
Hydricity of 3d Transition Metal Complexes from Density Functional Theory: A Benchmarking Study


Abstract
:1. Introduction

2. Methodology
2.1. Computational Details
2.2. Calculation of Hydricity
2.3. Acetonitrile Proton Clusters
3. Results and Discussion
3.1. Explicit Treatment of Solvent Molecules
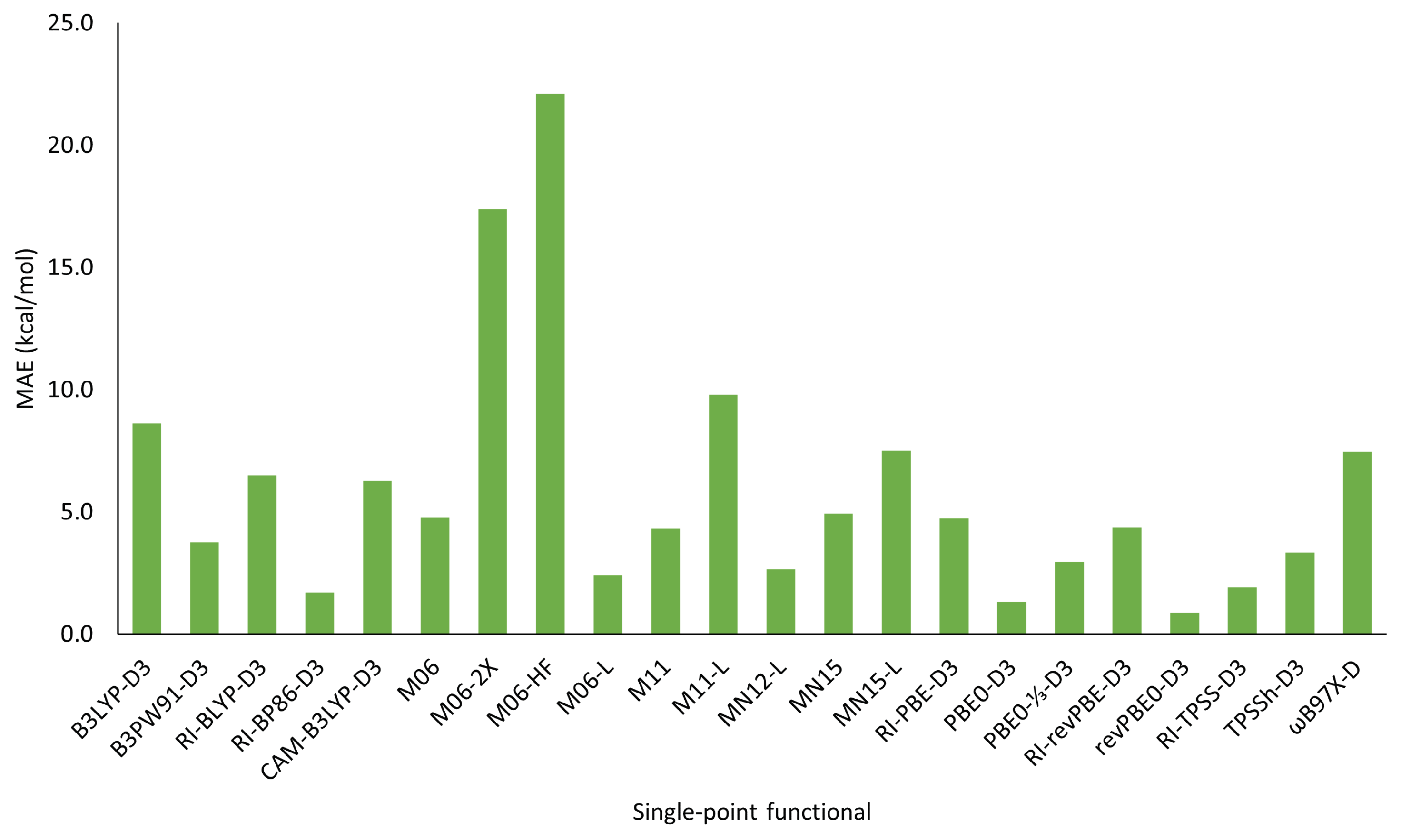
3.2. Choice of Single-Point Functional
3.3. Validation of Methodology
3.3.1. Choice of Basis Set for Single-Point Energies
3.3.2. Level of Geometry Optimisation
3.3.3. Choice of Solvation Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrmann, W.A.; Cornils, B. Organometallic Homogeneous Catalysis - Quo vadis? Angew. Chem. (Int. Ed. Engl.) 1997, 36, 1048–1067. [Google Scholar] [CrossRef]
- Reed-Berendt, B.G.; Polidano, K.; Morrill, L.C. Recent advances in homogeneous borrowing hydrogen catalysis using earth-abundant first row transition metals. Org. Biomol. Chem. 2019, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Carney, J.R.; Dillon, B.R.; Thomas, S.P. Recent Advances of Manganese Catalysis for Organic Synthesis. Eur. J. Org. Chem. 2016, 2016, 3912–3929. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Bender, T.A.; Dabrowski, J.A.; Gagné, M.R. Homogeneous catalysis for the production of low-volume, high-value chemicals from biomass. Nat. Rev. Chem. 2018, 2, 35–46. [Google Scholar] [CrossRef]
- Garbe, M.; Junge, K.; Beller, M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 2017, 4344–4362. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Vogiatzis, K.D.; Polynski, M.V.; Kirkland, J.K.; Townsend, J.; Hashemi, A.; Liu, C.; Pidko, E.A. Computational Approach to Molecular Catalysis by 3d Transition Metals: Challenges and Opportunities. Chem. Rev. 2019, 119, 2453–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elangovan, S.; Topf, C.; Fischer, S.; Jiao, H.; Spannenberg, A.; Baumann, W.; Ludwig, R.; Junge, K.; Beller, M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. [Google Scholar] [CrossRef]
- Wei, Z.; De Aguirre, A.; Junge, K.; Beller, M.; Jiao, H. Exploring the mechanisms of aqueous methanol dehydrogenation catalyzed by defined PNP Mn and Re pincer complexes under base-free as well as strong base conditions. Catal. Sci. Technol. 2018, 8, 3649–3665. [Google Scholar] [CrossRef]
- Junge, K.; Wendt, B.; Jiao, H.; Beller, M. Iridium-Catalyzed Hydrogenation of Carboxylic Acid Esters. ChemCatChem 2014, 6, 2810–2814. [Google Scholar] [CrossRef]
- Wei, Z.; Junge, K.; Beller, M.; Jiao, H. Hydrogenation of phenyl-substituted CN, CN, CC, CC and CO functional groups by Cr, Mo and W PNP pincer complexes–A DFT study. Catal. Sci. Technol. 2017, 7, 2298–2307. [Google Scholar] [CrossRef]
- Alberico, E.; Lennox, A.J.; Vogt, L.K.; Jiao, H.; Baumann, W.; Drexler, H.J.; Nielsen, M.; Spannenberg, A.; Checinski, M.P.; Junge, H.; et al. Unravelling the Mechanism of Basic Aqueous Methanol Dehydrogenation Catalyzed by Ru-PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 14890–14904. [Google Scholar] [CrossRef] [Green Version]
