
The Role of Organic Small Molecules in Pain Management


Abstract
:1. Introduction
Prostaglandins and the Cyclooxygenase Anti-Inflammatory Pathway
2. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
3. Multitarget Drugs
4. Cannabis
5. Calcium Channel
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrer, M.D.; Busquets-Cortés, C.; Capó, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2018, 26, 3225–3241. [Google Scholar] [CrossRef]
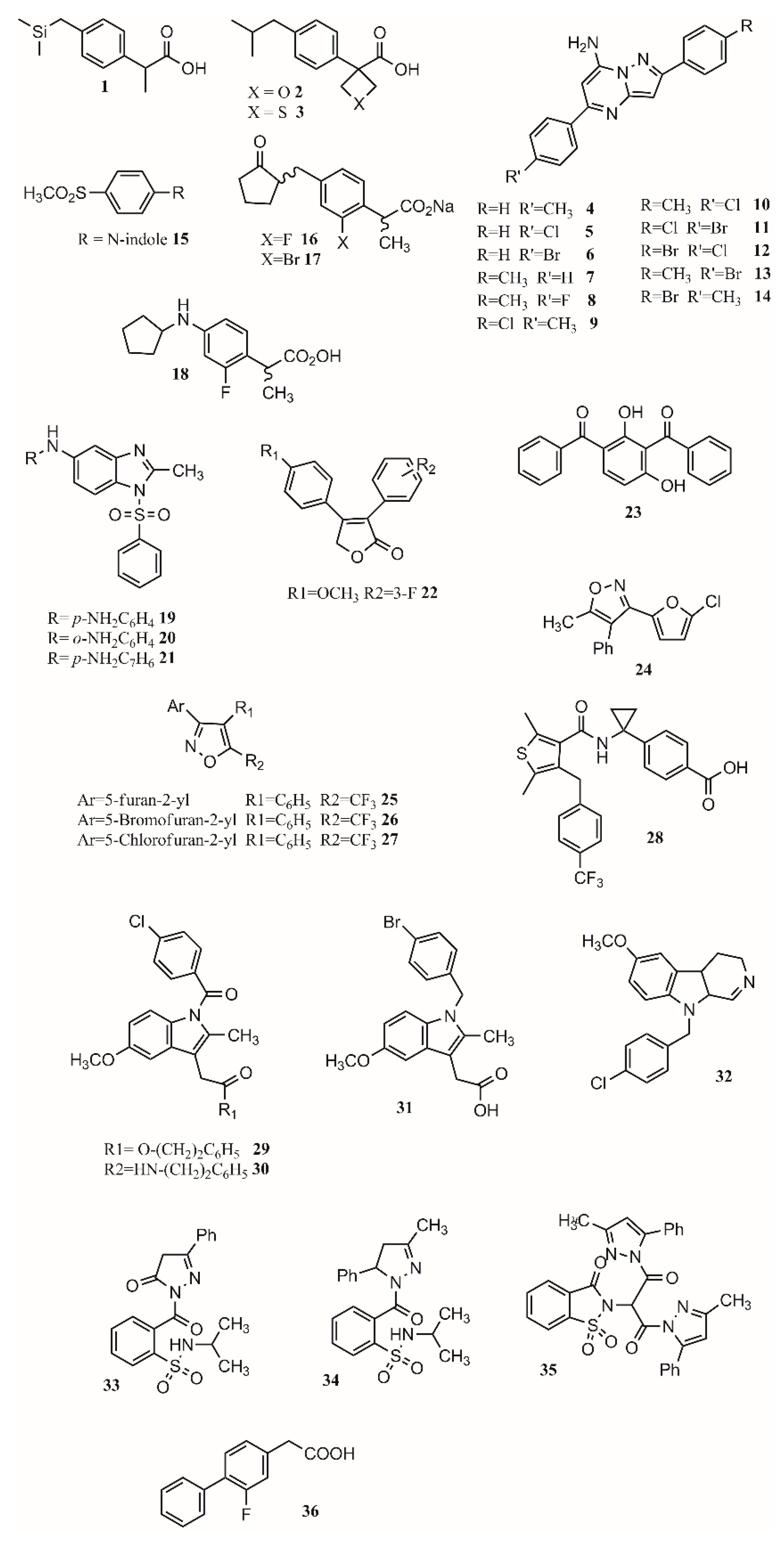
- Aggarwal, R.; Singh, G.; Kaushik, P.; Kaushik, D.; Paliwal, D.; Kumar, A. Molecular docking design and one-pot expeditious synthesis of novel 2,5-diarylpyrazolo [1,5-a] pyrimidin-7-amines as anti-inflammatory agents. Eur. J. Med. Chem. 2015, 101, 326–333. [Google Scholar] [CrossRef]
- Shenvi, S.; Kiran, K.R.; Kumar, K.; Diwakar, L.; Reddy, G.C. Synthesis and biological evaluation of boswellic acid-NSAID hybrid molecules as anti-inflammatory and anti-arthritic agents. Eur. J. Med. Chem. 2015, 98, 170–178. [Google Scholar] [CrossRef]
- Chan, K.L.; Cathomas, F.; Russo, S.J. Central and peripheral inflammation link metabolic syndrome and major depressive disorder. Physiology 2019, 34, 123–133. [Google Scholar] [CrossRef]
- Collins, K.H.; Herzog, W.; MacDonald, G.Z.; Reimer, R.A.; Rios, J.L.; Smith, I.C.; Zernicke, R.F.; Hart, D.A. Obesity, metabolic syndrome, and musculoskeletal disease: Common inflammatory pathways suggest a central role for loss of muscle integrity. Front. Physiol. 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Ramírez, B.; Becerril, S.; Salvador, J.; Portincasa, P.; Colina, I.; Gómez-Ambrosi, J. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci. Rep. 2017, 7, 6619. [Google Scholar] [CrossRef]
- Lopez-Candales, A.; Hernández Burgos, P.M.; Hernandez-Suarez, D.F.; Harris, D. Linking Chronic Inflammation with Cardiovascular Disease: From Normal Aging to the Metabolic Syndrome. J. Nat. Sci. 2017, 3, e341. [Google Scholar] [PubMed]
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic syndrome is an inflammatory disorder: A conspiracy between adipose tissue and phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef]
- Zanandrea, R.; Bonan, C.D.; Campos, M.M. Zebrafish as a model for inflammation and drug discovery. Drug Discov. Today 2020, 25, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.X.; Garneau-Tsodikova, S. What are the drugs of the future? MedChemComm 2018, 9, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Neha, K.; Wakode, S. Contemporary advances of cyclic molecules proposed for inflammation. Eur. J. Med. Chem. 2021, 221, 113493. [Google Scholar] [CrossRef] [PubMed]
- Demay, J.; Halary, S.; Knittel-Obrecht, A.; Villa, P.; Duval, C.; Hamlaoui, S.; Roussel, T.; Yéprémian, C.; Reinhardt, A.; Bernard, C.; et al. Anti-inflammatory, antioxidant, and wound-healing properties of cyanobacteria from thermal mud of balaruc-les-bains, France: A multi-approach study. Biomolecules 2021, 11, 28. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Li, W.-S.; Zhang, Z.-Y.; Luo, H.; Wang, J.-R.; Zhang, H.-Y.; Zeng, Z.-R.; Chen, B.; Li, X.-W.; Guo, Y.-W. Sinusiaetone A, an Anti-inflammatory Norditerpenoid with a Bicyclo [11.3.0] hexadecane Nucleus from the Hainan Soft Coral Sinularia siaesensis. Org. Lett. 2021. [Google Scholar] [CrossRef]
- Shi, Z.; Dun, B.; Wei, Z.; Liu, C.; Tian, J.; Ren, G.; Yao, Y. Peptides Released from Extruded Adzuki Bean Protein through Simulated Gastrointestinal Digestion Exhibit Anti-inflammatory Activity. J. Agric. Food Chem. 2021. [Google Scholar] [CrossRef]
- Burgos, R.A.; Alarcón, P.; Quiroga, J.; Manosalva, C.; Hancke, J. Andrographolide, an Anti-Inflammatory Multitarget Drug: All Roads Lead to Cellular Metabolism. Molecules 2020, 26, 5. [Google Scholar] [CrossRef]
