
A Compendium of the Most Promising Synthesized Organic Compounds against Several Fusarium oxysporum Species: Synthesis, Antifungal Activity, and Perspectives




Abstract
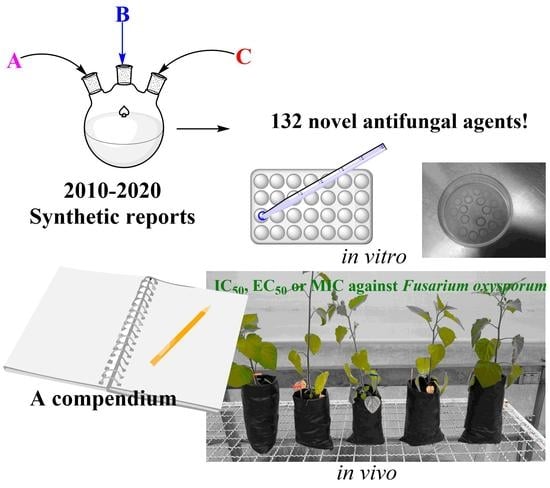
1. Introduction
2. Compendium of the Organic Molecules with the Highest Reported Antifungal Activity against FOX Species
3. Synthetic Methods for Highly Active Compounds against FOX Species
3.1. Open-Chain and Homoaromatic Core Molecules
3.2. Heterocyclic Ring Core Molecules
3.2.1. Monocyclic Systems
3.2.2. Polyheterocyclic Systems
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef]
- Flood, J. A review of Fusarium milt of oil palm caused by Fusarium oxysporum f. sp. elaeidis. Phytopathology 2007, 96, 660–662. [Google Scholar] [CrossRef]
- De Sain, M.; Rep, M. The role of pathogen-secreted proteins in fungal vascular wilt diseases. Int. J. Mol. Sci. 2015, 16, 23970–23993. [Google Scholar] [CrossRef]
- Ploetz, R.C. Management of Fusarium wilt of banana: A review with special reference to tropical race 4. Crop Prot. 2015, 73, 1–9. [Google Scholar] [CrossRef]
- Shirani Bidabadi, S.; Sijun, Z. Banana Fusarium wilt (Fusarium oxysporum f. sp. cubense) control and resistance, in the context of developing wilt-resistant bananas within sustainable production systems. Hortic. Plant J. 2018, 4, 208–218. [Google Scholar]
- Bubici, G.; Kaushal, M.; Prigigallo, M.I.; Cabanás, C.G.L.; Mercado-Blanco, J. Biological control agents against Fusarium wilt of banana. Front. Microbiol. 2019, 10, 616. [Google Scholar] [CrossRef] [PubMed]
- Pegg, K.G.; Moore, N.Y.; Bentley, S. Fusarium wilt of banana in Australia: A review. Aust. J. Agric. Res. 1996, 47, 637–650. [Google Scholar] [CrossRef]
- Ghag, S.B.; Shekhawat, U.K.S.; Ganapathi, T.R. Fusarium wilt of banana: Biology, epidemiology and management. Int. J. Pest Manag. 2015, 61, 250–263. [Google Scholar] [CrossRef]
- Srinivas, C.; Nirmala Devi, D.; Narasimha Murthy, K.; Mohan, C.D.; Lakshmeesha, T.R.; Singh, B.P.; Kalagatur, N.K.; Niranjana, S.R.; Hashem, A.; Alqarawi, A.A.; et al. Fusarium oxysporum f. sp. lycopersici causal agent of vascular wilt disease of tomato: Biology to diversity—A review. Saudi J. Biol. Sci. 2019, 26, 1315–1324. [Google Scholar] [CrossRef]
- Jeger, M.; Bragard, C.; Caffier, D.; Candresse, T.; Chatzivassiliou, E.; Dehnen-Schmutz, K.; Gilioli, G.; Grégoire, J.C.; Jaques Miret, J.A.; MacLeod, A.; et al. Pest categorisation of Fusarium oxysporum f. sp. albedinis. EFSA J. 2018, 16, 1–24. [Google Scholar]
- Lecomte, C.; Alabouvette, C.; Edel-Hermann, V.; Robert, F.; Steinberg, C. Biological control of ornamental plant diseases caused by Fusarium oxysporum: A review. Biol. Control 2016, 101, 17–30. [Google Scholar] [CrossRef]
- Sanogo, S.; Zhang, J. Resistance sources, resistance screening techniques and disease management for Fusarium wilt in cotton. Euphytica 2016, 207, 255–271. [Google Scholar] [CrossRef]
- Osorio-Guarín, J.A.; Enciso-Rodríguez, F.E.; González, C.; Fernández-Pozo, N.; Mueller, L.A.; Barrero, L.S. Association analysis for disease resistance to Fusarium oxysporum in cape gooseberry (Physalis peruviana L). BMC Genom. 2016, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Aybeke, M. Fusarium infection causes genotoxic disorders and antioxidant-based damages in Orobanche spp. Microbiol. Res. 2017, 201, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Munkvold, G. Fusarium species and their associated mycotoxins. In Mycotoxigenic Fungi: Methods and Protocols, Methods in Molecular Biology; Springer: Cham Switzerland, 2017; Volume 1542, pp. 51–106. ISBN 9781493967056. [Google Scholar]
- Singh, V.K.; Singh, H.B.; Upadhyay, R.S. Role of fusaric acid in the development of ‘Fusarium wilt’ symptoms in tomato: Physiological, biochemical and proteomic perspectives. Plant Physiol. Biochem. 2017, 118, 320–332. [Google Scholar] [CrossRef]
- Gupta, A.; Baran, R.; Summerbell, R. Fusarium infections of the skin. Curr. Opin. Infect. Dis. 2000, 13, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Guarro, J. Fusariosis, a complex infection caused by a high diversity of fungal species refractory to treatment. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Arnoni, M.V.; Paula, C.R.; Auler, M.E.; Simões, C.C.N.; Nakano, S.; Szeszs, M.W.; Melhem, M.D.S.C.; Pereira, V.B.R.; Garces, H.G.; Bagagli, E.; et al. Infections caused by Fusarium species in pediatric cancer patients and review of published literature. Mycopathologia 2018, 183, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Dignani, M.C.; Anaissie, E. Human fusariosis. Clin. Microbiol. Infect. 2004, 10, 67–75. [Google Scholar] [CrossRef]
- Vinale, F.; Sivasithamparam, K.; Ghisalberti, E.L.; Marra, R.; Woo, S.L.; Lorito, M. Trichoderma-plant-pathogen interactions. Soil Biol. Biochem. 2008, 40, 1–10. [Google Scholar] [CrossRef]
- Gajera, H.; Domadiya, R.; Patel, S.; Kapopara, M.; Golakiya, B. Molecular mechanism of Trichoderma as bio-control agents against phytopathogen system-a review. Curr. Res. Microbiol. Biotechnol. 2013, 1, 133–142. [Google Scholar]
- Antoun, H.; Prévost, D. Ecology of plant growth promoting rhizobacteria. In PGPR: Biocontrol and Biofertilization; Springer: Dordrecht, The Netherlands, 2005; pp. 1–38. ISBN 1402040024. [Google Scholar]
- Edel-Hermann, V.; Lecomte, C. Current status of Fusarium oxysporum formae speciales and races. Phytopathology 2019, 109, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Rep, M.; Kistler, H.C. The genomic organization of plant pathogenicity in Fusarium species. Curr. Opin. Plant Biol. 2010, 13, 420–426. [Google Scholar] [CrossRef]
- Dita, M.; Barquero, M.; Heck, D.; Mizubuti, E.S.G.; Staver, C.P. Fusarium wilt of banana: Current knowledge on epidemiology and research needs toward sustainable disease management. Front. Plant Sci. 2018, 9, 1468. [Google Scholar] [CrossRef]
- Oumouloud, A.; El-Otmani, M.; Chikh-Rouhou, H.; Claver, A.G.; Torres, R.G.; Perl-Treves, R.; Álvarez, J.M. Breeding melon for resistance to Fusarium wilt: Recent developments. Euphytica 2013, 192, 155–169. [Google Scholar] [CrossRef]
- Ngowi, A.V.F.; Mbise, T.J.; Ijani, A.S.M.; London, L.; Ajayi, O.C. Smallholder vegetable farmers in Northern Tanzania: Pesticides use practices, perceptions, cost and health effects. Crop Prot. 2007, 26, 1617–1624. [Google Scholar] [CrossRef]