- Jiao, H.; Junge, K.; Alberico, E.; Beller, M. A Comparative Computationally Study About the Defined M(II) Pincer Hydrogenation Catalysts (M = Fe, Ru, Os). J. Comput. Chem. 2016, 37, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Elangovan, S.; Garbe, M.; Jiao, H.; Spannenberg, A.; Junge, K.; Beller, M. Hydrogenation of Esters to Alcohols Catalyzed by Defined Manganese Pincer Complexes. Angew. Chem. 2016, 128, 15590–15594. [Google Scholar] [CrossRef]
- Ge, H.; Chen, X.; Yang, X. A mechanistic study and computational prediction of iron, cobalt and manganese cyclopentadienone complexes for hydrogenation of carbon dioxide. Chem. Commun. 2016, 52, 12422–12425. [Google Scholar] [CrossRef]
- Gámez, J.A.; Hölscher, M.; Leitner, W. On the applicability of density functional theory to manganese-based complexes with catalytic activity toward water oxidation. J. Comput. Chem. 2017, 38, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Chen, X.; Yang, X. DFT and AIMD prediction of a SNS manganese pincer complex for hydrogenation of acetophenone. Chem. Phys. Lett. 2019, 714, 37–44. [Google Scholar] [CrossRef]
- Chen, X.; Ge, H.; Yang, X. Newly designed manganese and cobalt complexes with pendant amines for the hydrogenation of CO2 to methanol: A DFT study. Catal. Sci. Technol. 2017, 7, 348–355. [Google Scholar] [CrossRef]
- Passera, A.; Mezzetti, A. Mn(I) and Fe(II)/PN(H)P Catalysts for the Hydrogenation of Ketones: A Comparison by Experiment and Calculation. Adv. Synth. Catal. 2019, 361, 4691–4706. [Google Scholar] [CrossRef]
- Zubar, V.; Lebedev, Y.; Azofra, L.M.; Cavallo, L.; El-Sepelgy, O.; Rueping, M. Hydrogenation of CO2-Derived Carbonates and Polycarbonates to Methanol and Diols by Metal–Ligand Cooperative Manganese Catalysis. Angew. Chem. Int. Ed. 2018, 57, 13439–13443. [Google Scholar] [CrossRef]
- Garbe, M.; Junge, K.; Walker, S.; Wei, Z.; Jiao, H.; Spannenberg, A.; Bachmann, S.; Scalone, M.; Beller, M. Manganese(I)-Catalyzed Enantioselective Hydrogenation of Ketones Using a Defined Chiral PNP Pincer Ligand. Angew. Chem. 2017, 129, 11389–11393. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Han, Z.; Ding, K. Manganese-Catalyzed anti -Selective Asymmetric Hydrogenation of α-Substituted β-Ketoamides. Angew. Chem. 2020, 132, 15695–15699. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, L.; Shao, Z.; Li, G.; Lan, Y.; Liu, Q. Unmasking the Ligand Effect in Manganese-Catalyzed Hydrogenation: Mechanistic Insight and Catalytic Application. J. Am. Chem. Soc. 2019, 141, 17337–17349. [Google Scholar] [CrossRef]
- Glatz, M.; Stöger, B.; Himmelbauer, D.; Veiros, L.F.; Kirchner, K. Chemoselective Hydrogenation of Aldehydes under Mild, Base-Free Conditions: Manganese Outperforms Rhenium. ACS Catal. 2018, 8, 4009–4016. [Google Scholar] [CrossRef]
- Van Putten, R.; Uslamin, E.A.; Garbe, M.; Liu, C.; Gonzalez-de Castro, A.; Lutz, M.; Junge, K.; Hensen, E.J.; Beller, M.; Lefort, L.; et al. Non-Pincer-Type Manganese Complexes as Efficient Catalysts for the Hydrogenation of Esters. Angew. Chem. Int. Ed. 2017, 56, 7531–7534. [Google Scholar] [CrossRef] [Green Version]