- Tian, H.; Wei, L.; Yao, Y.; Zeng, Z.; Liang, X.; Zhu, H. Analysis of the Anti-Inflammatory and Analgesic Mechanism of Shiyifang Vinum Based on Network Pharmacology. Evid. Based Complement. Altern. Med. 2021, 2021, 8871276. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.; Lüdtke, R.; Selbmann, H.K.; Kötter, I.; Tschirdewahn, B.; Schaffner, W.; Heide, L. Efficacy and tolerability of a standardized willow bark extract in patients with osteoarthritis: Randomized, placebo-controlled, double blind clinical trial. Z. Rheumatol. 2000, 59, 314–320. [Google Scholar] [CrossRef]
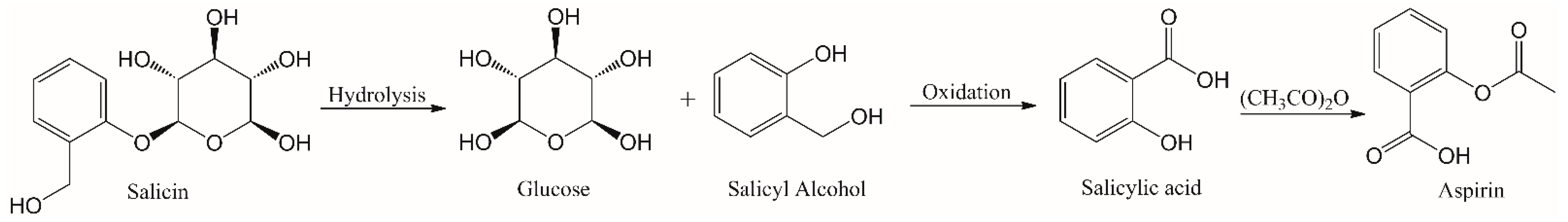
- Desborough, M.J.R.; Keeling, D.M. The aspirin story—From willow to wonder drug. Br. J. Haematol. 2017, 177, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Wood, E.J. Aspirin: The remarkable story of a wonder drug. Biochem. Mol. Biol. Educ. 2006, 34, 459–460. [Google Scholar] [CrossRef]
- Khayyal, M.T.; El-Ghazaly, M.A.; Abdallah, D.M.; Okpanyi, S.N.; Kelber, O.; Weiser, D. Mechanisms involved in the anti-inflammatory effect of a standardized willow bark extract. Arzneim. Forsch. /Drug Res. 2005, 55, 677–687. [Google Scholar] [CrossRef]
- Chrubasik, S.; Eisenberg, E.; Balan, E.; Weinberger, T.; Luzzati, R.; Conradt, C. Treatment of low back pain exacerbations with willow bark extract: A randomized double-blind study. Am. J. Med. 2000, 109, 9–14. [Google Scholar] [CrossRef]
- Vlachojannis, J.E.; Cameron, M.; Chrubasik, S. A systematic review on the effectiveness of willow bark for musculoskeletal pain. Phyther. Res. 2009, 23, 897–900. [Google Scholar] [CrossRef]
- Shara, M.; Stohs, S.J. Efficacy and Safety of White Willow Bark (Salix alba) Extracts. Phyther. Res. 2015, 29, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.K.; Gurjar, V.; Kumar, S.; Singh, N. Molecular basis for nonspecificity of nonsteroidal anti-inflammatory drugs (NSAIDs). Drug Discov. Today 2015, 20, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Structural and Chemical Biology of the Interaction of Cyclooxygenase with Substrates and Non-Steroidal Anti-Inflammatory Drugs. Chem. Rev. 2020, 120, 7592–7641. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, A.; Coluccia, M. Cyclooxygenase-1 (COX-1) and COX-1 inhibitors in cancer: A review of oncology and medicinal chemistry literature. Pharmaceuticals 2018, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Lolli, M.L.; Cena, C.; Medana, C.; Lazzarato, L.; Morini, G.; Coruzzi, G.; Manarini, S.; Fruttero, R.; Gasco, A. A new class of ibuprofen derivatives with reduced gastrotoxicity. J. Med. Chem. 2001, 44, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Gudis, K.; Sakamoto, C. The role of cyclooxygenase in gastric mucosal protection. Dig. Dis. Sci. 2005, 50, S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef]
- Vitale, P.; Tacconelli, S.; Perrone, M.G.; Malerba, P.; Simone, L.; Scilimati, A.; Lavecchia, A.; Dovizio, M.; Marcantoni, E.; Bruno, A.; et al. Synthesis, pharmacological characterization, and docking analysis of a novel family of diarylisoxazoles as highly selective cyclooxygenase-1 (COX-1) inhibitors. J. Med. Chem. 2013, 56, 4277–4299. [Google Scholar] [CrossRef] [PubMed]
