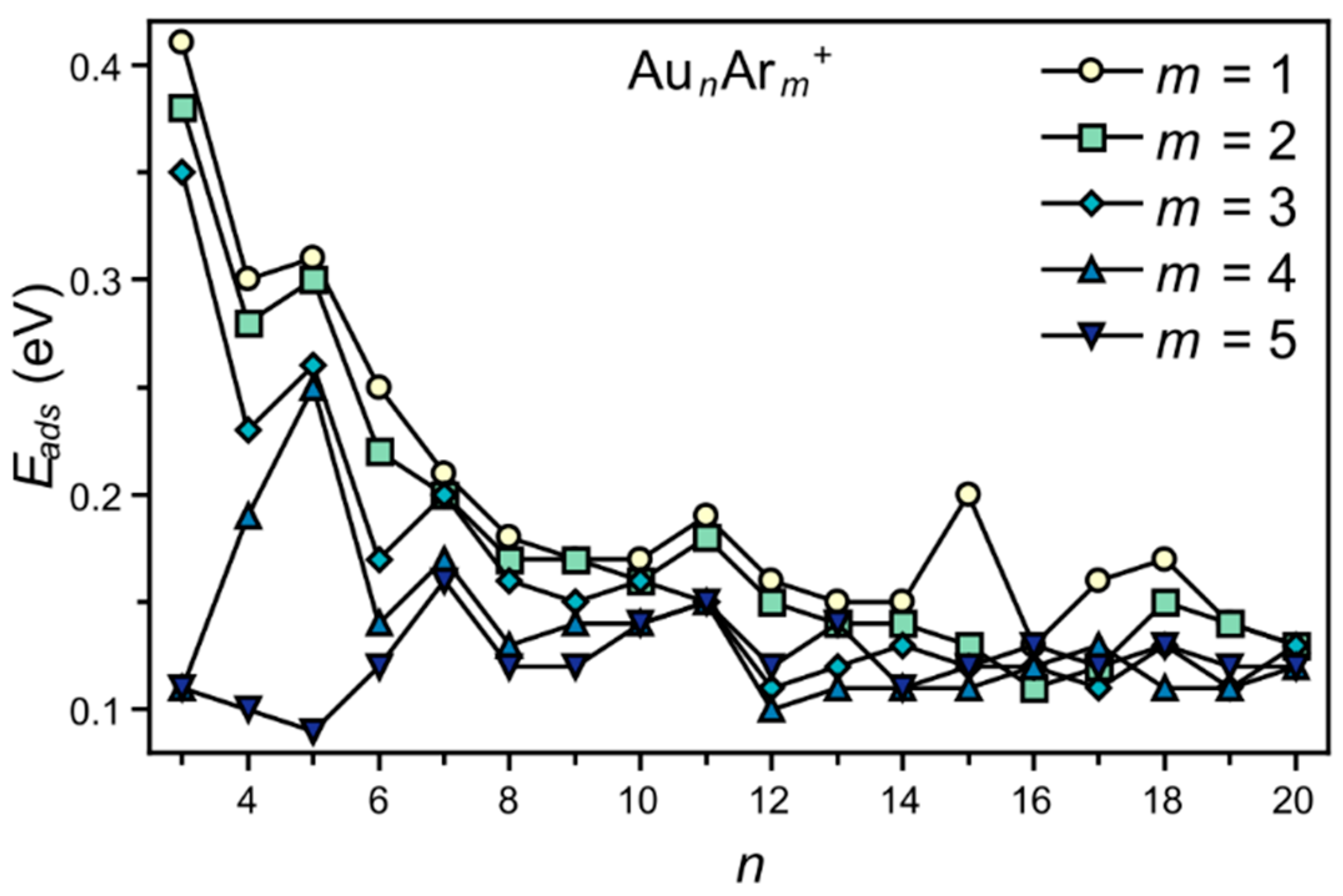
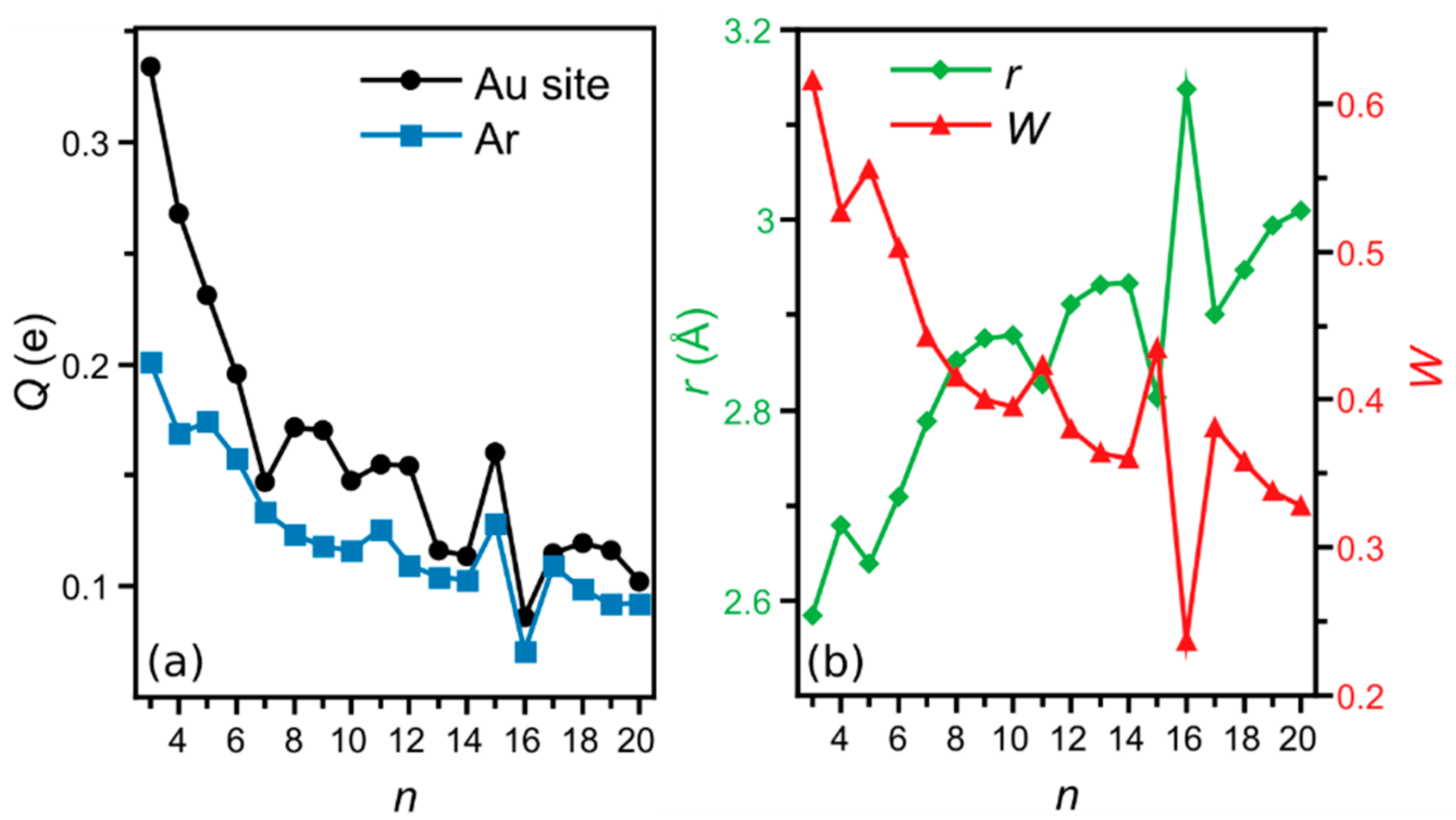
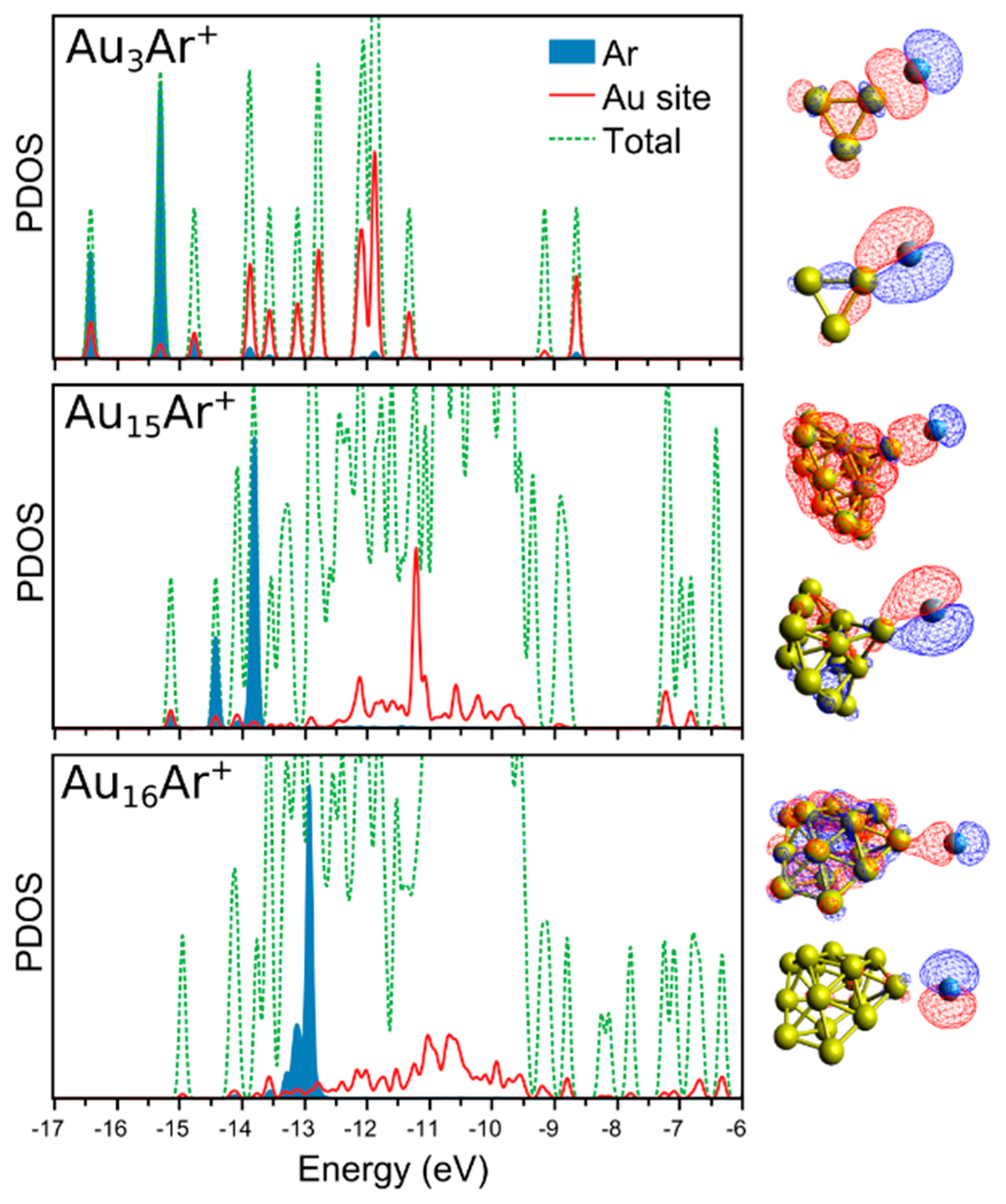
3. Results
4. Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hammer, B.; Norskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238–240. [Google Scholar] [CrossRef]
- Wallace, T.W.; Whetten, R.L. Coadsorption of CO and O2 on Selected Gold Clusters: Evidence for Efficient Room-Temperature CO2 Generation. J. Am. Chem. Soc. 2002, 124, 7499–7505. [Google Scholar] [CrossRef]
- Janssens, E.; Le, H.T.; Lievens, P. Adsorption of propene on neutral gold clusters in the gas phase. Chem. Eur. J. 2015, 21, 15256–15262. [Google Scholar] [CrossRef] [PubMed]
- Abdulhussein, H.A.; Ferrari, P.; Vanbuel, J.; Heard, C.; Fielicke, A.; Lievens, P.; Janssens, E.; Johnston, R.L. Altering CO binding on gold cluster cations by Pd-doping. Nanoscale 2019, 11, 16130–16141. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.M.; Bernhardt, T.M.C.V.; Bakker, J.M.; Barnett, R.N.L.U. Selective C− H bond cleavage in methane by small gold clusters. Angew. Chem. Int. Ed. 2017, 56, 13406–13410. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Jana, G.; Merino, B.; Chattaraj, P.K. Noble-noble strong union: Gold at its best to make a bond with a noble gas atom. ChemistryOpen 2019, 8, 173–187. [Google Scholar] [CrossRef]
- Pyykko, P. Predicted chemical bonds between rare gases and Au+. J. Am. Chem. Soc. 1995, 117, 2067–2070. [Google Scholar] [CrossRef]
- Read, J.P.; Buckingham, A.D. Covalency in ArAu+ and Related Species? J. Am. Chem. Soc. 1997, 119, 9010–9013. [Google Scholar] [CrossRef]