- Ryabchuk, P.; Stier, K.; Junge, K.; Checinski, M.P.; Beller, M. Molecularly Defined Manganese Catalyst for Low-Temperature Hydrogenation of Carbon Monoxide to Methanol. J. Am. Chem. Soc. 2019, 141, 16923–16929. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yang, H.; Zhao, M.; Wen, J.; Tucker, J.H.; Zhang, X. C1-Symmetric PNP Ligands for Manganese-Catalyzed Enantioselective Hydrogenation of Ketones: Reaction Scope and Enantioinduction Model. ACS Catal. 2020, 10, 13794–13799. [Google Scholar] [CrossRef]
- Liu, C.; van Putten, R.; Kulyaev, P.O.; Filonenko, G.A.; Pidko, E.A. Computational insights into the catalytic role of the base promoters in ester hydrogenation with homogeneous non-pincer-based Mn-P,N catalyst. J. Catal. 2018, 363, 136–143. [Google Scholar] [CrossRef]
- Van Putten, R.; Filonenko, G.A.; Gonzalez De Castro, A.; Liu, C.; Weber, M.; Müller, C.; Lefort, L.; Pidko, E. Mechanistic Complexity of Asymmetric Transfer Hydrogenation with Simple Mn-Diamine Catalysts. Organometallics 2019, 38, 3187–3196. [Google Scholar] [CrossRef] [Green Version]
- Zirakzadeh, A.; de Aguiar, S.R.; Stöger, B.; Widhalm, M.; Kirchner, K. Enantioselective Transfer Hydrogenation of Ketones Catalyzed by a Manganese Complex Containing an Unsymmetrical Chiral PNP’ Tridentate Ligand. ChemCatChem 2017, 9, 1744–1748. [Google Scholar] [CrossRef]
- Rawat, K.S.; Mahata, A.; Choudhuri, I.; Pathak, B. Catalytic hydrogenation of CO2 by manganese complexes: Role of π-acceptor ligands. J. Phys. Chem. C 2016, 120, 16478–16488. [Google Scholar] [CrossRef]
- Yang, X. Hydrogenation of carbon dioxide catalyzed by PNP pincer iridium, iron, and cobalt complexes: A computational design of base metal catalysts. ACS Catal. 2011, 1, 849–854. [Google Scholar] [CrossRef]
- Mondal, B.; Sengupta, K.; Rana, A.; Mahammed, A.; Botoshansky, M.; Dey, S.G.; Gross, Z.; Dey, A. Cobalt corrole catalyst for efficient hydrogen evolution reaction from H2O under ambient conditions: Reactivity, spectroscopy, and density functional theory calculations. Inorg. Chem. 2013, 52, 3381–3387. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Malakar, T.; Paul, A. Theoretical studies on the mechanism of homogeneous catalytic olefin hydrogenation and amine-borane dehydrogenation by a versatile boryl-ligand-based cobalt catalyst. ACS Catal. 2015, 5, 2754–2769. [Google Scholar] [CrossRef]
- Della Monica, F.; Vummaleti, S.V.; Buonerba, A.; Nisi, A.D.; Monari, M.; Milione, S.; Grassi, A.; Cavallo, L.; Capacchione, C. Coupling of Carbon Dioxide with Epoxides Efficiently Catalyzed by Thioether-Triphenolate Bimetallic Iron(III) Complexes: Catalyst Structure–Reactivity Relationship and Mechanistic DFT Study. Adv. Synth. Catal. 2016, 358, 3231–3243. [Google Scholar] [CrossRef]
- Murugesan, K.; Wei, Z.; Chandrashekhar, V.G.; Neumann, H.; Spannenberg, A.; Jiao, H.; Beller, M.; Jagadeesh, R.V. Homogeneous cobalt-catalyzed reductive amination for synthesis of functionalized primary amines. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Tong, L.P.; Liao, R.Z. Mechanism of Water Oxidation Catalyzed by a Mononuclear Iron Complex with a Square Polypyridine Ligand: A DFT Study. Inorg. Chem. 2018, 57, 4590–4601. [Google Scholar] [CrossRef] [PubMed]