- Migliore, M.; Habrant, D.; Sasso, O.; Albani, C.; Bertozzi, S.M.; Armirotti, A.; Piomelli, D.; Scarpelli, R. Potent multitarget FAAH-COX inhibitors: Design and structure-activity relationship studies. Eur. J. Med. Chem. 2016, 109, 216–237. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Velázquez, C.A.; Abdellatif, K.R.A.; Chowdhury, M.A.; Reisz, J.A.; Dumond, J.F.; King, S.B.; Knaus, E.E. Ethanesulfohydroxamic acid ester prodrugs of nonsteroidal anti-inflammatory drugs (NSAIDs): Synthesis, nitric oxide and nitroxyl release, cyclooxygenase inhibition, anti-inflammatory, and ulcerogenicity index studies. J. Med. Chem. 2011, 54, 1356–1364. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, B.; Li, J.; Liu, H.; Zhang, Y.; Yang, Z.; Liu, W. Design, synthesis, biological evaluation and docking study of novel indole-2-amide as anti-inflammatory agents with dual inhibition of COX and 5-LOX. Eur. J. Med. Chem. 2019, 180, 41–50. [Google Scholar] [CrossRef]
- Gedawy, E.M.; Kassab, A.E.; El Kerdawy, A.M. Design, synthesis and biological evaluation of novel pyrazole sulfonamide derivatives as dual COX-2/5-LOX inhibitors. Eur. J. Med. Chem. 2020, 189, 112066. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, J.R.; Pawiitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Glerse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Selinsky, B.S.; Gupta, K.; Sharkey, C.T.; Loll, P.J. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry 2001, 40, 5172–5180. [Google Scholar] [CrossRef]
- Viegas, A.; Manso, J.; Corvo, M.C.; Marques, M.M.B.; Cabrita, E.J. Binding of ibuprofen, ketorolac, and diclofenac to COX-1 and COX-2 studied by saturation transfer difference NMR. J. Med. Chem. 2011, 54, 8555–8562. [Google Scholar] [CrossRef]
- Uddin, M.J.; Elleman, A.V.; Ghebreselasie, K.; Daniel, C.K.; Crews, B.C.; Nance, K.D.; Huda, T.; Marnett, L.J. Design of fluorine-containing 3,4-diarylfuran-2(5H)-ones as selective COX-1 inhibitors. ACS Med. Chem. Lett. 2014, 5, 1254–1258. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, S.J.; Prickril, B.; Rasooly, A. Mechanisms of Phytonutrient Modulation of Cyclooxygenase-2 (COX-2) and Inflammation Related to Cancer. Nutr. Cancer 2018, 70, 350–375. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, N.; Suemasu, S.; Matoyama, M.; Kimoto, A.; Takeda, M.; Tanaka, K.I.; Ishihara, T.; Katsu, T.; Okamoto, Y.; Otsuka, M.; et al. Properties and synthesis of 2-{2-fluoro (or bromo)-4-[(2-oxocyclopentyl) methyl]phenyl}propanoic acid: Nonsteroidal anti-inflammatory drugs with low membrane permeabilizing and gastric lesion-producing activities. J. Med. Chem. 2010, 53, 7879–7882. [Google Scholar] [CrossRef] [PubMed]
- Smalley, W.E.; Ray, W.A.; Daugherty, J.R.; Griffin, M.R. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am. J. Epidemiol. 1995, 141, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Green, G.A. Understanding NSAIDs: From aspirin to COX-2. Clin. Cornerstone 2001, 3, 50–59. [Google Scholar] [CrossRef]
- Radwan, M.F.; Dalby, K.N.; Kaoud, T.S. Propyphenazone-based analogues as prodrugs and selective cyclooxygenase-2 inhibitors. ACS Med. Chem. Lett. 2014, 5, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Bjarnason, I.; Scarpignato, C.; Holmgren, E.; Olszewski, M.; Rainsford, K.D.; Lanas, A. Mechanisms of Damage to the Gastrointestinal Tract from Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology 2018, 154, 500–514. [Google Scholar] [CrossRef] [Green Version]
- Reddy, M.V.R.; Billa, V.K.; Pallela, V.R.; Mallireddigari, M.R.; Boominathan, R.; Gabriel, J.L.; Reddy, E.P. Design, synthesis, and biological evaluation of 1-(4-sulfamylphenyl)-3-trifluoromethyl-5-indolyl pyrazolines as cyclooxygenase-2 (COX-2) and lipoxygenase (LOX) inhibitors. Bioorg. Med. Chem. 2008, 16, 3907–3916. [Google Scholar] [CrossRef]
- Wired Release Global Ibuprofen Market Projected to Witness Robust Development by 2020–2026. Available online: https://www.pharmiweb.com/press-release/2020-01-17/ibuprofen-market-is-valued-at-2944-million-us-in-2020-is-expected-to-reach-4476-million-us-by-th (accessed on 14 February 2021).