- Bellert, D.; Breckenridge, W.H. Bonding in Ground-State and Excited-State A+·Rg van der Waals Ions (A = Atom, Rg = Rare-Gas Atom): A Model-Potential Analysis. Chem. Rev. 2002, 102, 1595–1622. [Google Scholar] [CrossRef] [PubMed]
- Belpassi, L.; Infante, I.; Tarantelli, F.; Visscher, L.J. The Chemical Bond between Au(I) and the Noble Gases. Comparative Study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by Density Functional and Coupled Cluster Methods. Am. Chem. Soc. 2008, 130, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Grandinetti, F. Noble Gas Chemistry: Structure, Bonding, and Gas-Phase Chemistry; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2018. [Google Scholar]
- Yin, B.; Luo, Z. Coinage metal clusters: From superatom chemistry to genetic materials. Coord. Chem. Rev. 2021, 429, 213643. [Google Scholar] [CrossRef]
- Mathew, A.; Pradeep, T. Noble metal clusters: Applications in energy, environment, and biology. Part. Part. Syst. Charact. 2014, 31, 1017–1053. [Google Scholar] [CrossRef]
- Ferrari, P.; Vanbuel, J.; Janssens, P.; Lievens, P. Tuning the reactivity of small metal clusters by heteroatom doping. Acc. Chem. Res. 2018, 51, 3174–3182. [Google Scholar] [CrossRef] [PubMed]
- de Heer, W.A. The physics of simple metal clusters: Experimental aspects and simple models. Rev. Mod. Phys. 1993, 65, 611. [Google Scholar] [CrossRef]
- Gruene, P.; Rayner, D.M.; Redlich, B.; van der Meer, A.F.G.; Lyon, J.T.; Meijer, G.; Fielicke, A. Structures of neutral Au7, Au19, and Au20 clusters in the gas phase. Science 2008, 321, 674–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayeghi, A.; Heard, C.J.; Johnston, R.L.; Schäfer, R. Optical