- Dias, M.C. Phytotoxicity: An overview of the physiological responses of plants exposed to fungicides. J. Bot. 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Lushchak, V.I.; Matviishyn, T.M.; Husak, V.V.; Storey, J.M.; Storey, K.B. Pesticide toxicity: A mechanistic approach. EXCLI J. 2018, 17, 1101–1136. [Google Scholar] [PubMed]
- Cedergreen, N.; Dalhoff, K.; Li, D.; Gottardi, M.; Kretschmann, A.C. Can toxicokinetic and toxicodynamic modeling be used to understand and predict synergistic interactions between chemicals? Environ. Sci. Technol. 2017, 51, 14379–14389. [Google Scholar] [CrossRef]
- Isaac, G.; Abu-Tahon, M. In vitro antifugal activity of medicinal plant extract against Fusarium oxysporum f. sp. lycopersici race 3 the causal agent of tomato wilt. Acta Biol. Hung. 2014, 65, 107–118. [Google Scholar] [CrossRef]
- Arif, T.; Bhosale, J.D.; Kumar, N.; Mandal, T.K.; Bendre, R.S.; Lavekar, G.S.; Dabur, R. Natural products—Antifungal agents derived from plants. J. Asian Nat. Prod. Res. 2009, 11, 621–638. [Google Scholar] [CrossRef] [PubMed]
- Demetzos, C.; Dimas, K.S. Labdane-type diterpenes: Chemistry and biological activity. Stud. Nat. Prod. Chem. 2001, 25, 235–292. [Google Scholar]
- González, M.A.; Mancebo-Aracil, J.; Tangarife-Castaño, V.; Agudelo-Goméz, L.; Zapata, B.; Mesa-Arango, A.; Betancur-Galvis, L. Synthesis and biological evaluation of (+)-labdadienedial, derivatives and precursors from (+)-sclareolide. Eur. J. Med. Chem. 2010, 45, 4403–4408. [Google Scholar] [CrossRef]
- Maheshwari, R.K.; Singh, A.K.; Gaddipati, J.; Srimal, R.C. Multiple biological activities of curcumin: A short review. Life Sci. 2006, 78, 2081–2087. [Google Scholar] [CrossRef]
- Radi, S.; Tighadouini, S.; Feron, O.; Riant, O.; Bouakka, M.; Benabbes, R.; Mabkhot, Y.N. Synthesis of novel β-keto-enol derivatives tethered pyrazole, pyridine and furan as new potential antifungal and anti-breast cancer agents. Molecules 2015, 20, 20186–20194. [Google Scholar] [CrossRef]
- Yang, H.; Du, Z.; Wang, W.; Song, M.; Sanidad, K.Z. Structure and activity relationship of curcumin: Role of methoxy group in anti-inflammatory and anti-colitis effects of curcumin. J. Agric. Food Chem. 2017, 65, 4509–4515. [Google Scholar] [CrossRef]
- Quiroga, D.; Becerra, L.D.; Coy-Barrera, E. Ultrasound-assisted synthesis, antifungal activity against Fusarium oxysporum, and three-dimensional quantitative structure−activity relationship of N,S-Dialkyl dithiocarbamates derived from 2-amino acids. ACS Omega 2019, 4, 13710–13720. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, Y. A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef]
- Dutta, S.; Saha, A. Iodine mediated direct coupling of benzylic alcohols with dithiocarbamate anions: An easy access of S-benzyl dithiocarbamate esters under neat reaction condition. Tetrahedron Lett. 2020, 61, 152382. [Google Scholar] [CrossRef]
- Lekkala, R.; Lekkala, R.; Moku, B.; Rakesh, K.P.; Qin, H.L. Applications of sulfuryl fluoride (SO2F2) in chemical transformations. Org. Chem. Front. 2019, 6, 3490–3516. [Google Scholar] [CrossRef]
- Ravindar, L.; Bukhari, S.N.A.; Rakesh, K.P.; Manukumar, H.M.; Vivek, H.K.; Mallesha, N.; Xie, Z.Z.; Qin, H.L. Aryl fluorosulfate analogues as potent antimicrobial agents: SAR, cytotoxicity and docking studies. Bioorg. Chem. 2018, 81, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Krasnova, L.; Finn, M.G.; Barry Sharpless, K. Sulfur(VI) fluoride exchange (SuFEx): Another good reaction for click chemistry. Angew. Chem. Int. Ed. 2014, 53, 9430–9448. [Google Scholar] [CrossRef] [PubMed]
- Baggio, C.; Udompholkul, P.; Gambini, L.; Salem, A.F.; Jossart, J.; Perry, J.J.P.; Pellecchia, M. Aryl-fluorosulfate-based Lysine covalent Pan-Inhibitors of apoptosis protein (IAP) antagonists with cellular efficacy. J. Med. Chem. 2019, 62, 9188–9200. [Google Scholar] [CrossRef] [PubMed]
- Pochampally, J.; Valeru, A.; Macha, R.; Bhavani, A.K.D.; Nalla, U.; Tigulla, P.; Gandu, B.; Gangagnirao, A. Synthesis and molecular modeling studies of novel tert-butyl 2, 4- disubstituted carboxamido phenylcarbamate derivatives and evaluation of their antimicrobial activity. Pharma Chem. 2014, 6, 61–76. [Google Scholar]
- Kumar, S.V.; Ma, D. Synthesis of N-(Hetero)aryl carbamates via CuI/MNAO catalyzed cross-coupling of (Hetero)aryl halides with potassium cyanate in alcohols. J. Org. Chem. 2018, 83, 2706–2713. [Google Scholar] [CrossRef]
- Dindarloo Inaloo, I.; Esmaeilpour, M.; Majnooni, S.; Reza Oveisi, A. Nickel-catalyzed synthesis of N-(Hetero)aryl carbamates from cyanate salts and phenols activated with cyanuric chloride. ChemCatChem 2020, 12, 5486–5491. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Soliman, A.M.; Alsaid, M.S.; Askar, A.A. Synthesis, antimicrobial activity and docking study of some novel 4-(4,4-dimethyl-2,6-dioxocyclohexylidene)methylamino derivatives carrying biologically active sulfonamide moiety. Arab. J. Chem. 2020, 13, 545–556. [Google Scholar] [CrossRef]
- Castro, M.Á.; Gamito, A.M.; Tangarife-Castaño, V.; Zapata, B.; Miguel Del Corral, J.M.; Mesa-Arango, A.C.; Betancur-Galvis, L.; San Feliciano, A. Synthesis and antifungal activity of terpenyl-1,4-naphthoquinone and 1,4-anthracenedione derivatives. Eur. J. Med. Chem. 2013, 67, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Pyta, K.; Blecha, M.; Janas, A.; Klich, K.; Pecyna, P.; Gajecka, M.; Przybylski, P. Synthesis, structure and antimicrobial evaluation of a new gossypol triazole conjugates functionalized with aliphatic chains and benzyloxy groups. Bioorg. Med. Chem. Lett. 2016, 26, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Fadda, A.A.; Afsah, E.S.M.; Awad, R.S. Synthesis and antimicrobial activity of some new benzo and naphthonitrile derivatives. Eur. J. Med. Chem. 2013, 60, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Tanaka, Y.; Miyamoto, M.; Takeda, T.; Hoshi, K.; Yamada, N.; Ohtake, A. Development of 3,4-dihydro-2H-benzo[1,4]oxazine derivatives as dual thromboxane A2 receptor antagonists and prostacyclin receptor agonists. Bioorg. Med. Chem. 2006, 14, 2005–2021. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Yadav, L.; Lal, J.; Jaiswal, P.K.; Mathur, M.; Swami, A.K.; Chaudhary, S. Synthesis, antimicrobial activity, structure-activity relationship and cytotoxic studies of a new series of functionalized (Z)-3-(2-oxo-2-substituted ethylidene)-3,4-dihydro-2H-benzo[b][1,4]oxazin-2-ones. Bioorg. Med. Chem. Lett. 2017, 27, 4393–4398. [Google Scholar] [CrossRef]