- Harrak, Y.; Casula, G.; Basset, J.; Rosell, G.; Plescia, S.; Raffa, D.; Cusimano, M.G.; Pouplana, R.; Pujol, M.D. Synthesis, anti-inflammatory activity, and in vitro antitumor effect of a novel class of cyclooxygenase inhibitors: 4-(Aryloyl)phenyl methyl sulfones. J. Med. Chem. 2010, 53, 6560–6571. [Google Scholar] [CrossRef] [PubMed]
- Taher, E.S.; Ibrahim, T.S.; Fares, M.; AL-Mahmoudy, A.M.M.; Radwan, A.F.; Orabi, K.Y.; El-Sabbagh, O.I. Novel benzenesulfonamide and 1,2-benzisothiazol-3(2H)-one-1,1-dioxide derivatives as potential selective COX-2 inhibitors. Eur. J. Med. Chem. 2019, 171, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Onder, G.; Pellicciotti, F.; Gambassi, G.; Bernabei, R. NSAID-related psychiatric adverse events: Who is at risk? Drugs 2004, 64, 2619–2627. [Google Scholar] [CrossRef]
- Machado, G.C.; Maher, C.G.; Ferreira, P.H.; Day, R.O.; Pinheiro, M.B.; Ferreira, M.L. Non-steroidal anti-inflammatory drugs for spinal pain: A systematic review and meta-analysis. Ann. Rheum. Dis. 2017, 76, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Chapurlat, R.; Al-Daghri, N.; Herrero-Beaumont, G.; Bruyère, O.; Rannou, F.; Roth, R.; Uebelhart, D.; Reginster, J.Y. Safety of Oral Non-Selective Non-Steroidal Anti-Inflammatory Drugs in Osteoarthritis: What Does the Literature Say? Drugs Aging 2019, 36, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baigent, C.; Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [Google Scholar]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosser, T.; Ricciotti, E.; FitzGerald, G.A. The Cardiovascular Pharmacology of Nonsteroidal Anti-Inflammatory Drugs. Trends Pharmacol. Sci. 2017, 38, 733–748. [Google Scholar] [CrossRef]
- Sisa, M.; Dvorakova, M.; Temml, V.; Jarosova, V.; Vanek, T.; Landa, P. Synthesis, inhibitory activity and in silico docking of dual COX/5-LOX inhibitors with quinone and resorcinol core. Eur. J. Med. Chem. 2020, 204, 112620. [Google Scholar] [CrossRef]
- Fiorucci, S.; Meli, R.; Bucci, M.; Cirino, G. Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem. Pharmacol. 2001, 62, 1433–1438. [Google Scholar] [CrossRef]
- Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar]
- Gaba, M.; Singh, D.; Singh, S.; Sharma, V.; Gaba, P. Synthesis and pharmacological evaluation of novel 5-substituted-1-(phenylsulfonyl)-2-methylbenzimidazole derivatives as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2010, 45, 2245–2249. [Google Scholar] [CrossRef]
- Lednicer, D. The Organic Chemistry of Drug Synthesis; John Wiley and Sons: Hoboken, NJ, USA, 2007; Volume 7, ISBN 9780470107508. [Google Scholar]
- Varrassi, G.; Pergolizzi, J.V.; Dowling, P.; Paladini, A. Ibuprofen Safety at the Golden Anniversary: Are all NSAIDs the Same? A Narrative Review. Adv. Ther. 2020, 37, 61–82. [Google Scholar] [CrossRef] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
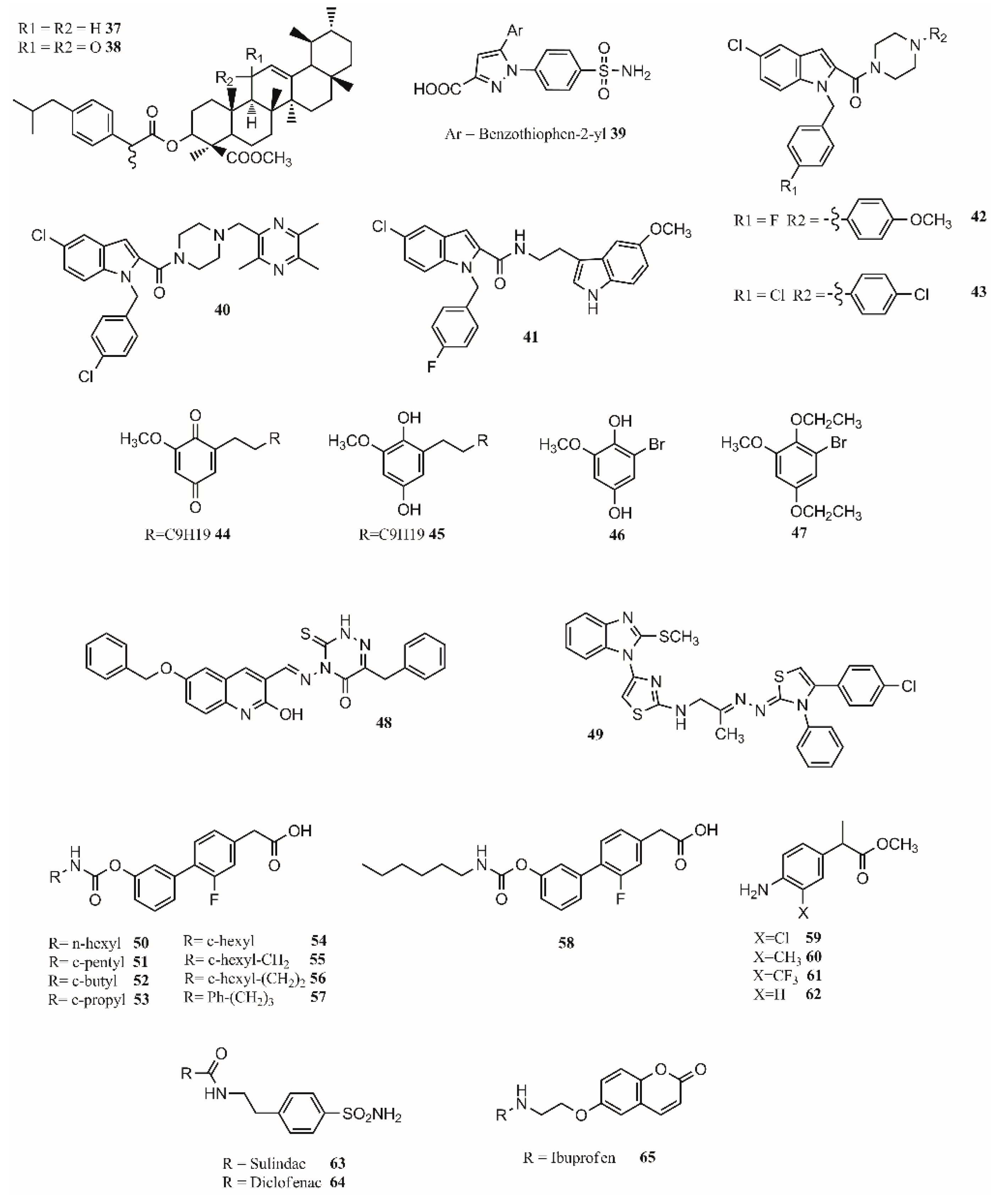
- Kleemiss, F.; Justies, A.; Duvinage, D.; Watermann, P.; Ehrke, E.; Sugimoto, K.; Fugel, M.; Malaspina, L.A.; Dittmer, A.; Kleemiss, T.; et al. Sila-Ibuprofen. J. Med. Chem. 2020, 63, 12614–12622. [Google Scholar] [CrossRef]
- Franz, A.K.; Wilson, S.O. Organosilicon molecules with medicinal applications. J. Med. Chem. 2013, 56, 388–405. [Google Scholar] [CrossRef]
- Sodano, F.; Lazzarato, L.; Rolando, B.; Spyrakis, F.; De Caro, C.; Magliocca, S.; Marabello, D.; Chegaev, K.; Gazzano, E.; Riganti, C.; et al. Paracetamol-Galactose Conjugate: A Novel Prodrug for an Old Analgesic Drug. Mol. Pharm. 2019, 16, 4181–4189. [Google Scholar] [CrossRef]
- Lassalas, P.; Oukoloff, K.; Makani, V.; James, M.; Tran, V.; Yao, Y.; Huang, L.; Vijayendran, K.; Monti, L.; Trojanowski, J.Q.; et al. Evaluation of Oxetan-3-ol, Thietan-3-ol, and Derivatives Thereof as Bioisosteres of the Carboxylic Acid Functional Group. ACS Med. Chem. Lett. 2017, 8, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Su, M.; Hong, J.; La Kim, E.; Moon, H.R.; Chung, H.Y.; Kim, S.; Jung, J.H. Design of balanced COX inhibitors based on anti-inflammatory and/or COX-2 inhibitory ascidian metabolites. Eur. J. Med. Chem. 2019, 180, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Neves, B.J.; Braga, R.C.; Melo-Filho, C.C.; Moreira-Filho, J.T.; Muratov, E.N.; Andrade, C.H. QSAR-based virtual screening: Advances and applications in drug discovery. Front. Pharmacol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeon, S.; Anuwongcharoen, N.; Shoombuatong, W.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.S.; Nantasenamat, C. Probing the origins of human acetylcholinesterase inhibition via QSAR modeling and molecular docking. PeerJ 2016, 4, e2322. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.C.; Márquez, E.A.; Mora, J.R. Molecular modeling studies of bromopyrrole alkaloids as potential antimalarial compounds: A DFT approach. Med. Chem. Res. 2018, 27, 844–856. [Google Scholar] [CrossRef]