and electronic properties of mixed Ag-Au tetramer cations. J. Chem. Phys. 2014, 140, 054312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häkkinen, H. Electronic shell structures in bare and protected metal nanoclusters. Adv. Phys. X 2016, 1, 467–491. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.; Ferrari, P.; Janssens, E.; Lievens, P. Thermal radiation of gold clusters on microsecond time scales. Phys. Rev. A 2017, 96, 022511. [Google Scholar] [CrossRef] [Green Version]
- Pyykko, P. Relativistic effects in structural chemistry. Chem. Rev. 1988, 88, 563–594. [Google Scholar] [CrossRef]
- Delgado-Callico, L.; Ferrari, P.; Bakker, J.M.; Baletto, F.; Janssens, E. Benchmarking density functional theory methods for modelling cationic metal–argon complexes. Theor. Chem. Acc. 2021, 140, 38. [Google Scholar] [CrossRef]
- Shayeghi, A.; Johnston, R.L.; Rayner, D.M.; Schäfer, R.; Fielicke, A. The nature of bonding between argon and mixed gold–silver trimers. Angew. Chem. Int. Ed. 2015, 54, 10675–10680. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, A.; Schäfer, R.; Rayner, D.M.; Johnston, R.L.; Fielicke, A. Charge-induced dipole vs. relativistically enhanced covalent interactions in Ar-tagged Au-Ag tetramers and pentamers. J. Chem. Phys. 2015, 143, 024310. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Hou, G.-L.; Lushchikova, O.L.; Calvo, F.; Bakker, J.M.; Janssens, E. The structures of cationic gold clusters probed by far-infrared spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 11572–11577. [Google Scholar] [CrossRef]