- Lv, P.; Chen, Y.; Zhao, Z.; Shi, T.; Wu, X.; Xue, J.; Li, Q.X.; Hua, R. Design, synthesis, and antifungal activities of 3-acyl thiotetronic acid derivatives: New fatty acid synthase inhibitors. J. Agric. Food Chem. 2018, 66, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Garzone, P.D.; Kroboth, P.D. Pharmacokinetics of the newer benzodiazepines. Clin. Pharmacokinet. 1989, 16, 337–364. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Investig. Drugs 2011, 20, 733–744. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Shankar, B.; Jalapathi, P.; Ramesh, M.; Kumar, A.K.; Ragavender, M.; Bharath, G. Synthesis, antimicrobial evaluation, and docking studies of some novel benzofuran based analogues of chalcone and 1,4-benzodiazepine. Russ. J. Gen. Chem. 2016, 86, 1711–1721. [Google Scholar] [CrossRef]
- Shankar, B.; Jalapathi, P.; Saikrishna, B.; Perugu, S.; Manga, V. Synthesis, anti-microbial activity, cytotoxicity of some novel substituted (5-(3-(1H-benzo[d]imidazol-2-yl)-4-hydroxybenzyl)benzofuran-2-yl)(phenyl)methanone analogs. Chem. Cent. J. 2018, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ajdačić, V.; Senerovic, L.; Vranić, M.; Pekmezovic, M.; Arsic-Arsnijevic, V.; Veselinovic, A.; Veselinovic, J.; Šolaja, B.A.; Nikodinovic-Runic, J.; Opsenica, I.M. Synthesis and evaluation of thiophene-based guanylhydrazones (iminoguanidines) efficient against panel of voriconazole-resistant fungal isolates. Bioorg. Med. Chem. 2016, 24, 1277–1291. [Google Scholar] [CrossRef] [PubMed]
- Persch, E.; Dumele, O.; Diederich, F. Molecular recognition in chemical and biological systems. Angew. Chem. Int. Ed. 2015, 54, 3290–3327. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.; Shanmugam, M.; Jothivel, S.; Kabilan, S. Design, synthesis, spectral and biological evaluation of novel 1-allyl substituted 2,6-diphenylpiperidin-4-ones and its derivatives of oximes/oxime ethers. Bioorg. Med. Chem. Lett. 2012, 22, 6602–6607. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, P.; Reddy, C.S.; Suresh Babu, K.; Reddy, P.M.; Srinivasa Rao, V.; Parthasarathy, T. Synthesis, characterization and molecular docking studies of novel 2-amino 3-cyano pyrano[2,3H]chrysin derivatives as potential antimicrobial agents. Med. Chem. Res. 2015, 24, 3696–3709. [Google Scholar] [CrossRef]
- Anuradha, V.; Srinivas, P.V.; Ranga Rao, R.; Manjulatha, K.; Purohit, M.G.; Madhusudana Rao, J. Isolation and synthesis of analgesic and anti-inflammatory compounds from Ochna squarrosa L. Bioorg. Med. Chem. 2006, 14, 6820–6826. [Google Scholar] [CrossRef] [PubMed]
- Ranga Rao, R.; Tiwari, A.K.; Prabhakar Reddy, P.; Suresh Babu, K.; Ali, A.Z.; Madhusudana, K.; Madhusudana Rao, J. New furanoflavanoids, intestinal α-glucosidase inhibitory and free-radical (DPPH) scavenging, activity from antihyperglycemic root extract of Derris indica (Lam.). Bioorg. Med. Chem. 2009, 17, 5170–5175. [Google Scholar] [CrossRef]
- Nishino, T.; Nishiyama, Y.; Sonoda, N. Deoxygenative dimerization of benzylic and allylic alcohols, and their ethers and esters using lanthanum metal and chlorotrimethylsilane in the presence of a catalytic amount of iodine and copper(I) iodide. Bull. Chem. Soc. Jpn. 2003, 76, 635–641. [Google Scholar] [CrossRef]
- Messanga, B.B.; Tih, R.G.; Kimbu, S.F.; Sondengam, B.L.; Martin, M.T.; Bodo, B. Calodenone, a new isobiflavonoid from ochna calodendron. J. Nat. Prod. 1992, 55, 245–248. [Google Scholar] [CrossRef]
- Fylaktakidou, K.; Hadjipavlou-Litina, D.; Litinas, K.; Nicolaides, D. Natural and synthetic coumarin derivatives with anti-inflammatory / antioxidant activities. Curr. Pharm. Des. 2005, 10, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, X.; Jin, H.; Yang, X.; Qin, B. Antifungal activity of umbelliferone derivatives: Synthesis and structure-activity relationships. Microb. Pathog. 2017, 104, 110–115. [Google Scholar] [CrossRef]
- Hoult, J.R.S.; Payá, M. Pharmacological and biochemical actions of simple coumarins: Natural products with therapeutic potential. Gen. Pharmacol. 1996, 27, 713–722. [Google Scholar] [CrossRef]
- Makandar, S.B.N.; Basanagouda, M.; Kulkarni, M.V.; Pranesha, V.P. Synthesis, antimicrobial and DNA cleavage studies of some 4-aryloxymethylcoumarins obtained by reaction of 4-bromomethylcoumarins with bidentate nucleophiles. Med. Chem. Res. 2012, 21, 2603–2614. [Google Scholar] [CrossRef]
- Kulkarni, M.V.; Patil, V.D. Studies on Coumarins, I. Arch. Der Pharm. 1981, 314, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, Y. FeCl3-catalyzed dimerization/elimination of 1,1-diarylalkenes: Efficient synthesis of functionalized 4H-chromenes. Org. Biomol. Chem. 2018, 16, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.P.; Deshmukh, T.R.; Sangshetti, J.N.; Khedkar, V.M.; Shingate, B.B. Ultrasound assisted rapid synthesis, biological evaluation, and molecular docking study of new 1,2,3-triazolyl pyrano[2,3-c]pyrazoles as antifungal and antioxidant agent. Synth. Commun. 2019, 49, 2521–2537. [Google Scholar] [CrossRef]
- Amin, K.M.; Abdel Rahman, D.E.; Abdelrasheed Allam, H.; El-Zoheiry, H.H. Design and synthesis of novel coumarin derivatives as potential acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorg. Chem. 2021, 110, 104792. [Google Scholar] [CrossRef]
- Chate, A.V.; Redlawar, A.A.; Bondle, G.M.; Sarkate, A.P.; Tiwari, S.V.; Lokwani, D.K. A new efficient domino approach for the synthesis of coumarin-pyrazolines as antimicrobial agents targeting bacterial D-alanine-D-alanine ligase. New J. Chem. 2019, 43, 9002–9011. [Google Scholar] [CrossRef]
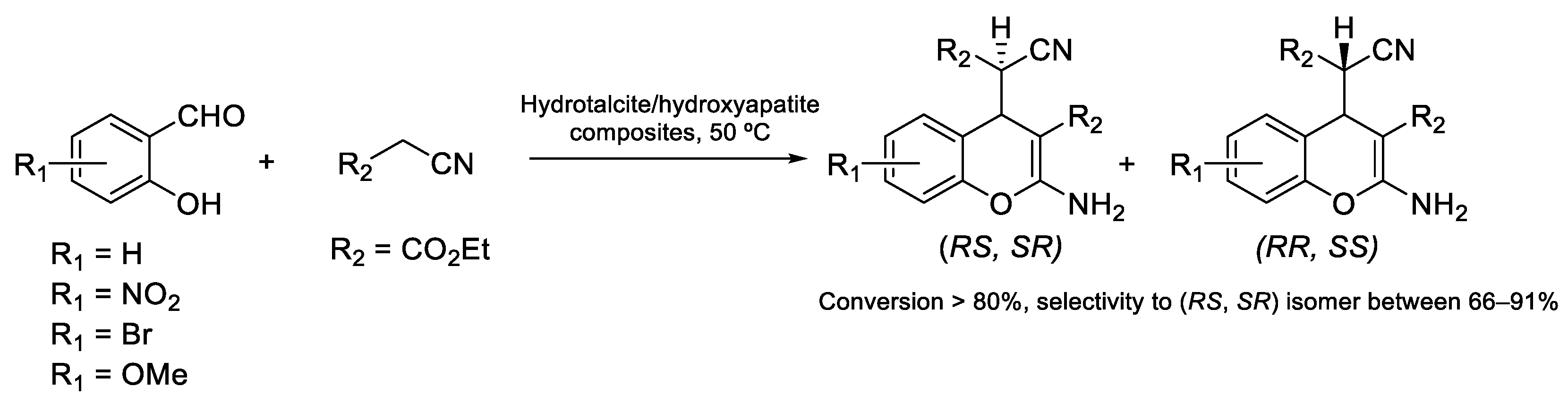
- Velázquez-Herrera, F.D.; González-Rodal, D.; Fetter, G.; Pérez-Mayoral, E. Towards highly efficient hydrotalcite/hydroxyapatite composites as novel catalysts involved in eco-synthesis of chromene derivatives. Appl. Clay Sci. 2020, 198, 105833. [Google Scholar] [CrossRef]