- Flores-Sumoza, M.; Alcázar, J.J.; Márquez, E.; Mora, J.R.; Lezama, J.; Puello, E. Classical QSAR and docking simulation of 4-pyridone derivatives for their antimalarial activity. Molecules 2018, 23, 3166. [Google Scholar] [CrossRef] [Green Version]
- Imran, S.; Taha, M.; Ismail, N.H.; Kashif, S.M.; Rahim, F.; Jamil, W.; Hariono, M.; Yusuf, M.; Wahab, H. Synthesis of novel flavone hydrazones: In-vitro evaluation of α-glucosidase inhibition, QSAR analysis and docking studies. Eur. J. Med. Chem. 2015, 105, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.R.; Márquez, E.A.; Calle, L. Computational molecular modelling of N-cinnamoyl and hydroxycinnamoyl amides as potential α-glucosidase inhibitors. Med. Chem. Res. 2018, 27, 2214–2223. [Google Scholar] [CrossRef]
- Lakhlili, W.; Yasri, A.; Ibrahimi, A. Structure-activity relationships study of mTOR kinase inhibition using QSAR and structure-based drug design approaches. OncoTargets Ther. 2016, 9, 7345–7353. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Cui, L.; Song, F.; Luan, M.; Ji, J.; Si, H.; Duan, Y.; Zhai, H. 3D-QSAR and Molecular Docking Studies on Design Anti-Prostate Cancer Curcumin Analogues. Curr. Comput. Aided. Drug Des. 2018, 16, 245–256. [Google Scholar] [CrossRef]
- Cabrera, N.; Mora, J.R.; Marquez, E.A. Computational Molecular Modeling of Pin1 Inhibition Activity of Quinazoline, Benzophenone, and Pyrimidine Derivatives. J. Chem. 2019, 2019, 2954250. [Google Scholar] [CrossRef] [Green Version]
- Suvannang, N.; Preeyanon, L.; Malik, A.A.; Schaduangrat, N.; Shoombuatong, W.; Worachartcheewan, A.; Tantimongcolwat, T.; Nantasenamat, C. Probing the origin of estrogen receptor alpha inhibition: Via large-scale QSAR study. RSC Adv. 2018, 8, 11344–11356. [Google Scholar] [CrossRef] [Green Version]
- Misaka, E.; Yamaguchi, T.; Iizuka, Y. Anti-inflammatory, analgesic and antipyretic activities of sodium 2-[4-(2-oxocyclopentan-1-ylmethyl) phenyl] propionate dihydrate (CS-600). Pharmacometrics 1981, 21, 753–771. [Google Scholar]
- Kawano, S.; Tsuji, S.; Hayashi, N.; Takei, Y.; Nagano, K.; Fusamoto, H.; Kamada, T. Effects of loxoprofen sodium, a newly synthesized non-steroidal anti-inflammatory drug, and indomethacin on gastric mucosal haemodynamics in the human. J. Gastroenterol. Hepatol. 1995, 10, 81–85. [Google Scholar] [CrossRef]
- Yamakawa, N.; Suemasu, S.; Okamoto, Y.; Tanaka, K.I.; Ishihara, T.; Asano, T.; Miyata, K.; Otsuka, M.; Mizushima, T. Synthesis and biological evaluation of derivatives of 2-{2-fluoro-4-[(2-oxocyclopentyl)methyl]phenyl}propanoic acid: Nonsteroidal anti-inflammatory drugs with low gastric ulcerogenic activity. J. Med. Chem. 2012, 55, 5143–5150. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.; Cai, D.; Damodaran, K.; Gomez, R.F.; Keck, J.G.; Laborde, E.; Lum, R.T.; Macke, T.J.; Martin, G.; Schow, S.R.; et al. Novel cyclooxygenase-1 inhibitors discovered using affinity fingerprints. J. Med. Chem. 2004, 47, 4875–4880. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.Y. Perspectives in Nonsteroidal Anti-inflammatory Agents. Angew. Chem. Int. Ed. Engl. 1972, 11, 460–472. [Google Scholar] [CrossRef]
- Curtis, E.; Fuggle, N.; Shaw, S.; Spooner, L.; Ntani, G.; Parsons, C.; Corp, N.; Honvo, G.; Baird, J.; Maggi, S.; et al. Safety of Cyclooxygenase-2 Inhibitors in Osteoarthritis: Outcomes of a Systematic Review and Meta-Analysis. Drugs Aging 2019, 36, 25–44. [Google Scholar] [CrossRef] [Green Version]
- Black, W.C.; Brideau, C.; Chan, C.C.; Charleson, S.; Chauret, N.; Claveau, D.; Ethier, D.; Gordon, R.; Greig, G.; Guay, J.; et al. 2,3-diarylcyclopentenones as orally active, highly selective cyclooxygenase-2 inhibitors. J. Med. Chem. 1999, 42, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Khanna, I.K.; Yu, Y.; Huff, R.M.; Weier, R.M.; Xu, X.; Koszyk, F.J.; Collins, P.W.; Cogburn, J.N.; Isakson, P.C.; Koboldt, C.M.; et al. Selective cyclooxygenase-2 inhibitors: Heteroaryl modified 1,2-diarylimidazoles are potent, orally active antiinflammatory agents. J. Med. Chem. 2000, 43, 3168–3185. [Google Scholar] [CrossRef]
- Leblanc, Y.; Gauthier, J.Y.; Ethier, D.; Guay, J.; Mancini, J.; Riendeau, D.; Tagari, P.; Vickers, P.; Wong, E.; Prasit, P. Synthesis and biological evaluation of 2,3-diarylthiophenes as selective Cox-2 and Cox-1 inhibitors. Bioorg. Med. Chem. Lett. 1995, 5, 2123–2128. [Google Scholar] [CrossRef]
- Talley, J.J.; Bertenshaw, S.R.; Brown, D.L.; Carter, J.S.; Graneto, M.J.; Koboldt, C.M.; Masferrer, J.L.; Norman, B.H.; Rogier, D.J.; Zweifel, B.S.; et al. 4,5-diaryloxazole inhibitors of cyclooxygenase-2 (COX-2). Med. Res. Rev. 1999, 19, 199–208. [Google Scholar] [CrossRef]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Docter, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)- 3(trifluoromethyl)-1h-pyrazol-1-yl]benzenesulfonamide (sc-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Nicoll-Griffith, D.A.; Yergey, J.A.; Trimble, L.A.; Silva, J.M.; Li, C.; Chauret, N.; Gauthier, J.Y.; Grimm, E.; Léger, S.; Roy, P.; et al. Synthesis, characterization, and activity of metabolites derived from the cyclooxygenase-2 inhibitor rofecoxib (MK-0966, Vioxx(TM)). Bioorg. Med. Chem. Lett. 2000, 10, 2683–2686. [Google Scholar] [CrossRef]