- Kaydashev, V.; Ferrari, P.; Heard, C.; Janssens, E.; Johnston, R.L.; Lievens, P. Optical absorption of small palladium-doped gold clusters. Part. Part. Syst. Charact. 2016, 33, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Wang, L.-S. Probing the 2D to 3D structural transition in gold cluster anions using argon tagging. Phys. Rev. Lett. 2009, 102, 153401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, R.; Florian, J.; Liu, J.-X.; Gruene, P.; Lyon, J.T.; Rayner, D.M.; Fielicke, A.; Scheffler, M.; Ghiringhelli, L.M. Two-to-three dimensional transition in neutral gold clusters: The crucial role of van der Waals interactions and temperature. Phys. Rev. Mater. 2019, 3, 016002. [Google Scholar] [CrossRef] [Green Version]
- Lang, S.M.; Claes, P.; Cuong, N.T.; Nguyen, M.T.; Lievens, P.; Janssens, E. Copper doping of small gold cluster cations: Influence on geometric and electronic structure. J. Chem. Phys. 2011, 135, 224305. [Google Scholar] [CrossRef]
- Kaydashev, V.E.; Janssens, E.; Lievens, P. Optical absorption spectra of palladium doped gold cluster cations. J. Chem. Phys. 2015, 142, 034310. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.A. Invited review article: Laser vaporization cluster sources. Rev. Sci. Instrum. 2012, 83, 041101. [Google Scholar] [CrossRef]
- Ferrari, P.; Vanbuel, J.; Li, Y.L.T.; Janssens, E.; Lievens, P. Modifications of gas aggregation sources: The double laser ablation source approach. In Gas Aggregation Synthesis of Nanoparticles; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2017; pp. 57–78. [Google Scholar]