- Gonzalez-Carrillo, G.; Gonzalez, J.; Emparan-Legaspi, M.J.; Lino-Lopez, G.J.; Aguayo-Villarreal, I.A.; Ceballos-Magaña, S.G.; Martinez-Martinez, F.J.; Muñiz-Valencia, R. Propylsulfonic acid grafted on mesoporous siliceous FDU-5 material: A high TOF catalyst for the synthesis of coumarins via Pechmann condensation. Microporous Mesoporous Mater. 2020, 307, 110458. [Google Scholar] [CrossRef]
- Basanagouda, M.; Shivashankar, K.; Kulkarni, M.V.; Rasal, V.P.; Patel, H.; Mutha, S.S.; Mohite, A.A. Synthesis and antimicrobial studies on novel sulfonamides containing 4-azidomethyl coumarin. Eur. J. Med. Chem. 2010, 45, 1151–1157. [Google Scholar] [CrossRef]
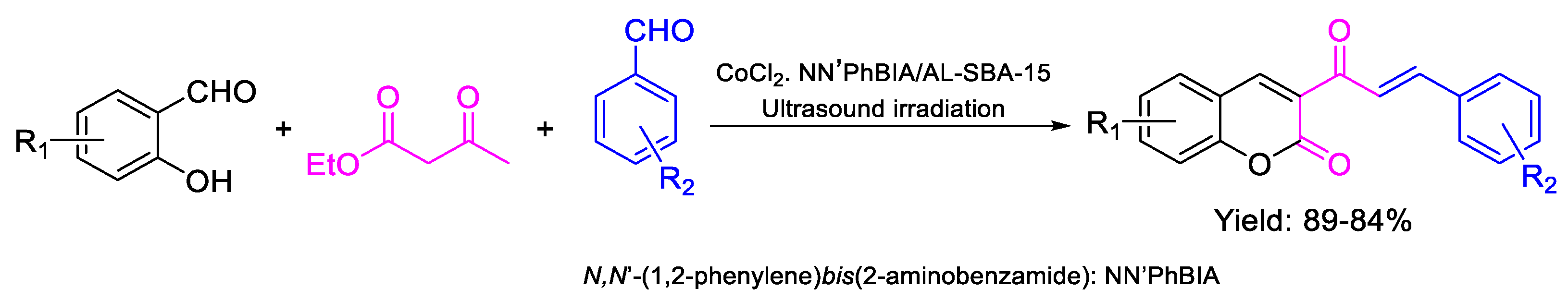
- Akbarzadeh, Z.; Safaei-Ghomi, J. Ultrasound assisted eco-friendly synthesis of 3-cinnamoyl coumarins using N,N′-(1,2-phenylene)bis(2-aminobenzamide) dichloro cobalt immobilized on mesoporous Al-SBA-15 as a new and recyclable catalyst. Green Chem. Lett. Rev. 2020, 13, 141–154. [Google Scholar] [CrossRef]
- Havsteen, B.H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 2002, 96, 67–202. [Google Scholar] [CrossRef]
- Chen, X.; Leng, J.; Rakesh, K.P.; Darshini, N.; Shubhavathi, T.; Vivek, H.K.; Mallesha, N.; Qin, H.L. Synthesis and molecular docking studies of xanthone attached amino acids as potential antimicrobial and anti-inflammatory agents. MedChemComm 2017, 8, 1706–1719. [Google Scholar] [CrossRef]
- Chauhan, M.; Saxena, A.; Saha, B. An insight in anti-malarial potential of indole scaffold: A review. Eur. J. Med. Chem. 2021, 218, 113400. [Google Scholar] [CrossRef] [PubMed]
- Dolšak, A.; Gobec, S.; Sova, M. Indoleamine and tryptophan 2,3-dioxygenases as important future therapeutic targets. Pharmacol. Ther. 2020, 221, 107746. [Google Scholar] [CrossRef]
- Arumugam, N.; Raghunathan, R.; Almansour, A.I.; Karama, U. An efficient synthesis of highly functionalized novel chromeno[4,3-b] pyrroles and indolizino[6,7-b]indoles as potent antimicrobial and antioxidant agents. Bioorg. Med. Chem. Lett. 2012, 22, 1375–1379. [Google Scholar] [CrossRef]
- Kokurkina, G.V.; Dutov, M.D.; Shevelev, S.A.; Ugrak, B.I. Intramolecular cyclization of O-(3,5-diaminophenyl)-substituted ketoximes as a route to 6-amino-4-hydroxyindoles. Mendeleev Commun. 2011, 21, 196–197. [Google Scholar] [CrossRef]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef]
- Liu, X.R.; Wu, H.; He, Z.Y.; Ma, Z.Q.; Feng, J.T.; Zhang, X. Design, synthesis and fungicidal activities of some novel pyrazole derivatives. Molecules 2014, 19, 14036–14051. [Google Scholar] [CrossRef] [PubMed]
- Rajanarendar, E.; Nagi Reddy, M.; Rama Krishna, S.; Rama Murthy, K.; Reddy, Y.N.; Rajam, M.V. Design, synthesis, antimicrobial, anti-inflammatory and analgesic activity of novel isoxazolyl pyrimido[4,5-b]quinolines and isoxazolyl chromeno[2,3-d]pyrimidin-4-ones. Eur. J. Med. Chem. 2012, 55, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Solanki, I. Synthesis and antimicrobial studies of furyl based new bispyrazolines linked via aliphatic chains. J. Saudi Chem. Soc. 2017, 21, 251–261. [Google Scholar] [CrossRef]
- Yusuf, M.; Nisa, S.; Paul, K. New biphenyl-based bispyrazolines: Synthesis, antimicrobial, and docking studies. J. Heterocycl. Chem. 2019, 56, 2659–2670. [Google Scholar] [CrossRef]
- Bondock, S.; Khalifa, W.; Fadda, A.A. Synthesis and antimicrobial activity of some new 4-hetarylpyrazole and furo[2,3-c]pyrazole derivatives. Eur. J. Med. Chem. 2011, 46, 2555–2561. [Google Scholar] [CrossRef]
- Upadhyay, A.; Srivastava, S.K.; Srivastava, S.D. Conventional and microwave assisted synthesis of some new N-[(4-oxo-2-substituted aryl-1,3-thiazolidine)-acetamidyl]-5-nitroindazoles and its antimicrobial activity. Eur. J. Med. Chem. 2010, 45, 3541–3548. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Paramashivappa, R.; Phani Kumar, P.; Subba Rao, P.V.; Srinivasa Rao, A. Design, synthesis and biological evaluation of benzimidazole/benzothiazole and benzoxazole derivatives as cyclooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 657–660. [Google Scholar] [CrossRef]
- Nikalje, A.P.G.; Tiwari, S.V.; Sarkate, A.P.; Karnik, K.S. Imidazole-thiazole coupled derivatives as novel lanosterol 14-α demethylase inhibitors: Ionic liquid mediated synthesis, biological evaluation and molecular docking study. Med. Chem. Res. 2018, 27, 592–606. [Google Scholar] [CrossRef]
- Abrigach, F.; Karzazi, Y.; Benabbes, R.; El Youbi, M.; Khoutoul, M.; Taibi, N.; Karzazi, N.; Benchat, N.; Bouakka, M.; Saalaoui, E.; et al. Synthesis, biological screening, POM, and 3D-QSAR analyses of some novel pyrazolic compounds. Med. Chem. Res. 2017, 26, 1784–1795. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Khedkar, V.M.; Jha, P.C.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1,2,3-Triazole tethered acetophenones: Synthesis, bioevaluation and molecular docking study. Chin. Chem. Lett. 2016, 27, 1058–1063. [Google Scholar] [CrossRef]
- Darandale, S.N.; Mulla, N.A.; Pansare, D.N.; Sangshetti, J.N.; Shinde, D.B. A novel amalgamation of 1,2,3-triazoles, piperidines and thieno pyridine rings and evaluation of their antifungal activity. Eur. J. Med. Chem. 2013, 65, 527–532. [Google Scholar] [CrossRef]
- Hajós, G.; Riedl, Z.; Kollenz, G. Recent advances in ring transformations of five-membered heterocycles and their fused derivatives. Eur. J. Org. Chem. 2001, 3405–3414. [Google Scholar] [CrossRef]
- Kadi, A.A.; Al-Abdullah, E.S.; Shehata, I.A.; Habib, E.E.; Ibrahim, T.M.; El-Emam, A.A. Synthesis, antimicrobial and anti-inflammatory activities of novel 5-(1-adamantyl)-1,3,4-thiadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5006–5011. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.Y.; Wang, X.M.; Yang, Y.H.; Zhu, H.L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef]
- Yusuf, M.; Jain, P. Synthesis and biological significances of 1,3,4-thiadiazolines and related heterocyclic compounds. Arab. J. Chem. 2014, 7, 525–552. [Google Scholar] [CrossRef]
- Yusuf, M.; Kaur, M.; Jain, P. Synthesis and antimicrobial evaluations of 1,3,4-thiadiazoline-based bisheterocyclics. J. Heterocycl. Chem. 2014, 52, 692–700. [Google Scholar] [CrossRef]
- El-Atawy, M.A.; Omar, A.Z.; Hagar, M.; Shashira, E.M. Transalkylidation reaction: Green, catalyst-free synthesis of thiosemicarbazones and solving the NMR conflict between their acyclic structure and intramolecular cycloaddition products. Green Chem. Lett. Rev. 2019, 12, 364–376. [Google Scholar] [CrossRef]