- Ahmed, E.M.; Kassab, A.E.; El-Malah, A.A.; Hassan, M.S.A. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019, 171, 25–37. [Google Scholar] [CrossRef]
- Palomer, A.; Cabré, F.; Pascual, J.; Campos, J.; Trujillo, M.A.; Entrena, A.; Gallo, M.A.; García, L.; Mauleón, D.; Espinosa, A. Identification of novel cyclooxygenase-2 selective inhibitors using pharmacophore models. J. Med. Chem. 2002, 45, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Rowlinson, S.W.; Crews, B.C.; Marnett, L.J. Amide derivatives of meclofenamic acid as selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 521–524. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Marnett, A.B.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J. Med. Chem. 2000, 43, 2860–2870. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Xu, S.; Crews, B.C.; Aleem, A.M.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J. Harmaline Analogs as Substrate-Selective Cyclooxygenase-2 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1881–1885. [Google Scholar] [CrossRef]
- Prusakiewicz, J.J.; Duggan, K.C.; Rouzer, C.A.; Marnett, L.J. Differential sensitivity and mechanism of inhibition of COX-2 oxygenation of arachidonic acid and 2-arachidonoylglycerol by ibuprofen and mefenamic acid. Biochemistry 2009, 48, 7353–7355. [Google Scholar] [CrossRef] [Green Version]
- Windsor, M.A.; Hermanson, D.J.; Kingsley, P.J.; Xu, S.; Crews, B.C.; Ho, W.; Keenan, C.M.; Banerjee, S.; Sharkey, K.A.; Marnett, L.J. Substrate-selective inhibition of cyclooxygenase-2: Development and evaluation of achiral profen probes. ACS Med. Chem. Lett. 2012, 3, 759–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thipparaboina, R.; Kumar, D.; Chavan, R.B.; Shastri, N.R. Multidrug co-crystals: Towards the development of effective therapeutic hybrids. Drug Discov. Today 2016, 21, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Godman, B.; McCabe, H.; Leong, T.D.; Mueller, D.; Martin, A.P.; Hoxha, I.; Mwita, J.C.; Rwegerera, G.M.; Massele, A.; de Oliveira Costa, J.; et al. Fixed dose drug combinations–are they pharmacoeconomically sound? Findings and implications especially for lower- and middle-income countries. Exp. Rev. Pharmacoecon. Outcomes Res. 2020, 20, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.L.C.S.; Fernandes, R.P.; Charpentier, M.D.; ter Horst, J.H.; Caires, F.J.; Chorilli, M. Co-crystals of non-steroidal anti-inflammatory drugs (NSAIDs): Insight toward formation, methods, and drug enhancement. Particuology 2021, 58, 227–241. [Google Scholar] [CrossRef]
- Kumari, P.; Singh, P.; Kaur, J.; Bhatti, R. Design, Synthesis, and Activity Evaluation of Stereoconfigured Tartarate Derivatives as Potential Anti-inflammatory Agents In Vitro and In Vivo. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Abdelazeem, A.H.; El-Din, A.G.S.; Arab, H.H.; El-Saadi, M.T.; El-Moghazy, S.M.; Amin, N.H. Design, synthesis and anti-inflammatory/analgesic evaluation of novel di-substituted urea derivatives bearing diaryl-1,2,4-triazole with dual COX-2/sEH inhibitory activities. J. Mol. Struct. 2021, 1240, 130565. [Google Scholar] [CrossRef]
- Liu, K.; Li, D.; Zheng, W.; Shi, M.; Chen, Y.; Tang, M.; Yang, T.; Zhao, M.; Deng, D.; Zhang, C.; et al. Discovery, Optimization, and Evaluation of Quinazolinone Derivatives with Novel Linkers as Orally Efficacious Phosphoinositide-3-Kinase Delta Inhibitors for Treatment of Inflammatory Diseases. J. Med. Chem. 2021. [Google Scholar] [CrossRef]
- Melagraki, G.; Afantitis, A.; Igglessi-Markopoulou, O.; Detsi, A.; Koufaki, M.; Kontogiorgis, C.; Hadjipavlou-Litina, D.J. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur. J. Med. Chem. 2009, 44, 3020–3026. [Google Scholar] [CrossRef]
- Abdellatif, K.R.A.; Chowdhury, M.A.; Dong, Y.; Das, D.; Yu, G.; Velázquez, C.A.; Suresh, M.R.; Knaus, E.E. Dinitroglyceryl and diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of aspirin, indomethacin and ibuprofen: Synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett. 2009, 19, 3014–3018. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.A.; Abdellatif, K.R.A.; Dong, Y.; Das, D.; Suresh, M.R.; Knaus, E.E. Synthesis of celecoxib analogues possessing a N-difluoromethyl-1,2- dihydropyrid-2-one 5-lipoxygenase pharmacophore: Biological evaluation as dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity. J. Med. Chem. 2009, 52, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Al-Ostoot, F.H.; Zabiulla; Salah, S.; Khanum, S.A. Recent investigations into synthesis and pharmacological activities of phenoxy acetamide and its derivatives (chalcone, indole and quinoline) as possible therapeutic candidates. J. Iran. Chem. Soc. 2021. [Google Scholar] [CrossRef]
- Jacob, Í.T.T.; Gomes, F.O.S.; de Miranda, M.D.S.; de Almeida, S.M.V.; da Cruz-Filho, I.J.; Peixoto, C.A.; da Silva, T.G.; Moreira, D.R.M.; de Melo, C.M.L.; de Oliveira, J.F.; et al. Anti-inflammatory activity of novel thiosemicarbazone compounds indole-based as COX inhibitors. Pharmacol. Rep. 2021, 73, 907–925. [Google Scholar] [CrossRef]