- Wiedner, E.S.; Chambers, M.B.; Pitman, C.L.; Bullock, R.M.; Miller, A.J.; Appel, A.M. Thermodynamic Hydricity of Transition Metal Hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Chin, B.; Lough, A.J.; Morris, R.H.; Schweitzer, C.T.; D’Agostino, C. Influence of Chloride versus Hydride on H-H Bonding and Acidity of the Trans Dihydrogen Ligand in the Complexes trans-[Ru(H2)X(PR2CH2CH2PR2)2]+, X = Cl, H, R = Ph, Et. Crystal Structure Determinations of [RuCl(dppe)2]PF6 and trans-[Ru(H2)Cl(dppe)2]PF6. Inorg. Chem. 1994, 33, 6278–6288. [Google Scholar] [CrossRef]
- Guerchais, V.; Astruc, D.; Nunn, C.M.; Cowley, A.H. Generation, Characterization, and Chemistry of the Methylene Complexes [Fe(η5-C5Me5)(CO)(L)(=CH2)]+ (L = CO, PPh3) and the X-ray Crystal Structure of [Fe(η5-C5Me5)(CO)2(CH2PPh3)]+BF4-. Organometallics 1990, 9, 1036–1041. [Google Scholar] [CrossRef]
- Klug, C.M.; Dougherty, W.G.; Kassel, W.S.; Wiedner, E.S. Electrocatalytic Hydrogen Production by a Nickel Complex Containing a Tetradentate Phosphine Ligand. Organometallics 2019, 38, 1269–1279. [Google Scholar] [CrossRef]
- Neale, S.E. Computational Investigations of Homogeneous Catalysis and Spin- State Energetics in Fe ( II ) and Co ( III ) Complexes. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2019. [Google Scholar]
- Bühl, M.; Kabrede, H. Geometries of transition-metal complexes from density-functional theory. J. Chem. Theory Comput. 2006, 2, 1282–1290. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1457–1465. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Tao, J.; Staroverov, V.N.; Scuseria, G.E.; Csonka, G.I. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. J. Chem. Phys. 2005, 123, 062201. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533–16539. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.M.; Peeters, F.M.; Hai, G.Q.; Devreese, J.T. Erratum: Donor transition energy in GaAs superlattices in a magnetic field along the growth axis. Phys. Rev. B 1993, 48, 4978. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Truhlar, D.G. Comparative DFT study of van der Waals complexes: Rare-gas dimers, alkaline-earth dimers, zinc dimer and zinc-rare-gas dimers. J. Phys. Chem. A 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. Density functional for spectroscopy: No long-range self-interaction error, good performance for Rydberg and charge-transfer states, and better performance on average than B3LYP for ground states. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peverati, R.; Truhlar, D.G. Improving the accuracy of hybrid meta-GGA density functionals by range separation. J. Phys. Chem. Lett. 2011, 2, 2810–2817. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. M11-L: A local density functional that provides improved accuracy for electronic structure calculations in chemistry and physics. J. Phys. Chem. Lett. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Peverati, R.; Truhlar, D.G. An improved and broadly accurate local approximation to the exchange-correlation density functional: The MN12-L functional for electronic structure calculations in chemistry and physics. Phys. Chem. Chem. Phys. 2012, 14, 13171–13174. [Google Scholar] [CrossRef]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; He, X.; Truhlar, D.G. MN15-L: A New Local Exchange-Correlation Functional for Kohn-Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theory Comput. 2016, 12, 1280–1293. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Errata: Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Guido, C.A.; Brémond, E.; Adamo, C.; Cortona, P. Communication: One third: A new recipe for the PBE0 paradigm. J. Chem. Phys. 2013, 138, 021104. [Google Scholar] [CrossRef] [Green Version]
- Ernzerhof, M.; Perdew, J.P. Generalized gradient approximation to the angle- and system-averaged exchange hole. J. Chem. Phys. 1998, 109, 3313–3320. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [Green Version]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137, Erratum in J. Chem. Phys. 2004, 121, 11507. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Estes, D.P.; Vannucci, A.K.; Hall, A.R.; Lichtenberger, D.L.; Norton, J.R. Thermodynamics of the metal-hydrogen bonds in (η5-C5H5)M(CO)2H (M = Fe, Ru, Os). Organometallics 2011, 30, 3444–3447. [Google Scholar] [CrossRef]
- Ciancanelli, R.; Noll, B.C.; DuBois, D.L.; Rakowski DuBois, M. Comprehensive thermodynamic characterization of the metal-hydrogen bond in a series of cobalt-hydride complexes. J. Am. Chem. Soc. 2002, 124, 2984–2992. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Appel, A.M.; Dubois, D.L.; Bullock, R.M. Thermochemical and mechanistic studies of electrocatalytic hydrogen production by cobalt complexes containing pendant amines. Inorg. Chem. 2013, 52, 14391–14403. [Google Scholar] [CrossRef] [PubMed]
- Moltved, K.A.; Kepp, K.P. Chemical Bond Energies of 3d Transition Metals Studied by Density Functional Theory. J. Chem. Theory Comput. 2018, 14, 3479–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmel, D.; Goll, S.K.; Leito, I.; Krossing, I. A unified pH scale for all phases. Angew. Chem. Int. Ed. 2010, 49, 6885–6888. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Reimann, C.; Pantazis, D.A.; Bredow, T.; Neese, F. Geometries of third-row transition-metal complexes from density-functional theory. J. Chem. Theory Comput. 2008, 4, 1449–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariyaratne, J.K.P.; Bierrum, A.M.; Green, M.L.H.; Ishaq, M.; Prout, C.K.; Swanwick, M.G. Evidence for near-neighbour interactions in some substituted methyl derivatives of transition metals including the molecular crystal structure determinations of (π-C5H5)Fe(CO)2CH2CO2H and (π-C5H5)Mo(CO)3CH2CO2H. J. Chem. Soc. A 1969, 1309–1321. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| System | Methodology a | Dispersion b,c | Solvent Model c | References |
|---|---|---|---|---|
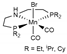 | B3PW91/TZVP | [9,10,15] | ||
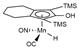 | M06/6-31++G(d,p) | M | PCM | [16] |
 | PBE0/def2-TZVP | [17] | ||
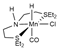 | B97X-D/ECP (Mn)/6-31++G(d,p) | GD2 | PCM | [18] |