- Gomha, S.M.; Abdel-aziz, H.M.; Badrey, M.G.; Abdulla, M.M. Efficient synthesis of some new 1,3,4-Thiadiazoles and 1,2,4-Triazoles kinked to pyrazolylcoumarin ring system as potent 5α-reductase inhibitors. J. Heterocycl. Chem. 2019, 56, 1275–1282. [Google Scholar] [CrossRef]
- Yusuf, M.; Thakur, S. Synthesis, characterization & in vitro antimicrobial-antioxidant studies of novel N,1-diphenyl-4,5-dihydro-1H-1,2,4-triazol-3-amine derivatives. J. Heterocycl. Chem. 2019, 56, 3403–3413. [Google Scholar]
- Bhatia, M.S.; Zarekar, B.E.; Choudhari, P.B.; Ingale, K.B.; Bhatia, N.M. Combinatorial approach: Identification of potential antifungals from triazole minilibraries. Med. Chem. Res. 2011, 20, 116–120. [Google Scholar] [CrossRef]
- Xu, W.; He, J.; He, M.; Han, F.; Chen, X.; Pan, Z.; Wang, J.; Tong, M. Synthesis and antifungal activity of novel sulfone derivatives containing 1,3,4-oxadiazole moieties. Molecules 2011, 16, 9129–9141. [Google Scholar] [CrossRef]
- Wang, S.; Qi, L.; Liu, H.; Lei, K.; Wang, X.; Liu, R. Synthesis of 1,3,4-oxadiazoles derivatives with antidepressant activity and their binding to the 5-HT1Areceptor. RSC Adv. 2020, 10, 30848–30857. [Google Scholar] [CrossRef]
- Lu, F.; Gong, F.; Li, L.; Zhang, K.; Li, Z.; Zhang, X.; Yin, Y.; Wang, Y.; Gao, Z.; Zhang, H.; et al. Electrochemical synthesis of 2,5-disubstituted 1,3,4-oxadiazoles from α-keto acids and acylhydrazines under mild conditions. Eur. J. Org. Chem. 2020, 2020, 3257–3260. [Google Scholar] [CrossRef]
- Ningaiah, S.; Bhadraiah, U.K.; Doddaramappa, S.D.; Keshavamurthy, S.; Javarasetty, C. Novel pyrazole integrated 1,3,4-oxadiazoles: Synthesis, characterization and antimicrobial evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 245–248. [Google Scholar] [CrossRef]
- Kumari, M.; Tahlan, S.; Narasimhan, B.; Ramasamy, K.; Lim, S.M.; Shah, S.A.A.; Mani, V.; Kakkar, S. Synthesis and biological evaluation of heterocyclic 1,2,4-triazole scaffolds as promising pharmacological agents. BMC Chem. 2021, 15, 1–16. [Google Scholar] [CrossRef]
- Rishikesan, R.; Karuvalam, R.P.; Muthipeedika, N.J.; Sajith, A.M.; Eeda, K.R.; Pakkath, R.; Haridas, K.R.; Bhaskar, V.; Narasimhamurthy, K.H.; Muralidharan, A. Synthesis of some novel piperidine fused 5-thioxo-1H-1,2,4-triazoles as potential antimicrobial and antitubercular agents. J. Chem. Sci. 2021, 133, 1–12. [Google Scholar] [CrossRef]
- Deshmukh, T.R.; Khare, S.P.; Krishna, V.S.; Sriram, D.; Sangshetti, J.N.; Bhusnure, O.; Khedkar, V.M.; Shingate, B.B. Design and synthesis of new aryloxy-linked dimeric 1,2,3-triazoles via click chemistry approach: Biological evaluation and molecular docking study. J. Heterocycl. Chem. 2019, 56, 2144–2162. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1,2,3-Triazole incorporated coumarin derivatives as potential antifungal and antioxidant agents. Chin. Chem. Lett. 2016, 27, 295–301. [Google Scholar] [CrossRef]
- Gazit, A.; App, H.; McMahon, G.; Chen, J.; Levitzki, A.; Bohmer, F.D. Tyrphostins. 5. Potent inhibitors of platelet-derived growth factor receptor tyrosine kinase: Structure-activity relationships in quinoxalines, quinolines, and indole tyrphostins. J. Med. Chem. 1996, 39, 2170–2177. [Google Scholar] [CrossRef]
- Anzenbacher, P.; Try, A.C.; Miyaji, H.; Jursikova, K.; Lynch, V.M.; Marquez, M.; Sessler, J.L. Fluorinated calix[4]pyrrole and dipyrrolylquinoxaline: Neutral anion receptors with augmented affinities and enhanced selectivities. J. Am. Chem. Soc. 2000, 122, 10268–10272. [Google Scholar] [CrossRef]
- Jaso, A.; Zarranz, B.; Aldana, I.; Monge, A. Synthesis of new quinoxaline-2-carboxylate 1,4-dioxide derivatives as anti-Mycobacterium tuberculosis agents. J. Med. Chem. 2005, 48, 2019–2025. [Google Scholar] [CrossRef] [PubMed]
- Ammar, Y.A.; Farag, A.A.; Ali, A.M.; Hessein, S.A.; Askar, A.A.; Fayed, E.A.; Elsisi, D.M.; Ragab, A. Antimicrobial evaluation of thiadiazino and thiazolo quinoxaline hybrids as potential DNA gyrase inhibitors; design, synthesis, characterization and morphological studies. Bioorg. Chem. 2020, 99, 103841. [Google Scholar] [CrossRef]
- Pathak, N.; Rathi, E.; Kumar, N.; Kini, S.G.; Rao, C.M. A Review on anticancer potentials of benzothiazole derivatives. Mini Rev. Med. Chem. 2019, 20, 12–23. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, M.R.; Patil, S.A.; Budagupi, S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. [Google Scholar] [CrossRef]
- Banerjee, S.; Payra, S.; Saha, A. A Review on synthesis of benzothiazole derivatives. Curr. Organocatal. 2017, 4, 164–181. [Google Scholar] [CrossRef]
- Ballari, M.S.; Herrera Cano, N.; Lopez, A.G.; Wunderlin, D.A.; Feresín, G.E.; Santiago, A.N. Green Synthesis of Potential Antifungal Agents: 2-Benzyl Substituted Thiobenzoazoles. J. Agric. Food Chem. 2017, 65, 10325–10331. [Google Scholar] [CrossRef]
- Ballari, M.S.; Herrera Cano, N.; Wunderlin, D.A.; Feresin, G.E.; Santiago, A.N. One-pot sequential synthesis and antifungal activity of 2-(benzylsulfonyl)benzothiazole derivatives. RSC Adv. 2019, 9, 29405–29413. [Google Scholar] [CrossRef]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Enaminonitrile in heterocyclic synthesis: Synthesis and antimicrobial evaluation of some new pyrazole, isoxazole and pyrimidine derivatives incorporating a benzothiazole moiety. Eur. J. Med. Chem. 2009, 44, 4813–4818. [Google Scholar] [CrossRef]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety. Eur. J. Med. Chem. 2010, 45, 3692–3701. [Google Scholar] [CrossRef]
- Fadda, A.A.; Soliman, N.N.; Bayoumy, N.M. Antimicrobial properties of some new synthesized benzothiazole linked carboxamide, acetohydrazide, and sulfonamide systems. J. Heterocycl. Chem. 2019, 56, 2369–2378. [Google Scholar] [CrossRef]
- Zha, G.F.; Leng, J.; Darshini, N.; Shubhavathi, T.; Vivek, H.K.; Asiri, A.M.; Marwani, H.M.; Rakesh, K.P.; Mallesha, N.; Qin, H.L. Synthesis, SAR and molecular docking studies of benzo[d]thiazole-hydrazones as potential antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2017, 27, 3148–3155. [Google Scholar] [CrossRef]