- Qian, S.; Huang, Y.; Li, J.; Zhang, Y.; Zhang, B.; Jin, F. Synthesis and Anti-proliferative Activity of Indole-2-amide Derivatives as Cyclooxygenase-2/5-lipoxygenase (COX-2/5-LOX) Dual Inhibitors. Chin. J. Org. Chem. 2021, 41, 1631–1638. [Google Scholar] [CrossRef]
- Ghanim, A.M.; Rezq, S.; Ibrahim, T.S.; Romero, D.G.; Kothayer, H. Novel 1,2,4-triazine-quinoline hybrids: The privileged scaffolds as potent multi-target inhibitors of LPS-induced inflammatory response via dual COX-2 and 15-LOX inhibition. Eur. J. Med. Chem. 2021, 219, 113457. [Google Scholar] [CrossRef]
- Maghraby, M.T.E.; Abou-Ghadir, O.M.F.; Abdel-Moty, S.G.; Ali, A.Y.; Salem, O.I.A. Novel class of benzimidazole-thiazole hybrids: The privileged scaffolds of potent anti-inflammatory activity with dual inhibition of cyclooxygenase and 15-lipoxygenase enzymes. Bioorg. Med. Chem. 2020, 28, 115403. [Google Scholar] [CrossRef]
- Berrino, E.; Carradori, S.; Angeli, A.; Carta, F.; Supuran, C.T.; Guglielmi, P.; Coletti, C.; Paciotti, R.; Schweikl, H.; Maestrelli, F.; et al. Dual carbonic anhydrase ix/xii inhibitors and carbon monoxide releasing molecules modulate LPS-mediated inflammation in mouse macrophages. Antioxidants 2021, 10, 56. [Google Scholar] [CrossRef]
- Akgul, O.; Di Cesare Mannelli, L.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Alfawaz Altamimi, A.S.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of Novel Nonsteroidal Anti-Inflammatory Drugs and Carbonic Anhydrase Inhibitors Hybrids (NSAIDs-CAIs) for the Management of Rheumatoid Arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef]
- Bua, S.; Di Cesare Mannelli, L.; Vullo, D.; Ghelardini, C.; Bartolucci, G.; Scozzafava, A.; Supuran, C.T.; Carta, F. Design and Synthesis of Novel Nonsteroidal Anti-Inflammatory Drugs and Carbonic Anhydrase Inhibitors Hybrids (NSAIDs-CAIs) for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2017, 60, 1159–1170. [Google Scholar] [CrossRef]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955-2007. Arthritis Rheum. 2010, 62, 1576–1582. [Google Scholar] [CrossRef] [Green Version]
- Placha, D.; Jampilek, J. Chronic inflammatory diseases, anti-inflammatory agents and their delivery nanosystems. Pharmaceutics 2021, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.M.; Angeli, A.; El-Azab, A.S.; Hammouda, M.E.A.; El-Sherbeny, M.A.; Supuran, C.T. Synthesis and anti-inflammatory activity of sulfonamides and carboxylates incorporating trimellitimides: Dual cyclooxygenase/carbonic anhydrase inhibitory actions. Bioorg. Chem. 2019, 84, 260–268. [Google Scholar] [CrossRef]
- Vučković, S.; Savić Vujović, K.; Srebro, D.; Medić, B.; Ilic-Mostic, T. Prevention of Renal Complications Induced by Non- Steroidal Anti-Inflammatory Drugs. Curr. Med. Chem. 2016, 23, 1953–1964. [Google Scholar]
- Vučković, S.; Srebro, D.; Vujović, K.S.; Vučetić, Č.; Prostran, M. Cannabinoids and pain: New insights from old molecules. Front. Pharmacol. 2018, 9, 1259. [Google Scholar] [CrossRef] [Green Version]
- Baron, E.P. Medicinal Properties of Cannabinoids, Terpenes, and Flavonoids in Cannabis, and Benefits in Migraine, Headache, and Pain: An Update on Current Evidence and Cannabis Science. Headache 2018, 58, 1139–1186. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Johnston, B.; Englesakis, M.; Moulin, D.E.; Bhatia, A. Selective Cannabinoids for Chronic Neuropathic Pain: A Systematic Review and Meta-Analysis. Anesth. Analg. 2017, 125, 1638–1652. [Google Scholar] [CrossRef]
- Khurana, L.; Mackie, K.; Piomelli, D.; Kendall, D.A. Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology 2017, 124, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Slivicki, R.A.; Xu, Z.; Kulkarni, P.M.; Pertwee, R.G.; Mackie, K.; Thakur, G.A.; Hohmann, A.G. Positive Allosteric Modulation of Cannabinoid Receptor Type 1 Suppresses Pathological Pain Without Producing Tolerance or Dependence. Biol. Psychiatry 2018, 84, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Starowicz, K.; Finn, D.P. Cannabinoids and Pain: Sites and Mechanisms of Action. In Advances in Pharmacology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 80, pp. 437–475. [Google Scholar]
- Bai, R.; Yao, C.; Zhong, Z.; Ge, J.; Bai, Z.; Ye, X.; Xie, T.; Xie, Y. Discovery of natural anti-inflammatory alkaloids: Potential leads for the drug discovery for the treatment of inflammation. Eur. J. Med. Chem. 2021, 213, 113165. [Google Scholar] [CrossRef]
- Andre, C.M.; Hausman, J.F.; Guerriero, G. Cannabis sativa: The plant of the thousand and one molecules. Front. Plant Sci. 2016, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Friedman, D.; French, J.A.; Maccarrone, M. Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurol. 2019, 18, 504–512. [Google Scholar] [CrossRef]
- Rahn, E.J.; Hohmann, A.G. Cannabinoids as Pharmacotherapies for Neuropathic Pain: From the Bench to the Bedside. Neurotherapeutics 2009, 6, 713–737. [Google Scholar] [CrossRef]