- Neese, F. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; Hussein, H.A.; Heard, C.J.; Vanbuel, J.; Lievens, P.; Johnston, R.L.; Janssens, E. Effect of palladium doping on the stability and fragmentation patterns of cationic gold clusters. Phys. Rev. A 2018, 97, 052508. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyser. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ferrari, P.; Delgado-Callico, L.; Lievens, P.; Baletto, F.; Janssens, E. Stability of cationic silver doped gold clusters and the subshell-closed electronic configuration of AgAu14+. J. Chem. Phys. 2020, 153, 244304. [Google Scholar] [CrossRef]
- Delgado-Callico, L.; Rossi, K.; Pinto-Miles, R.; Salzbrenner, P.; Baletto, F. A universal signature in the melting of metallic nanoparticles. Nanoscale 2021, 13, 1172–1180. [Google Scholar] [CrossRef]
- Koumpouras, K.; Larsson, J.A. Distinguishing between chemical bonding and physical binding using electron localization function (ELF). J. Phys. Condens. Matter. 2020, 32, 315502. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, P.; Janssens, E. Argon Adsorption on Cationic Gold Clusters Aun+ (n ≤ 20). Molecules 2021, 26, 4082. https://doi.org/10.3390/molecules26134082
Ferrari P, Janssens E. Argon Adsorption on Cationic Gold Clusters Aun+ (n ≤ 20). Molecules. 2021; 26(13):4082. https://doi.org/10.3390/molecules26134082
Chicago/Turabian StyleFerrari, Piero, and Ewald Janssens. 2021. "Argon Adsorption on Cationic Gold Clusters Aun+ (n ≤ 20)" Molecules 26, no. 13: 4082. https://doi.org/10.3390/molecules26134082
APA StyleFerrari, P., & Janssens, E. (2021). Argon Adsorption on Cationic Gold Clusters Aun+ (n ≤ 20). Molecules, 26(13), 4082. https://doi.org/10.3390/molecules26134082