- Puttaraju, K.B.; Shivashankar, K.; Mahendra, M.; Rasal, V.P.; Venkata Vivek, P.N.; Rai, K.; Chanu, M.B. Microwave assisted synthesis of dihydrobenzo[4,5]imidazo[1,2-a]pyrimidin-4- ones; Synthesis, in vitro antimicrobial and anticancer activities of novel coumarin substituted dihydrobenzo[4,5]imidazo[1,2-a]pyrimidin-4-ones. Eur. J. Med. Chem. 2013, 69, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Aouali, M.; Mhalla, D.; Allouche, F.; El Kaim, L.; Tounsi, S.; Trigui, M.; Chabchoub, F. Synthesis, antimicrobial and antioxidant activities of imidazotriazoles and new multicomponent reaction toward 5-amino-1-phenyl[1,2,4]triazole derivatives. Med. Chem. Res. 2015, 24, 2732–2741. [Google Scholar] [CrossRef]
- Salem, M.S.; Ali, M.A.M. Novel pyrazolo[3,4-b]pyridine derivatives: Synthesis, characterization, antimicrobial and antiproliferative profile. Biol. Pharm. Bull. 2016, 39, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.R.; Shankar, K.; Reddy, C.S. Synthesis, biological evaluation and docking studies of a new series of tris-heterocycles containing pyrazole, triazole and oxadiazole. Indian J. Chem. Sect. B Org. Med. Chem. 2018, 57, 687–699. [Google Scholar]
- Hassan, A.S.; Masoud, D.M.; Sroor, F.M.; Askar, A.A. Synthesis and biological evaluation of pyrazolo[1,5-a]pyrimidine-3-carboxamide as antimicrobial agents. Med. Chem. Res. 2017, 26, 2909–2919. [Google Scholar] [CrossRef]
- Rajanarendar, E.; Govardhan Reddy, K.; Rama Krishna, S.; Shireesha, B.; Reddy, Y.N.; Rajam, M.V. Design, synthesis, antimicrobial, anti-inflammatory, and analgesic activity of novel dihydrobenzo furo[3,2-e]isoxazolo[4,5-b]azepin-5(5aH)-ones. Med. Chem. Res. 2013, 22, 6143–6153. [Google Scholar] [CrossRef]
- Patel, M.A.; Bhila, V.G.; Patel, N.H.; Patel, A.K.; Brahmbhatt, D.I. Synthesis, characterization and biological evaluation of some pyridine and quinoline fused chromenone derivatives. Med. Chem. Res. 2012, 21, 4381–4388. [Google Scholar] [CrossRef]
- Aryan, R.; Beyzaei, H.; Nojavan, M.; Pirani, F.; Samareh Delarami, H.; Sanchooli, M. Expedient multicomponent synthesis of a small library of some novel highly substituted pyrido[2,3-d]pyrimidine derivatives mediated and promoted by deep eutectic solvent and in vitro and quantum mechanical study of their antibacterial and antifungal activities. Mol. Divers. 2019, 23, 93–105. [Google Scholar]
- Koudad, M.; El Hamouti, C.; Elaatiaoui, A.; Dadou, S.; Oussaid, A.; Abrigach, F.; Pilet, G.; Benchat, N.; Allali, M. Synthesis, crystal structure, antimicrobial activity and docking studies of new imidazothiazole derivatives. J. Iran. Chem. Soc. 2020, 17, 297–306. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Chabukswar, A.R.; Shinde, D.B. Microwave assisted one pot synthesis of some novel 2,5-disubstituted 1,3,4-oxadiazoles as antifungal agents. Bioorg. Med. Chem. Lett. 2011, 21, 444–448. [Google Scholar] [CrossRef]
- Thirupathi, G.; Venkatanarayana, M.; Dubey, P.K.; Bharathi Kumari, Y. Eco-friendly synthesis and antimicrobial activities of substituted-5-(1H- indol-3-yl)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione derivatives. Med. Chem. Res. 2014, 23, 1569–1580. [Google Scholar] [CrossRef]
- Arumugam, N.; Raghunathan, R.; Shanmugaiah, V.; Mathivanan, N. Synthesis of novel β-lactam fused spiroisoxazolidine chromanones and tetralones as potent antimicrobial agent for human and plant pathogens. Bioorg. Med. Chem. Lett. 2010, 20, 3698–3702. [Google Scholar] [CrossRef]
- Subhedar, D.D.; Shaikh, M.H.; Nawale, L.; Yeware, A.; Sarkar, D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. Novel tetrazoloquinoline-rhodanine conjugates: Highly efficient synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2016, 26, 2278–2283. [Google Scholar] [CrossRef]
- Ahmed, W.; Yan, X.; Hu, D.; Adnan, M.; Tang, R.Y.; Cui, Z.N. Synthesis and fungicidal activity of novel pyrazole derivatives containing 5-Phenyl-2-Furan. Bioorg. Med. Chem. 2019, 27, 115048. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Nikalje, A.; Lokwani, D.K.; Sarkate, A.P.; Jamir, K. Synthesis, biological evaluation, molecular docking study and acute oral toxicity study of coupled omidazole-pyrimidine derivatives. Lett. Drug Des. Discov. 2018, 15, 475–487. [Google Scholar] [CrossRef]
- Strus, P.; Borensztejn, K.; Szczepankiewicz, A.A.; Lisiecki, K.; Czarnocki, Z.; Nieznanska, H.; Wojcik, C.; Bialy, L.P.; Mlynarczuk-Bialy, I. Novel podophyllotoxin and benzothiazole derivative induces transitional morphological and functional changes in HaCaT cells. Toxicol. Vitr. 2021, 73, 105144. [Google Scholar] [CrossRef]
- Kumar, H.S.; Choi, C.-S.; Lee, K.H. Synthesis, Photophysical Properties, and Cytotoxicity of Rhodamine Based Fluorescent Probes. Russ. J. Bioorg. Chem. 2021, 47, 691–699. [Google Scholar] [CrossRef]
- Yang, T.; Mai, J.; Wu, S.; Liu, C.; Tang, L.; Mo, Z.; Zhang, M.; Guo, L.; Liu, M.; Ma, J. UV/chlorine process for degradation of benzothiazole and benzotriazole in water: Efficiency, mechanism and toxicity evaluation. Sci. Total Environ. 2021, 760, 144304. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Zou, T.; Chen, M.; Song, Y.; Yang, C.; Qiu, B.; Chen, Z.-F.; Tsang, S.Y.; Qi, Z.; Cai, Z. Contamination profiles and health impact of benzothiazole and its derivatives in PM2.5 in typical Chinese cities. Sci. Total Environ. 2021, 755, 142617. [Google Scholar] [CrossRef]
- Jiang, P.; Qiu, J.; Gao, Y.; Stefan, M.I.; Li, X.-F. Nontargeted identification and predicted toxicity of new byproducts generated from UV treatment of water containing micropollutant 2-mercaptobenzothiazole. Water Res. 2021, 188, 116542. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, C.; de Cássia Campos Pena, A.; Dallegrave, A.; Fernandes, A.N.; Gutterres, M. Photodegradation of Emerging Contaminant 2-(tiocyanomethylthio) Benzothiazole (TCMTB) in Aqueous Solution: Kinetics and Transformation Products. Bull. Environ. Contam. Toxicol. 2020, 105, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Golzadeh, N.; Barst, B.D.; Baker, J.M.; Auger, J.C.; McKinney, M.A. Alkylated polycyclic aromatic hydrocarbons are the largest contributor to polycyclic aromatic compound concentrations in traditional foods of the Bigstone Cree Nation in Alberta, Canada. Environ. Pollut. 2021, 275, 116625. [Google Scholar] [CrossRef] [PubMed]
- Catharina, L.; Carels, N. Specific enzyme functionalities of Fusarium oxysporum compared to host plants. Gene 2018, 676, 219–226. [Google Scholar] [CrossRef]