- Hill, K.P.; Palastro, M.D.; Johnson, B.; Ditre, J.W. Cannabis and Pain: A Clinical Review. Cannabis Cannabinoid Res. 2017, 2, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, P.; Hurst, D.P.; Reggio, P.H. Molecular Targets of the Phytocannabinoids: A Complex Picture. Prog. Chem. Org. Nat. Prod. 2017, 103, 103–131. [Google Scholar]
- Bakas, T.; van Nieuwenhuijzen, P.S.; Devenish, S.O.; McGregor, I.S.; Arnold, J.C.; Chebib, M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol. Res. 2017, 119, 358–370. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar]
- Romero-Sandoval, E.A.; Kolano, A.L.; Alvarado-Vázquez, P.A. Cannabis and Cannabinoids for Chronic Pain. Curr. Rheumatol. Rep. 2017, 19, 67. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockings, E.; Campbell, G.; Hall, W.D.; Nielsen, S.; Zagic, D.; Rahman, R.; Murnion, B.; Farrell, M.; Weier, M.; Degenhardt, L. Cannabis and cannabinoids for the treatment of people with chronic noncancer pain conditions: A systematic review and meta-analysis of controlled and observational studies. Pain 2018, 159, 1932–1954. [Google Scholar] [CrossRef]
- Stott, C.G.; White, L.; Wright, S.; Wilbraham, D.; Guy, G.W. A phase i study to assess the effect of food on the single dose bioavailability of the THC/CBD oromucosal spray. Eur. J. Clin. Pharmacol. 2013, 69, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Abrams, D.I.; Guzman, M. Cannabis in Cancer Care. Clin. Pharmacol. Ther. 2015, 97, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Kiran Vemuri, V.; Makriyannis, A. Medicinal Chemistry of Cannabinoids. Clin. Pharmacol. Ther. 2015, 97, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.E.; Ware, M.A. Cannabinoids for the Treatment of Chronic Non-Cancer Pain: An Updated Systematic Review of Randomized Controlled Trials. J. Neuroimmune Pharmacol. 2015, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Field, M.J.; Cox, P.J.; Stott, E.; Melrose, H.; Offord, J.; Su, T.Z.; Bramwell, S.; Corradini, L.; England, S.; Winks, J.; et al. Identification of the α2-δ-1 subunit of voltage-calcium calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc. Natl. Acad. Sci. USA 2006, 103, 17537–17542. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.A.; Straube, S.; Wiffen, P.J.; Derry, S.; McQuay, H.J.; Moore, M. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Bouhassira, D.; Lantéri-Minet, M.; Attal, N.; Laurent, B.; Touboul, C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008, 136, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Dworkin, R.H.; Kirkpatrick, P. Pregabalin. Nat. Rev. Drug Discov. 2005, 4, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Gajraj, N.M. Pregabalin: Its pharmacology and use in pain management. Anesth. Analg. 2007, 105, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.P. Mechanisms of analgesia by gabapentin and pregabalin—Calcium channel α2-δ [Cavα2-δ] ligands. Pain 2009, 142, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Kavoussi, R. Pregabalin: From molecule to medicine. Eur. Neuropsychopharmacol. 2006, 16, S128–S133. [Google Scholar] [CrossRef] [PubMed]
- Sills, G.J. The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 2006, 6, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Micó, J.-A.; Prieto, R. Elucidating the Mechanism of Action of Pregabalin. CNS Drugs 2012, 26, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Ben-Menachem, E. Pregabalin pharmacology and its relevance to clinical practice. Epilepsia 2004, 45, 13–18. [Google Scholar] [CrossRef]
- Armstrong, C.T.; Mason, P.E.; Anderson, J.L.R.; Dempsey, C.E. Arginine side chain interactions and the role of arginine as a gating charge carrier in voltage sensitive ion channels. Sci. Rep. 2016, 6, 21759. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.P.; Angelotti, T.; Fauman, E. Pharmacology and mechanism of action of pregabalin: The calcium channel α2-δ (alpha2-delta) subunit as a target for antiepileptic drug discovery. Epilepsy Res. 2007, 73, 137–150. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition (%) |
|---|---|
| 4 | 54.76 |
| 5 | 51.16 |
| 6 | 66.66 |
| 7 | 34.52 |
| 8 | 64.28 |
| 9 | 80.95 |
| 10 | 67.85 |
| 11 | 76.19 |
| 12 | 65.47 |
| 13 | 66.66 |
| 14 | 64.28 |
| Indomethacin | 84.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuesta, S.A.; Meneses, L. The Role of Organic Small Molecules in Pain Management. Molecules 2021, 26, 4029. https://doi.org/10.3390/molecules26134029
Cuesta SA, Meneses L. The Role of Organic Small Molecules in Pain Management. Molecules. 2021; 26(13):4029. https://doi.org/10.3390/molecules26134029
Chicago/Turabian StyleCuesta, Sebastián A., and Lorena Meneses. 2021. "The Role of Organic Small Molecules in Pain Management" Molecules 26, no. 13: 4029. https://doi.org/10.3390/molecules26134029
APA StyleCuesta, S. A., & Meneses, L. (2021). The Role of Organic Small Molecules in Pain Management. Molecules, 26(13), 4029. https://doi.org/10.3390/molecules26134029