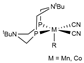 | M06/ECP (Mn,Co)/6-31(+)G(d,p) | M | PCM | [19] |
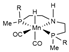 | B97X-D/ECP (Mn)/cc-PVTZ | GD2 | PCM | [20] |
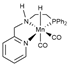 | M06/TZVP// B97X-D/TZVP (Mn)/SVP | M sp GD2 opt | PCM | [21] |
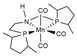 | B3PW91/TZVP | [22] | ||
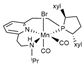 | B97X-D/ECP (Mn)/cc-pVDZ | GD2 | PCM | [23] |
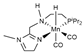 | M06L/def2-TZVP// B3LYP/def2-SVP | M sp | SMD sp | [24] |
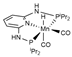 | PBE0/ECP (Mn)/6-31G(d,p) | PCM | [25] | |
 | PBE0/6-311+G(d,p)// PBE0/6-31G(d,p) | [26] | ||
 | DSD-PBE96-NL/def2-QZVP// PBE0-D3/def2-TZVP | GD3BJ opt | [27] | |
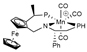 | M06/6-311++G(d,p)// M06/ECP (Mn)/6-31G(d) | M | SMD sp | [28] |
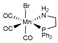 | PBE0/6-311+G(d)// PBE0/6-31G(d) | PCM sp | [29] | |
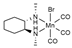 | PBE0/def2-QZVPP// PBE0-D3/def2-TZVP | GD3BJ opt | SMD opt | [30] |
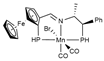 | PBE0/ECP (Mn)/6-31G(d,p) | [31] | ||
 | B3LYP/ECP (Mn)/6-311++G(d,p) | CPCM | [32] | |
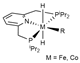 | B97X/6-31++G(d,p) | PCM | [33] | |
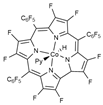 | BP86/6-311+G(d)// BP86/6-311G(d) (Co)/6-31G(d) | PCM sp | [34] | |
 | B3PW91/ECP (Co)/6-31++G(d,p)// B3PW91/ECP (Co)/6-31++G(d,p) (B,N,P)/6-31G(d) | GD sp GD opt | CPCM sp | [35] |
 | M06/TZVP// BP86/ECP (Fe)/SVP | M sp | PCM sp | [36] |
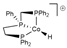 | B3PW91/def2-TZVP// B3PW91/TZVP | GD3BJ sp | SMD sp | [37] |
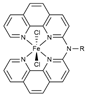 | B3LYP*/ECP (Fe)/6-311+G(2df,2p)// B3LYP/ECP (Fe)/6-31G(d,p) | GD3BJ sp GD3BJ opt | SMD sp | [38] |
| Complex | FeCp(CO)2H | Co(dppe)2H | Co(P4N2)H |
|---|---|---|---|
(kcal/mol) | 61.7 [39,80] | 49.9 [81,82] | 31.8 [82] |
 | 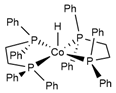 |  | |
 |  |  | |
| Positively charged cationic complex fragment after hydride dissociation a | 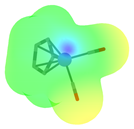 | 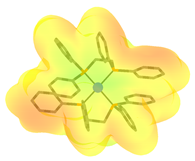 | 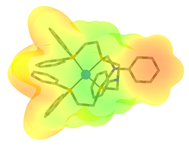 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodfellow, A.S.; Bühl, M. Hydricity of 3d Transition Metal Complexes from Density Functional Theory: A Benchmarking Study. Molecules 2021, 26, 4072. https://doi.org/10.3390/molecules26134072
Goodfellow AS, Bühl M. Hydricity of 3d Transition Metal Complexes from Density Functional Theory: A Benchmarking Study. Molecules. 2021; 26(13):4072. https://doi.org/10.3390/molecules26134072
Chicago/Turabian StyleGoodfellow, Alister S., and Michael Bühl. 2021. "Hydricity of 3d Transition Metal Complexes from Density Functional Theory: A Benchmarking Study" Molecules 26, no. 13: 4072. https://doi.org/10.3390/molecules26134072
APA StyleGoodfellow, A. S., & Bühl, M. (2021). Hydricity of 3d Transition Metal Complexes from Density Functional Theory: A Benchmarking Study. Molecules, 26(13), 4072. https://doi.org/10.3390/molecules26134072