- Behr, J. Chitin Synthase as an Antifungal Target: Recent Advances. Curr. Med. Chem. Anti-Infect. Agents 2003, 2, 173–189. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, S.; Dang, L.; Wang, S.; Chen, H.; Wu, Y.; Jiang, X.; Wu, P. A Novel Short-Root Gene Encodes a Glucosamine-6-Phosphate Acetyltransferase Required for Maintaining Normal Root Cell Shape in Rice. Plant Physiol. 2005, 138, 232–242. [Google Scholar] [CrossRef]
- Li, Y.; Kiledjian, M. Regulation of mRNA decapping. Wiley Interdiscip. Rev. RNA 2010, 1, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Lorenz, M.C. Carnitine acetyltransferases are required for growth on non-fermentable carbon sources but not for pathogenesis in Candida albicans. Microbiology 2008, 154, 500–509. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Colabroy, K.L.; Begley, T.P.; Ealick, S.E. Structural Studies on 3-Hydroxyanthranilate-3,4-dioxygenase: The Catalytic Mechanism of a Complex Oxidation Involved in NAD Biosynthesis. Biochemistry 2005, 44, 7632–7643. [Google Scholar] [CrossRef]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef]
- McIninch, J.K.; McIninch, J.D.; May, S.W. Catalysis, Stereochemistry, and Inhibition of Ureidoglycolate Lyase. J. Biol. Chem. 2003, 278, 50091–50100. [Google Scholar] [CrossRef]
- Mayer, A.; Neupert, W.; Lill, R. Translocation of Apocytochrome c across the Outer Membrane of Mitochondria. J. Biol. Chem. 1995, 270, 12390–12397. [Google Scholar] [CrossRef]
- Yang, C.; Hamel, C.; Vujanovic, V.; Gan, Y. Fungicide: Modes of Action and Possible Impact on Nontarget Microorganisms. ISRN Ecol. 2011, 2011. [Google Scholar] [CrossRef]
- Bean, T.P.; Cools, H.J.; Lucas, J.A.; Hawkins, N.D.; Ward, J.L.; Shaw, M.W.; Fraaije, B.A. Sterol content analysis suggests altered eburicol 14α-demethylase (CYP51) activity in isolates of Mycosphaerella graminicola adapted to azole fungicides. FEMS Microbiol. Lett. 2009, 296, 266–273. [Google Scholar] [CrossRef]
- Monk, B.C.; Sagatova, A.A.; Hosseini, P.; Ruma, Y.N.; Wilson, R.K.; Keniya, M.V. Fungal Lanosterol 14α-demethylase: A target for next-generation antifungal design. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140206. [Google Scholar] [CrossRef] [PubMed]
- Cob-Calan, N.N.; Chi-Uluac, L.A.; Ortiz-Chi, F.; Cerqueda-García, D.; Navarrete-Vázquez, G.; Ruiz-Sánchez, E.; Hernández-Núñez, E. Molecular Docking and Dynamics Simulation of Protein β-Tubulin and Antifungal Cyclic Lipopeptides. Molecules 2019, 24, 3387. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, B.P.; Jindal, B.; Srivastava, S.; Panda, D. Microtubules as antifungal and antiparasitic drug targets. Expert Opin. Ther. Pat. 2011, 21, 167–186. [Google Scholar] [CrossRef]
- Zheng, D.; Köller, W. Characterization of the mitochondrial cytochrome b gene from Venturia inaequalis. Curr. Genet. 1997, 32, 361–366. [Google Scholar] [CrossRef]
- Hunte, C.; Koepke, J.; Lange, C.; Roßmanith, T.; Michel, H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 2000, 8, 669–684. [Google Scholar] [CrossRef]
- Hily, J.-M.; Singer, S.D.; Villani, S.M.; Cox, K.D. Characterization of the cytochrome b (cyt b) gene from Monilinia species causing brown rot of stone and pome fruit and its significance in the development of QoI resistance. Pest Manag. Sci. 2011, 67, 385–396. [Google Scholar] [CrossRef]
- Shen, L.L.; Fostel, J.M. DNA Topoisomerase Inhibitors as Antifungal Agents. Adv. Pharmacol. 1994, 29, 227–244. [Google Scholar]
- Jarolim, K.; del Favero, G.; Ellmer, D.; Stark, T.D.; Hofmann, T.; Sulyok, M.; Humpf, H.-U.; Marko, D. Dual effectiveness of Alternaria but not Fusarium mycotoxins against human topoisomerase II and bacterial gyrase. Arch. Toxicol. 2017, 91, 2007–2016. [Google Scholar] [CrossRef]
- Prasad, J.K.; Pandey, P.; Anand, R.; Raghuwanshi, R. Drought Exposed Burkholderia seminalis JRBHU6 Exhibits Antimicrobial Potential through Pyrazine-1,4-Dione Derivatives Targeting Multiple Bacterial and Fungal Proteins. Front. Microbiol. 2021, 12, 513. [Google Scholar] [CrossRef]
- Amine Khodja, I.; Boulebd, H.; Bensouici, C.; Belfaitah, A. Design, synthesis, biological evaluation, molecular docking, DFT calculations and in silico ADME analysis of (benz)imidazole-hydrazone derivatives as promising antioxidant, antifungal, and anti-acetylcholinesterase agents. J. Mol. Struct. 2020, 1218, 128527. [Google Scholar] [CrossRef]
- Kundu, A.; Mandal, A.; Saha, S.; Prabhakaran, P.; Walia, S. Fungicidal activity and molecular modeling of fusarubin analogues from Fusarium oxysporum. Toxicol. Environ. Chem. 2020, 102, 78–91. [Google Scholar] [CrossRef]
- Salem, M.A.; Ragab, A.; El-Khalafawy, A.; Makhlouf, A.H.; Askar Ahmed, A.; Ammar, Y.A. Design, synthesis, in vitro antimicrobial evaluation and molecular docking studies of indol-2-one tagged with morpholinosulfonyl moiety as DNA gyrase inhibitors. Bioorg. Chem. 2020, 96, 103619. [Google Scholar] [CrossRef]







































































{kind=link}
{kind=link}
{kind=link}
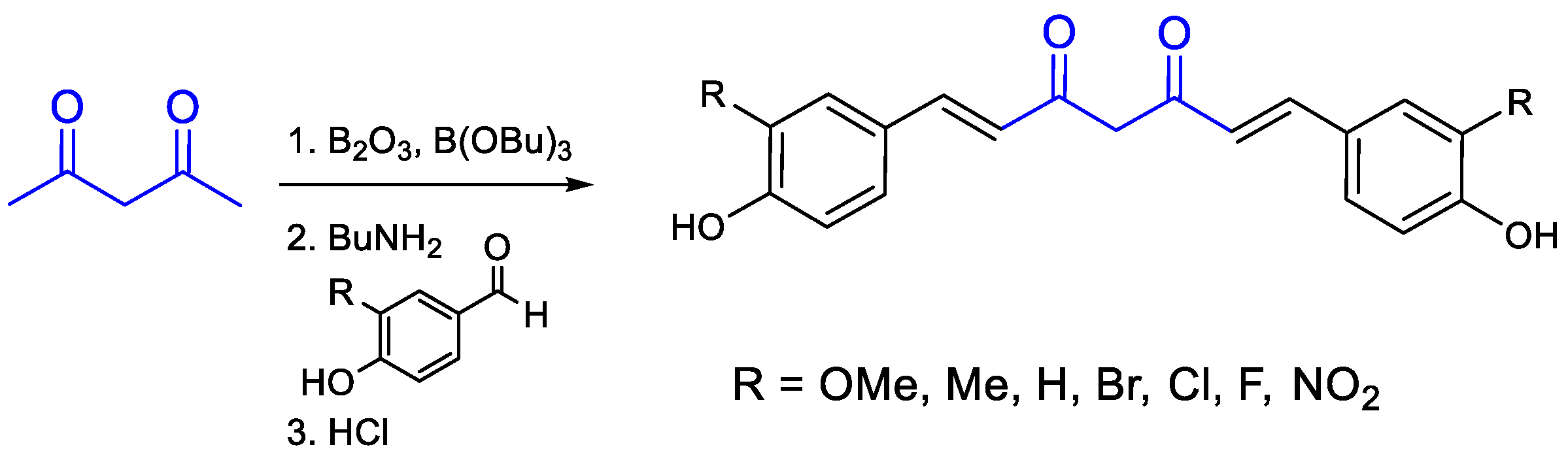{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
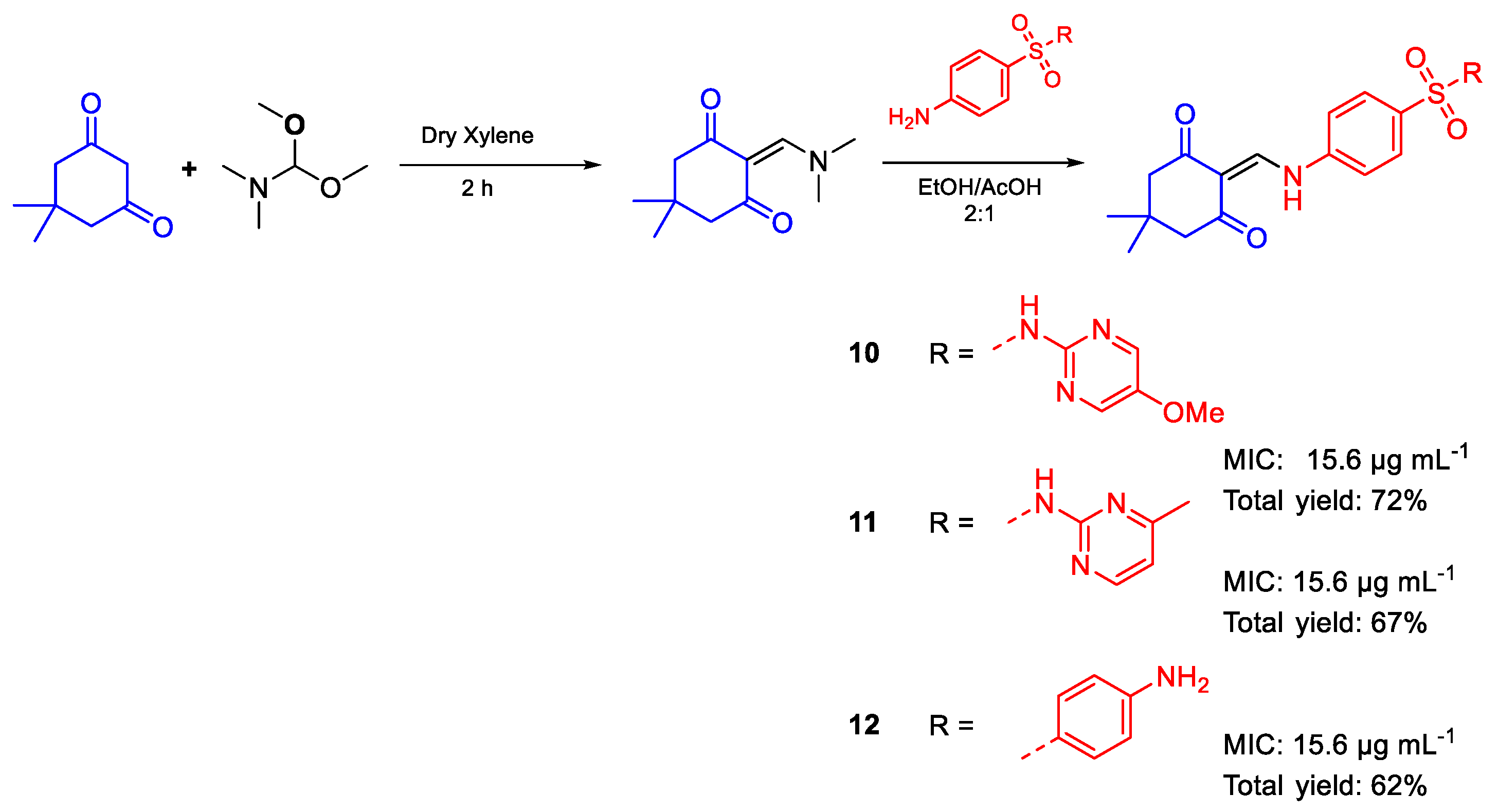{kind=link}
{kind=link}
{kind=link}
{kind=link}
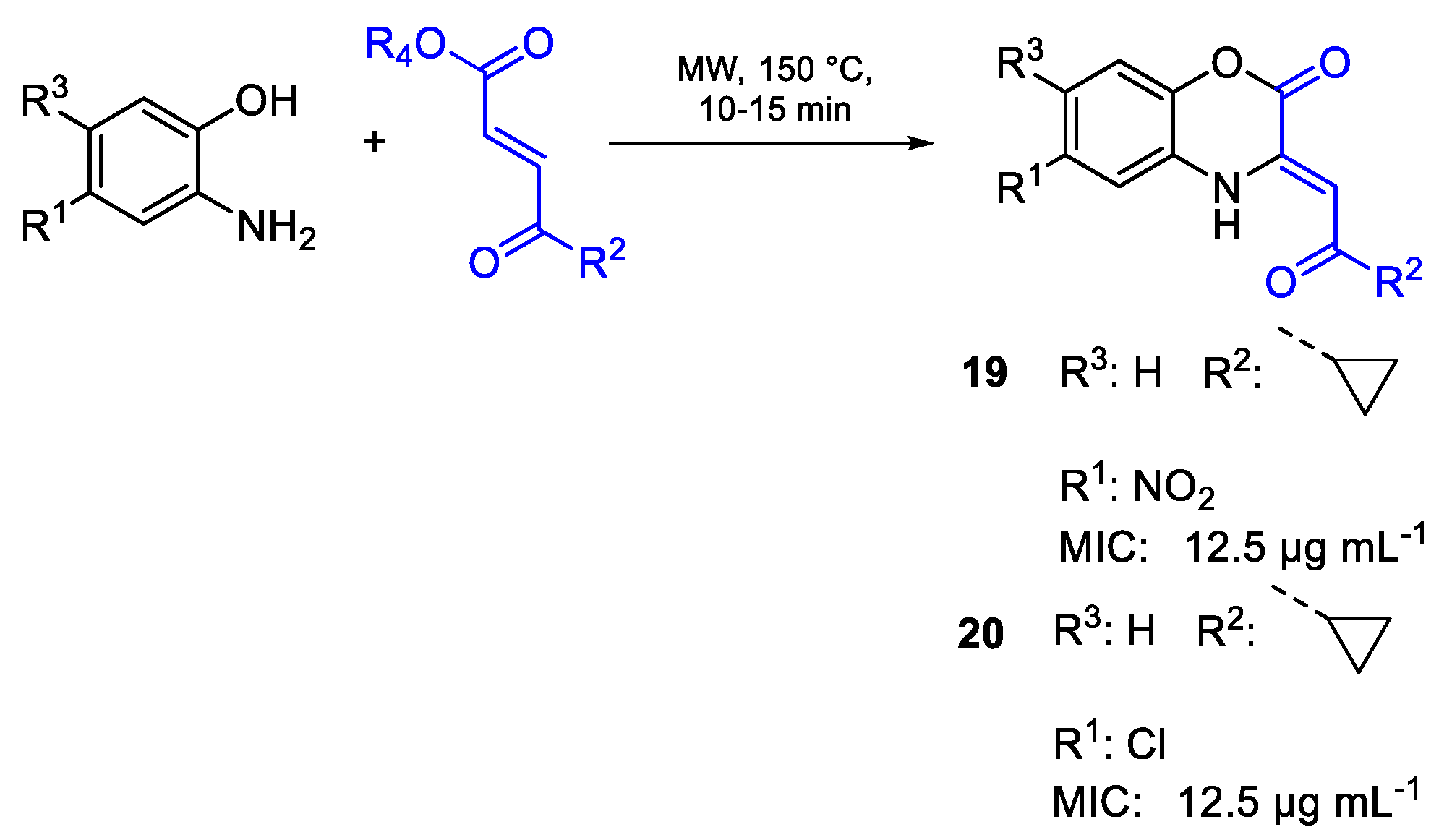{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Bioactivity Measure | Reported Value | Tested Microbial Strain |
|---|---|---|---|
| (Units in µM) | |||
| 90 | IC50 | 0.0067 ± 0.0005 µM | FOX M15-Pa |
| 91 | 0.23 ± 0.05 µM | ||
| 2 | IC50 | 0.055 µM | FOXf. sp Albedinis |
| 3 | 0.079 µM | ||
| 4 | 0.092 µM | ||
| 104 | 0.2 µg mL−1 (0.45) | FOX | |
| 105 | MIC | 0.2 µg mL−1 (0.45) | |
| 106 | 0.2 µg mL−1 (0.61) | ||
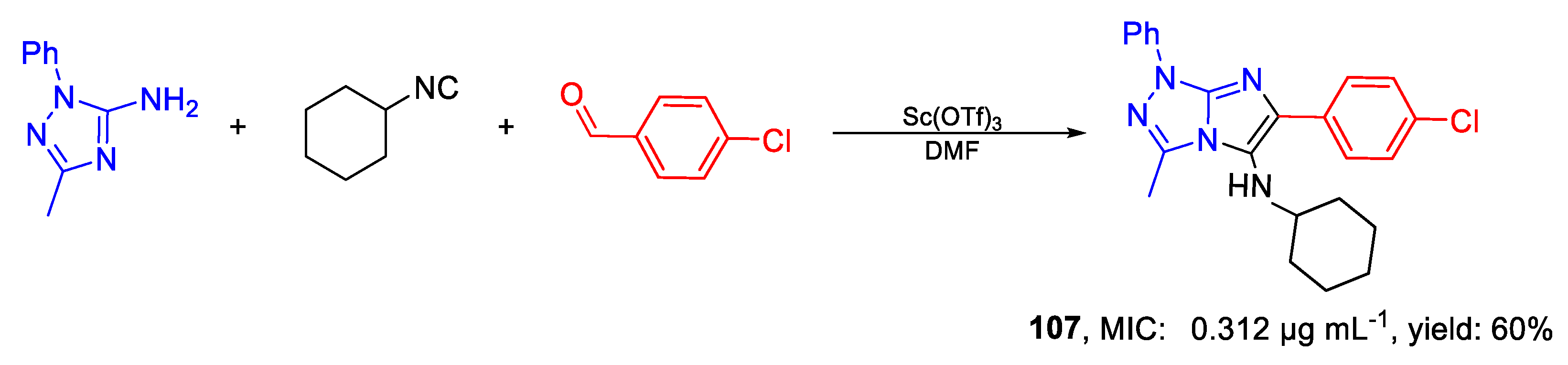
| 107 | MIC | 0.312 µg mL−1 | FOX CTM 10402 |
| (0.77) | |||
| MFC | 2.5 µg mL−1 | ||
| (6.16) | |||
| 108 | MIC | 0.98 µg mL−1 (2.21) | FOX ATCC 7601 |
| 109 | 1.95 µg mL−1 (4.43) | ||
| 110 | 1.95 µg mL−1 (5.29) | ||
| 40 | MIC | 1 µg mL−1 | FOX |
| (2.22) | |||
| 41 | 1 µg mL−1 | ||
| (2.22) | |||
| 39 | 1 µg mL−1 | ||
| (2.84) | |||
| 92 | IC50 | 2.3 ± 1.0 µM | FOX M15-Pa |
| 93 | 15.2 ± 1.4 µM | ||
| 94 | 23 ± 8 µM | ||
| 77 | MIC | 3.12 µg mL−1 (8.68) | FOX |
| 78 | 3.12 µg mL−1 (8.68) | ||
| 111 | MIC | 3.12 µg mL−1 (8.73) | FOX |
| 112 | 6.25 µg mL−1 (17.78) | ||
| 26 | MIC | 3.9 µg mL−1 | FOX AB18 |
| (10.84) | |||
| 27 | 7.8 µg mL−1 | ||
| (26.46) | |||
| 75 | MIC | 8 µg mL−1 (12.11) | FOX MTCC 2480 |
| 76 | 8 µg mL−1 (12.11) | ||
| 21 | EC50 | 4.1 µg mL−1 (13.04) | FOXf. sp. lycopersici |
| 22 | 7.4 µg mL−1 (25.40) | ||
| 28 | MIC | 6.25 µg mL−1 (13.28) | FOX |
| 95 | MIC | 6.25 µg mL−1 | FOX |
| (24.13) | |||
| 96 | 6.25 µg mL−1 | ||
| (21.83) | |||
| 97 | 6.25 µg mL−1 | ||
| (20.01) | |||
| 98 | 6.25 µg mL−1 | ||
| (13.34) | |||
| 99 | 12.5 µg mL−1 | ||
| (32.51) | |||
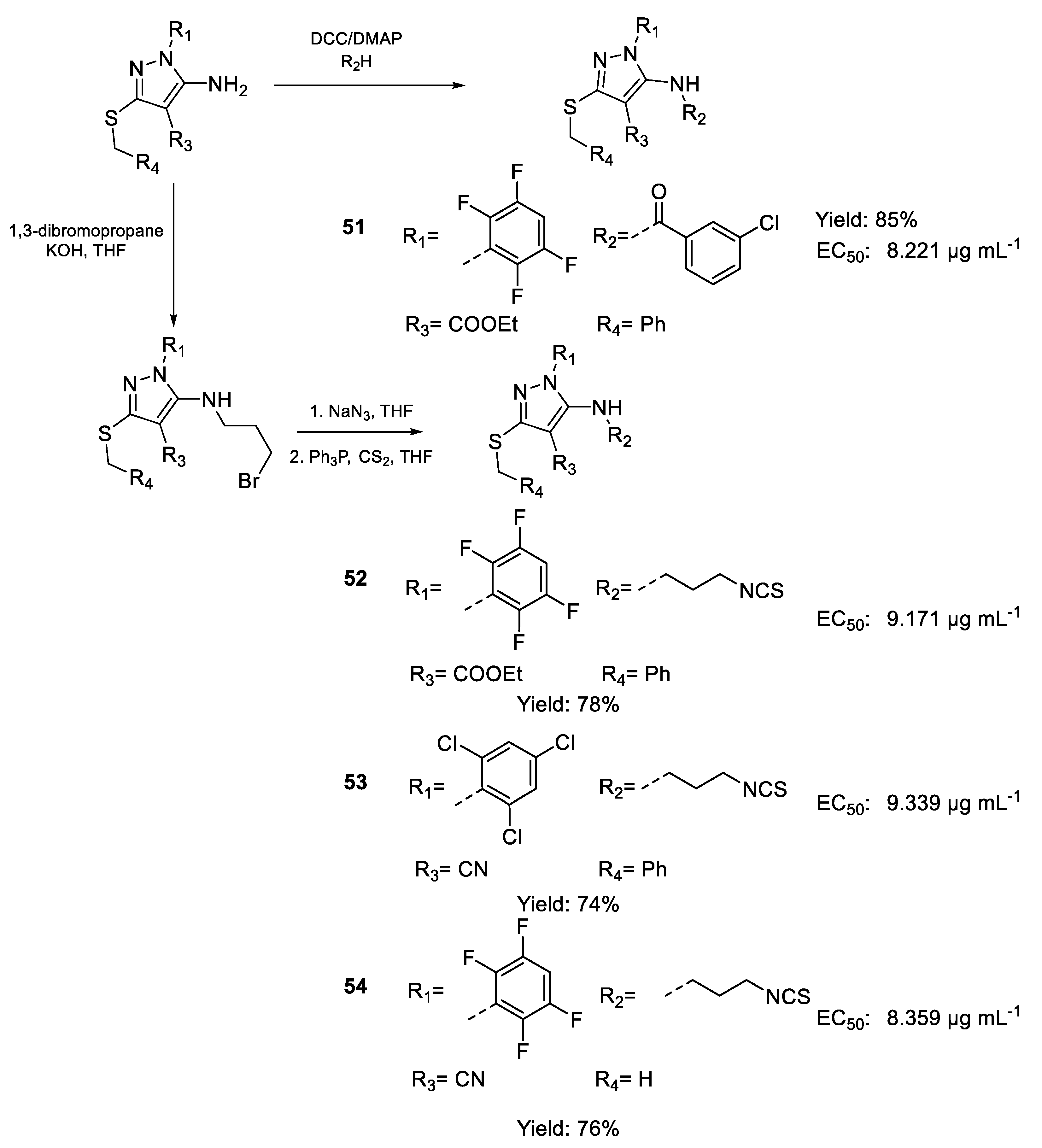
| 51 | EC50 | 8.221 µg mL−1 (14.58) | FOX (S-chl) f.sp |
| 52 | 9.171 µg mL−1 (17.48) | ||
| 53 | 9.339 µg mL−1 (18.36) | ||
| 54 | 8.359 µg mL−1 (20.82) | ||
| 15 | MIC | 16 µg mL−1 (16.41) | FOXf. sp. Betae & FOX f. sp. lycopersici |
| 16 | 16 µg mL−1 (16.89) | ||
| 56 | MIC | 9 µg mL−1 | FOX |
| (16.63) | |||
| 55 | 8 µg mL−1 | ||
| (18.65) | |||
| 57 | 9 µg mL−1 | ||
| (20.84) | |||
| 61 | MIC | 16 µg mL−1 (17.40) | FOX |
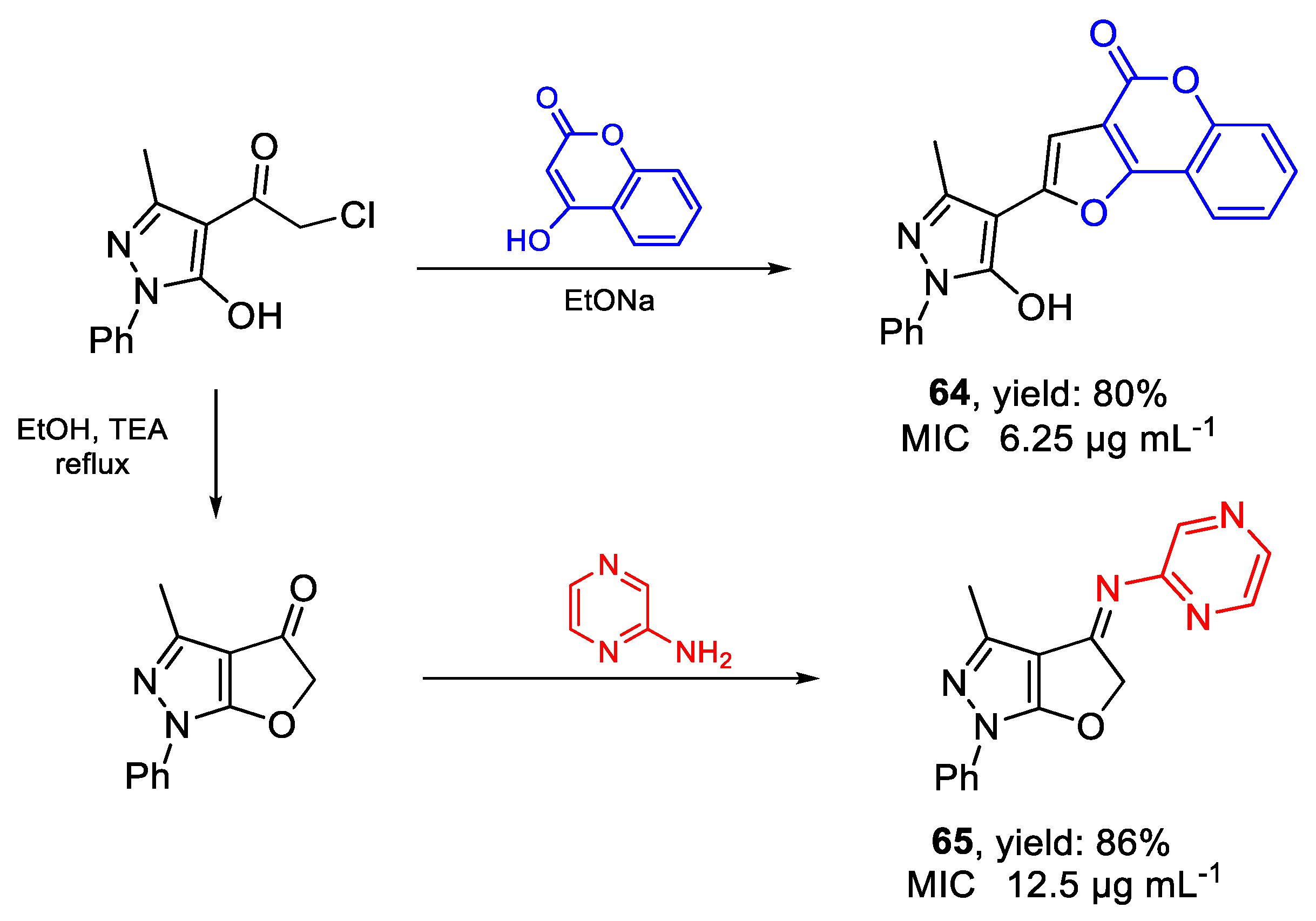
| 64 | MIC | 6.25 µg mL−1 | FOX ATCC 16417 |
| (17.44) | |||
| 65 | 12.5 µg mL−1 | ||
| (42.91) | |||
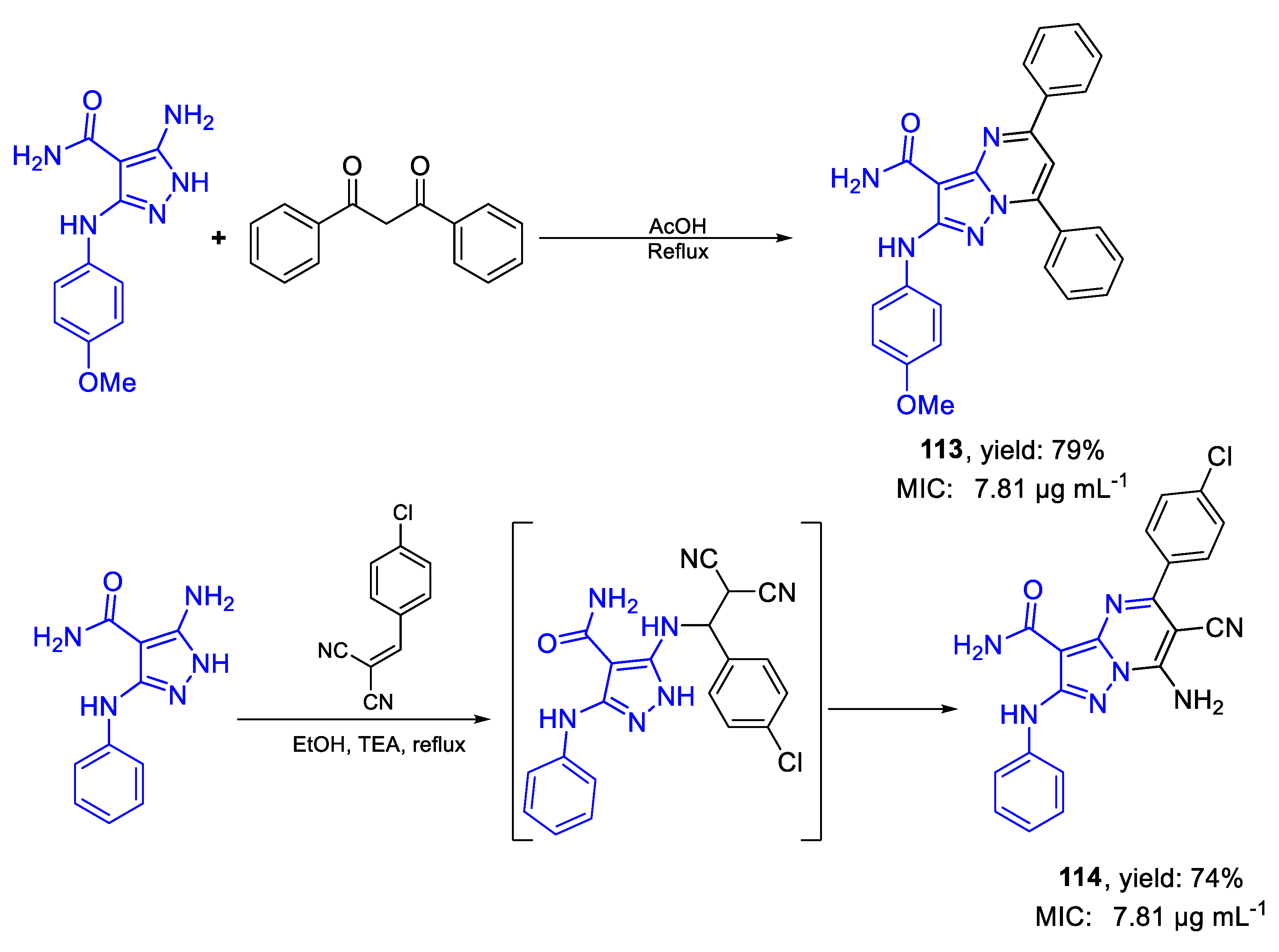
| 113 | MIC | 7.81 µg mL−1 | FOX RCMB 008002 |
| (17.93) | |||
| 114 | 7.81 µg mL−1 | ||
| (19.34) | |||
| 66 | MIC | 8 µg mL−1 (18.08) | FOX |
| 67 | 9 µg mL−1 (20.34) | ||
| 17 | MIC | 6.25 µg mL−1 (21.46) | FOX |
| 18 | 6.25 µg mL−1 (22.26) | ||
| 83 | MIC | 12.5 µg mL−1 | FOX NCIM 1332 |
| (24.58) | |||
| 85 | 25 µg mL−1 | ||
| (49.16) | |||
| 84 | 25 µg mL−1 | ||
| (51.59) | |||
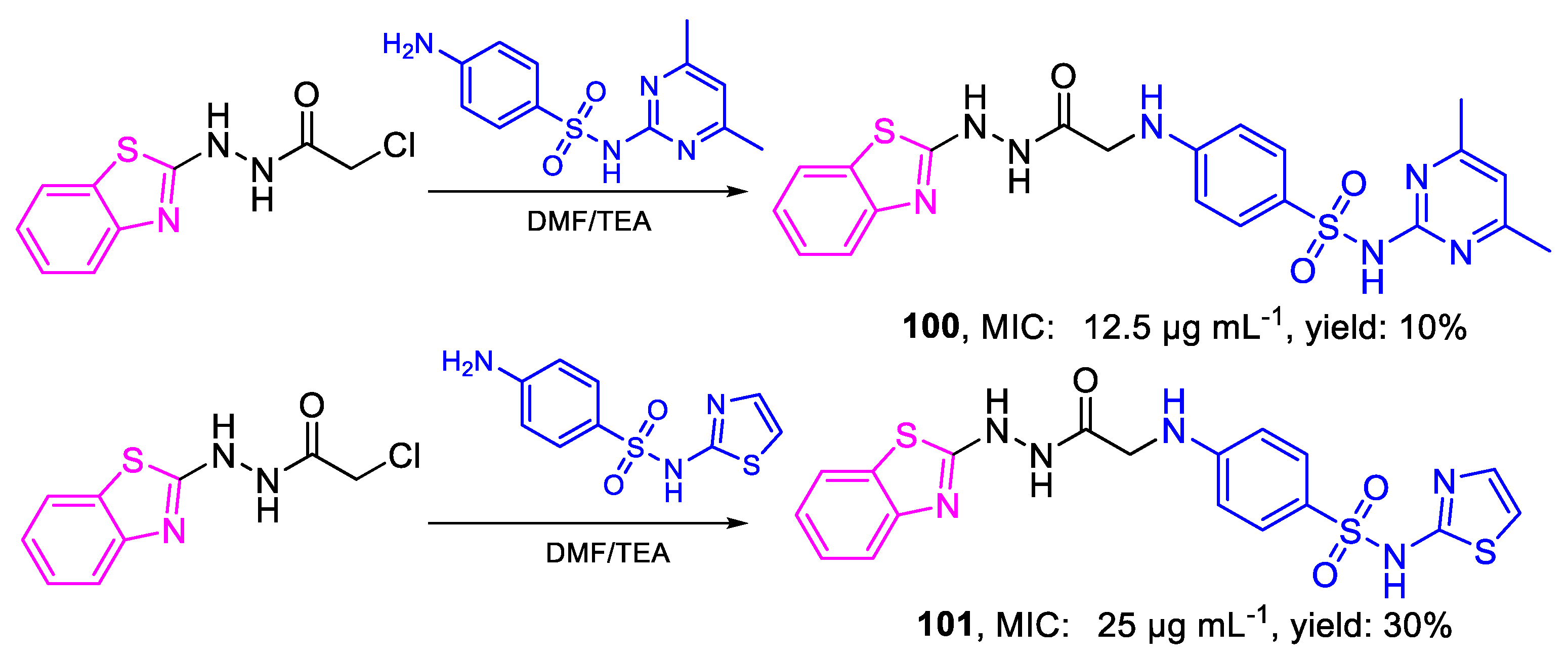
| 100 | MIC | 12.5 µg mL−1 | FOX |
| (25.85) | |||
| 101 | 25 µg mL−1 | ||
| (54.28) | |||
| 29 | MIC | 12.5 µg mL−1 (27.69) | FOX ATCC 16417 |
| 30 | 12.5 µg mL−1 (30.61) | ||
| 31 | EC50 | 10.1 µg mL−1 (27.79) | FOX |
| 32 | 10.6 µg mL−1 (29.33) | ||
| 115 | MIC | 8 µg mL−1 (27.94) | FOX |
| 116 | 8 µg mL−1 (29.60) | ||
| 86 | MIC | 12.5 µg mL−1 (28.42) | FOX NCIM 1332 |
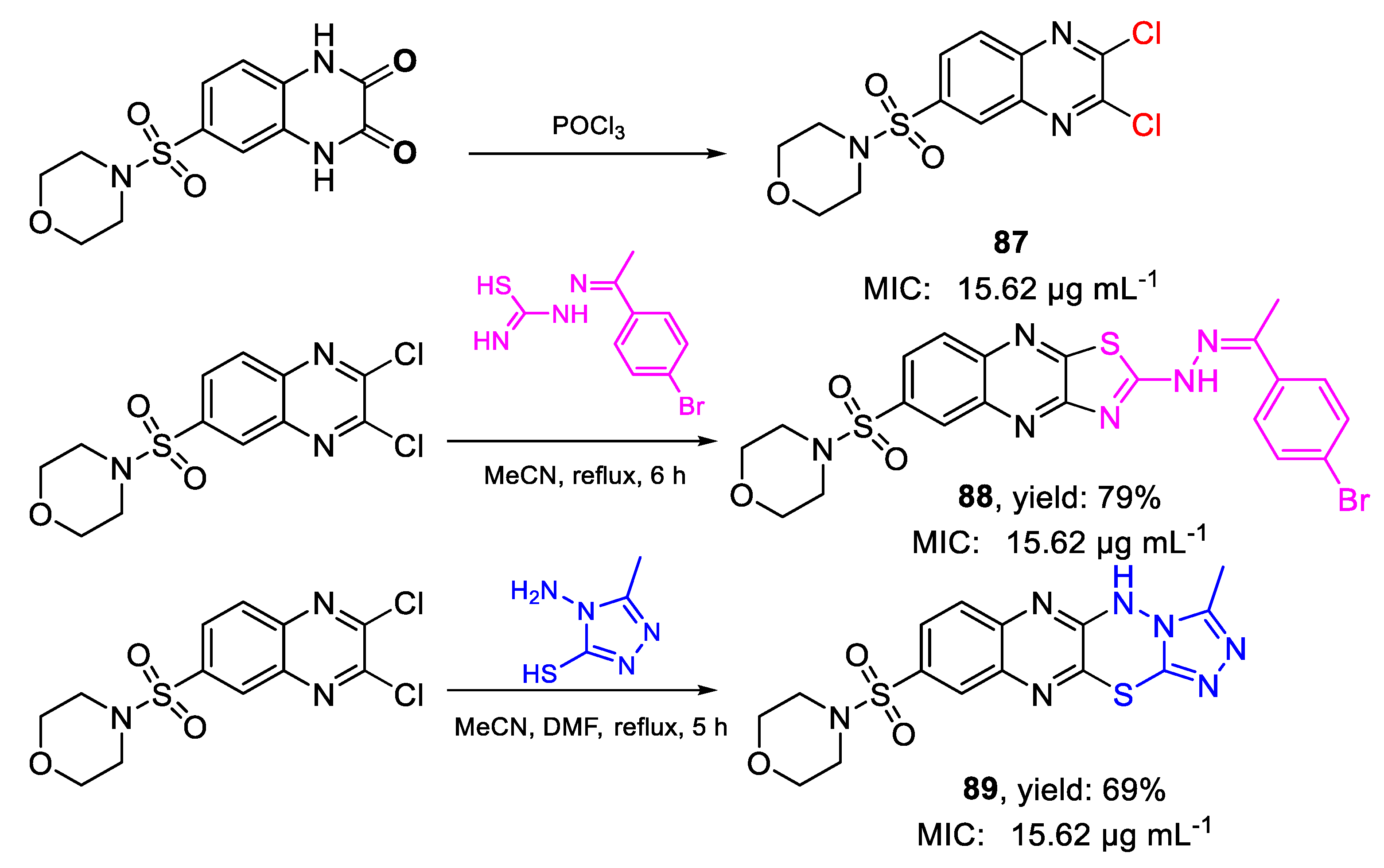
| 88 | MIC | 15.62 µg mL−1 | FOX RCMB 008002 |
| (28.53) | |||
| 89 | 15.62 µg mL−1 | ||
| (38.52) | |||
| 87 | 15.62 µg mL−1 | ||
| (44.86) | |||
| 10 | MIC | 15.6 µg mL−1 | FOX RCMB |
| (36.24) | 8002 | ||
| 11 | 15.6 µg mL−1 | ||
| (37.64) | |||
| 12 | 15.6 µg mL−1 | ||
| (39.15) | |||
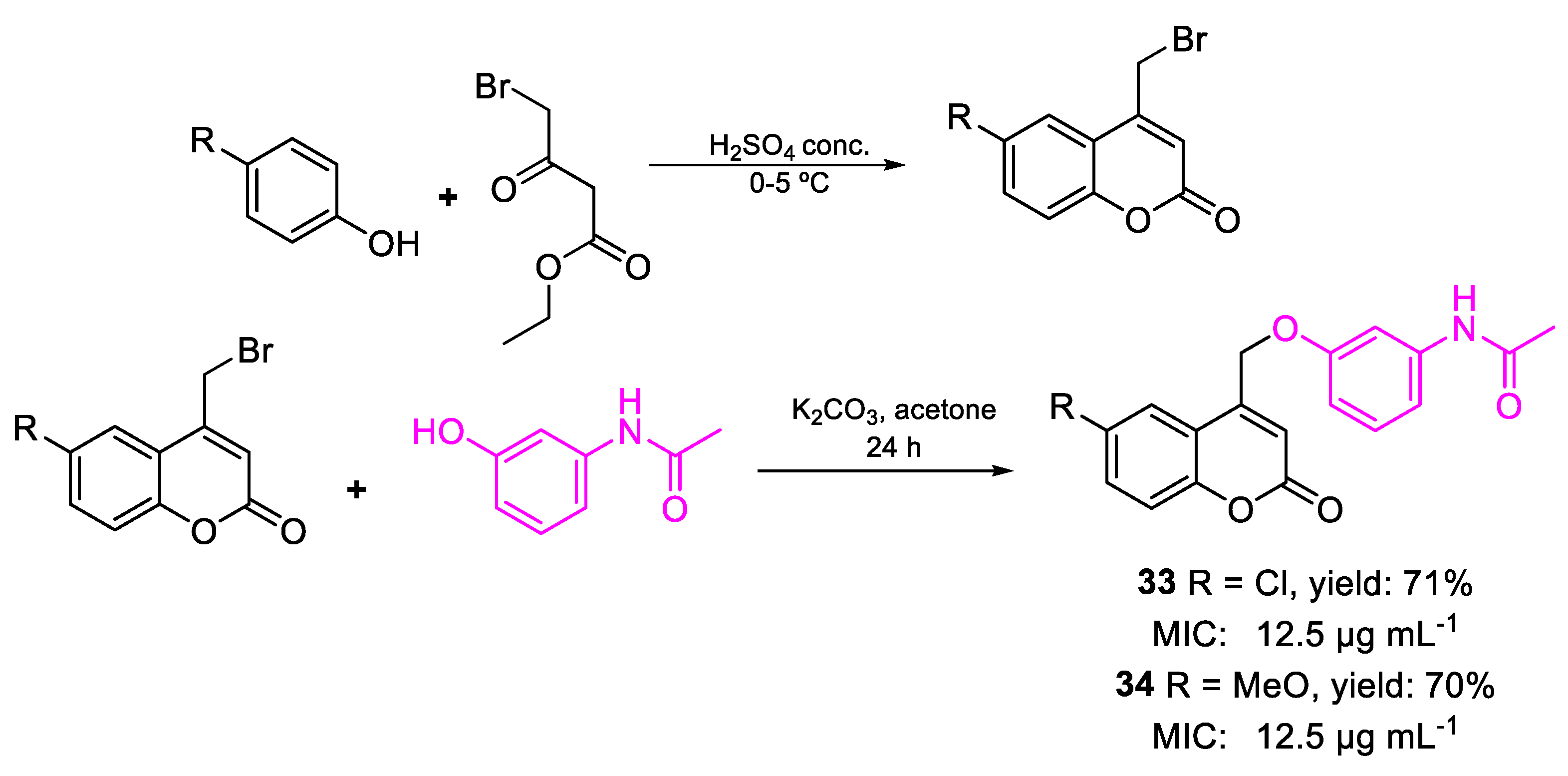
| 33 | MIC | 12.5 µg mL−1 (36.36) | FOX |
| 34 | 12.5 µg mL−1 (36.83) | ||
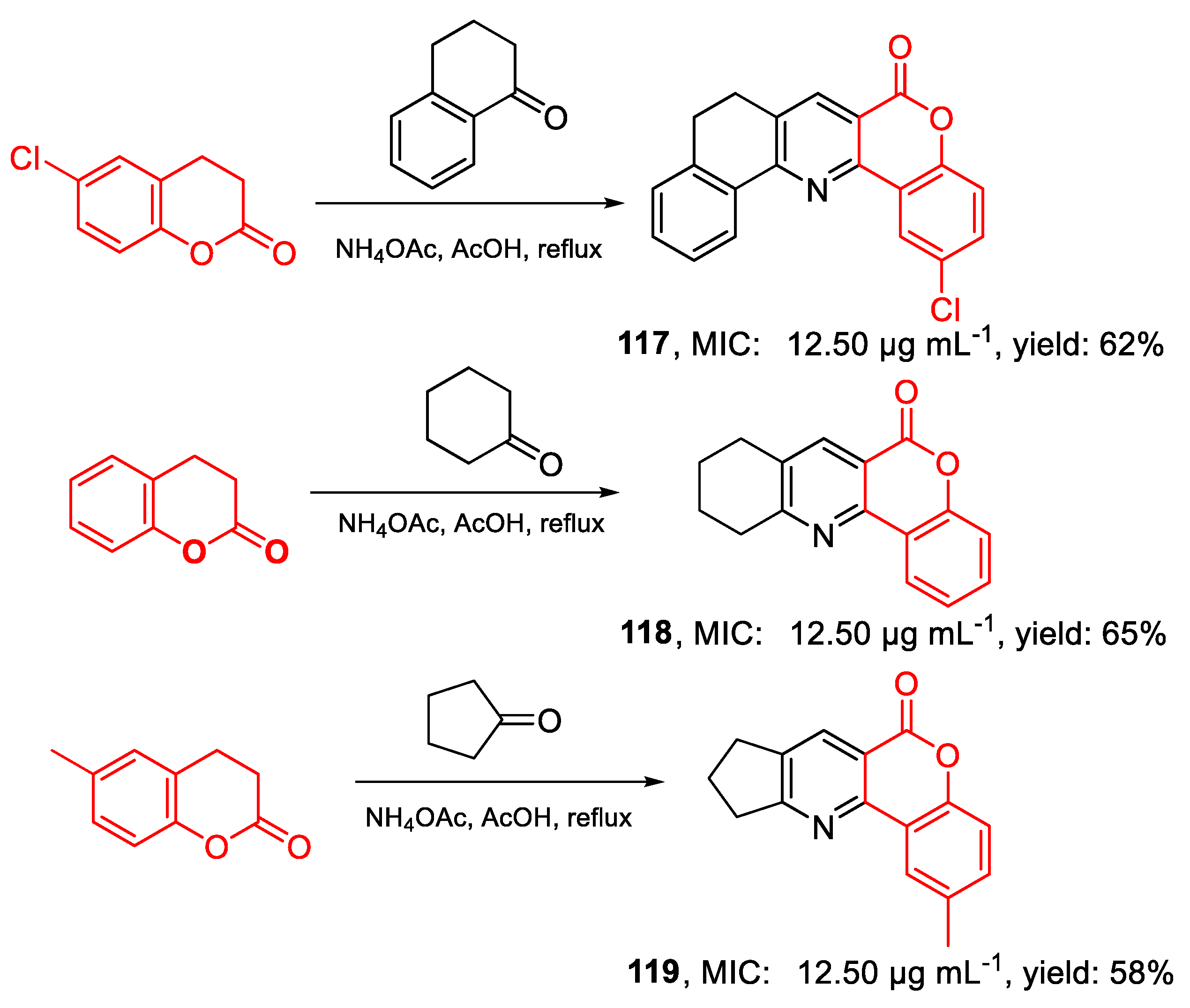
| 117 | MIC | 12.50 µg mL−1 (37.45) | FOX ATCC 16417 |
| 118 | 12.50 µg mL−1 (49.74) | ||
| 119 | 12.50 µg mL−1 (49.74) | ||
| 102 | MIC | 16 ± 1 µg mL−1 (37.63) | FOX |
| 103 | 18 ± 2 µg mL−1 (59.34) | ||
| 7 | MIC | 21 ± 3 µg mL−1 (39.14) | FOX |
| 8 | 23 ± 0 µg mL−1 (41.63) | ||
| 23 | MIC | 25 µg mL−1 (39.84) | FOX |
| 24 | 25 µg mL−1 (47.40) | ||
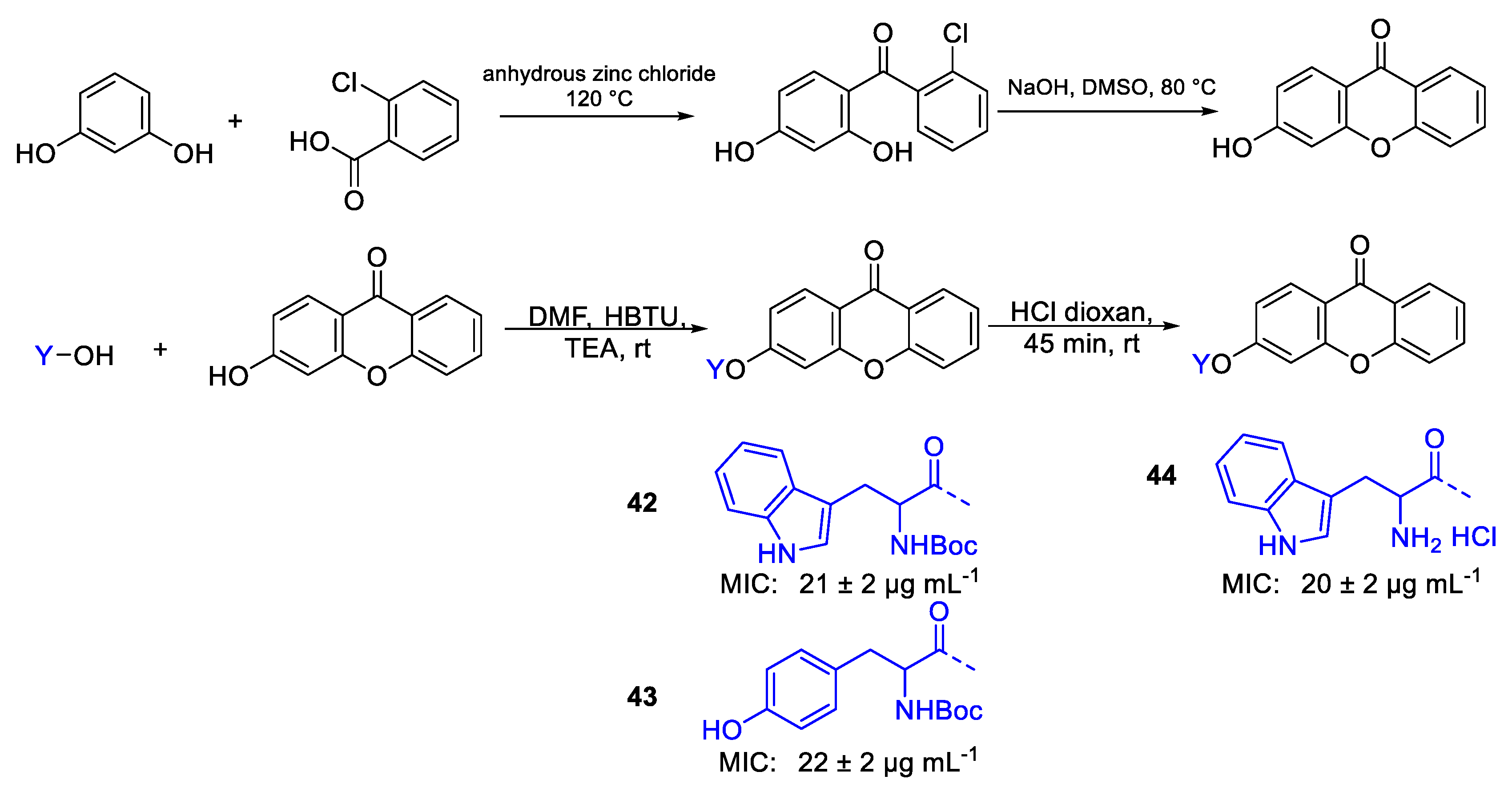
| 42 | MIC | 21 ± 2 µg mL−1 | FOX |
| (42.12) | |||
| 43 | 22 ± 2 µg mL−1 | ||
| (46.27) | |||
| 44 | 20 ± 2 µg mL−1 | ||
| (45.99) | |||
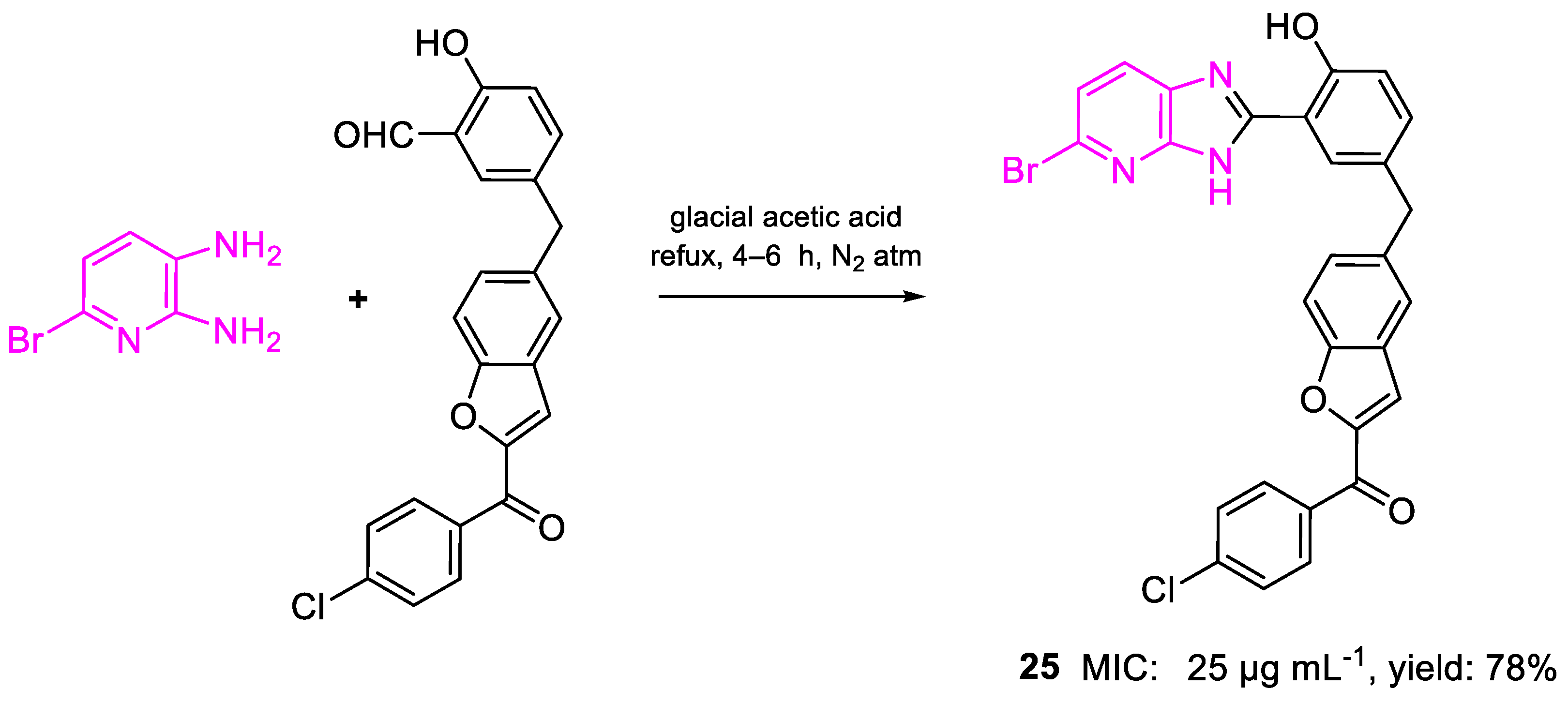
| 25 | MIC | 25 µg mL−1 (44.74) | FOX |
| 81 | MIC | 25 µg mL−1 (44.75) | FOX MTCC 284 |
| 82 | 25 µg mL−1 (57.01) | ||
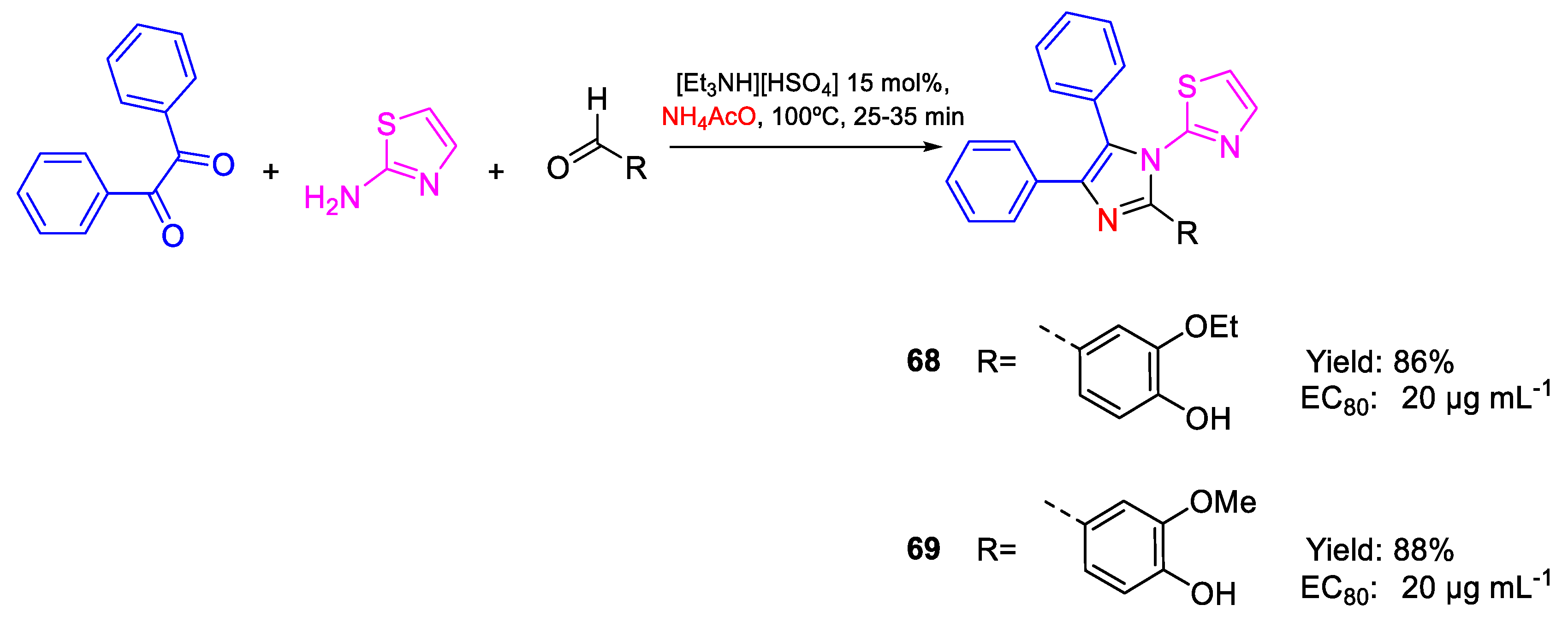
| 68 | EC80 | 20 µg mL−1 (45.50) | FOX NCIM 1332 |
| 69 | 20 µg mL−1 (47.00) | ||
| 19 | MIC | 12.5 µg mL−1 | FOX |
| (45.58) | |||
| 20 | 12.5 µg mL−1 | ||
| (47.40) | |||
| 120 | MIC | 16 µg mL−1 | FOX PTCC 5115 |
| (46.82) | |||
| 5 | IC50 | 52 ± 5 µM | FOX |
| 6 | 56 ± 3 µM | ||
| 121 | IC50 | 0.02 mg mL−1 | FOX |
| (53.69) | |||
| 9 | MIC | 25 µg mL−1 | FOX |
| (57.27) | |||
| 13 | MIC | 16 µg mL−1 | FOX ATCC 48112 |
| (58.24) | |||
| 14 | 32 µg mL−1 | ||
| (96.44) | |||
| 45 | MIC | 30 µg mL−1 (59.93) | FOX |
| 46 | 35 µg mL−1 (64.15) | ||
| 47 | 30 µg mL−1 | ||
| (66.59) | |||
| 48 | 35 µg mL−1 (72.83) | ||
| 122 | MIC | 25 ± 1.443 µg mL−1 | FOX |
| (61.14) | |||
| 123 | 25 ± 2.500 µg mL−1 | ||
| (64.03) | |||
| 1 | MIC | 19.8 ± 6.4 | FOX ATCC 48112 |
| µg mL−1 | |||
| (65.46) | |||
| 71 | MIC | 25 µg mL−1 (66.45) | FOX NCIM 1332 |
| 72 | 25 µg mL−1 (66.45) | ||
| 49 | EC50 | 18.0 µg mL−1 (67.10) | FOX |
| 50 | 21.5 µg mL−1 (84.56) | ||
| 35 | MIC | 25 µg mL−1 (70.67) | FOX NCIM 1332 |
| 36 | 25 µg mL−1 (71.56) | ||
| 124 | MIC | 25 µg mL−1 (71.39) | FOX |
| 125 | 25 µg mL−1 (79.04) | ||
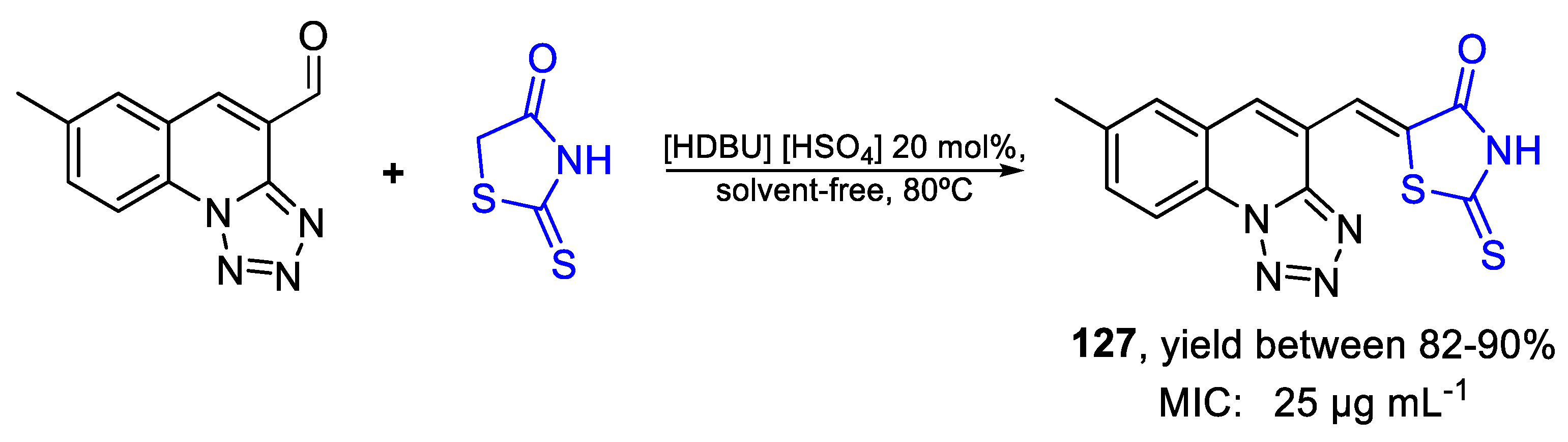
| 127 | MIC | 25 µg mL−1 | FOX |
| (76.36) | |||
| 79 | MIC | 32 µg mL−1 | FOX NCIM 1008 |
| (77.19) | |||
| 73 | MIC | 30 µg mL−1 | FOX NCIM 1332 |
| (78.63) | |||
| 74 | 40 µg mL−1 | ||
| (90.50) | |||
| 37 | MIC | 30 µg mL−1 | FOX NCIM 1332 |
| (78.86) | |||
| 38 | 28 µg mL−1 | ||
| (92.00) | |||
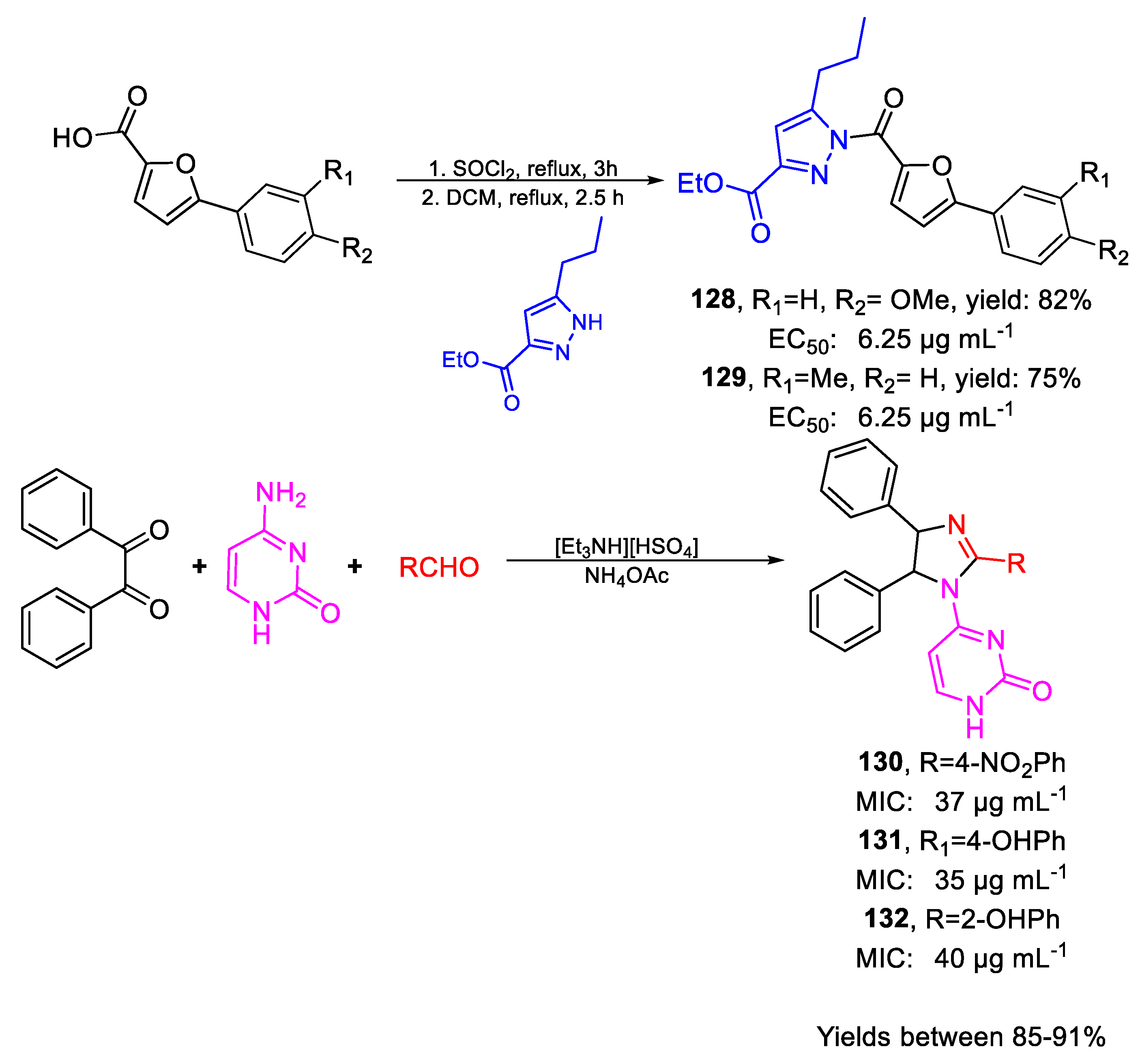
| 128 | EC50 | 6.25 µg mL−1 (80.54) | FOX |
| 129 | 6.25 µg mL−1 (98.52) | ||
| 130 | MIC80 | 37 µg mL−1 | FOX NCIM 1332 |
| (84.97) | |||
| 131 | 35 µg mL−1 | ||
| (86.11) | |||
| 132 | 40 µg mL−1 | ||
| (98.41) | |||
| 70 | IC50 | 0.086 mM (86.00) | FOX f. sp. albedinis |
| 126 | MIC | 50 µg mL−1 | FOX |
| (87.66) | |||
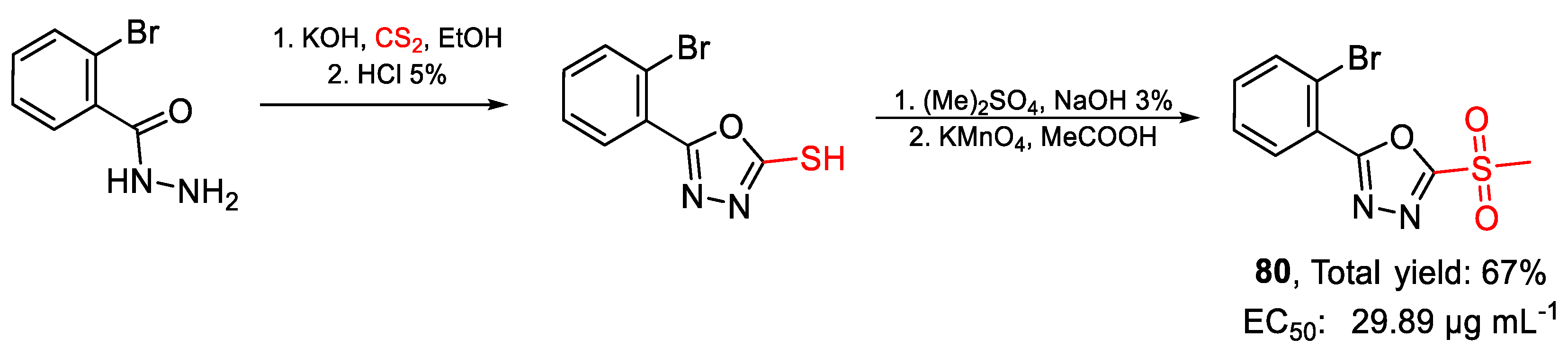
| 80 | EC50 | 29.89 µg mL−1 (98.60) | FOX |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borrego-Muñoz, P.; Ospina, F.; Quiroga, D. A Compendium of the Most Promising Synthesized Organic Compounds against Several Fusarium oxysporum Species: Synthesis, Antifungal Activity, and Perspectives. Molecules 2021, 26, 3997. https://doi.org/10.3390/molecules26133997
Borrego-Muñoz P, Ospina F, Quiroga D. A Compendium of the Most Promising Synthesized Organic Compounds against Several Fusarium oxysporum Species: Synthesis, Antifungal Activity, and Perspectives. Molecules. 2021; 26(13):3997. https://doi.org/10.3390/molecules26133997
Chicago/Turabian StyleBorrego-Muñoz, Paola, Felipe Ospina, and Diego Quiroga. 2021. "A Compendium of the Most Promising Synthesized Organic Compounds against Several Fusarium oxysporum Species: Synthesis, Antifungal Activity, and Perspectives" Molecules 26, no. 13: 3997. https://doi.org/10.3390/molecules26133997
APA StyleBorrego-Muñoz, P., Ospina, F., & Quiroga, D. (2021). A Compendium of the Most Promising Synthesized Organic Compounds against Several Fusarium oxysporum Species: Synthesis, Antifungal Activity, and Perspectives. Molecules, 26(13), 3997. https://doi.org/10.3390/molecules26133997