
Densely Functionalized 2-Methylideneazetidines: Evaluation as Antibacterials

 , ,
, , 
 , , ,
, , ,  , and
, and 
Abstract
:
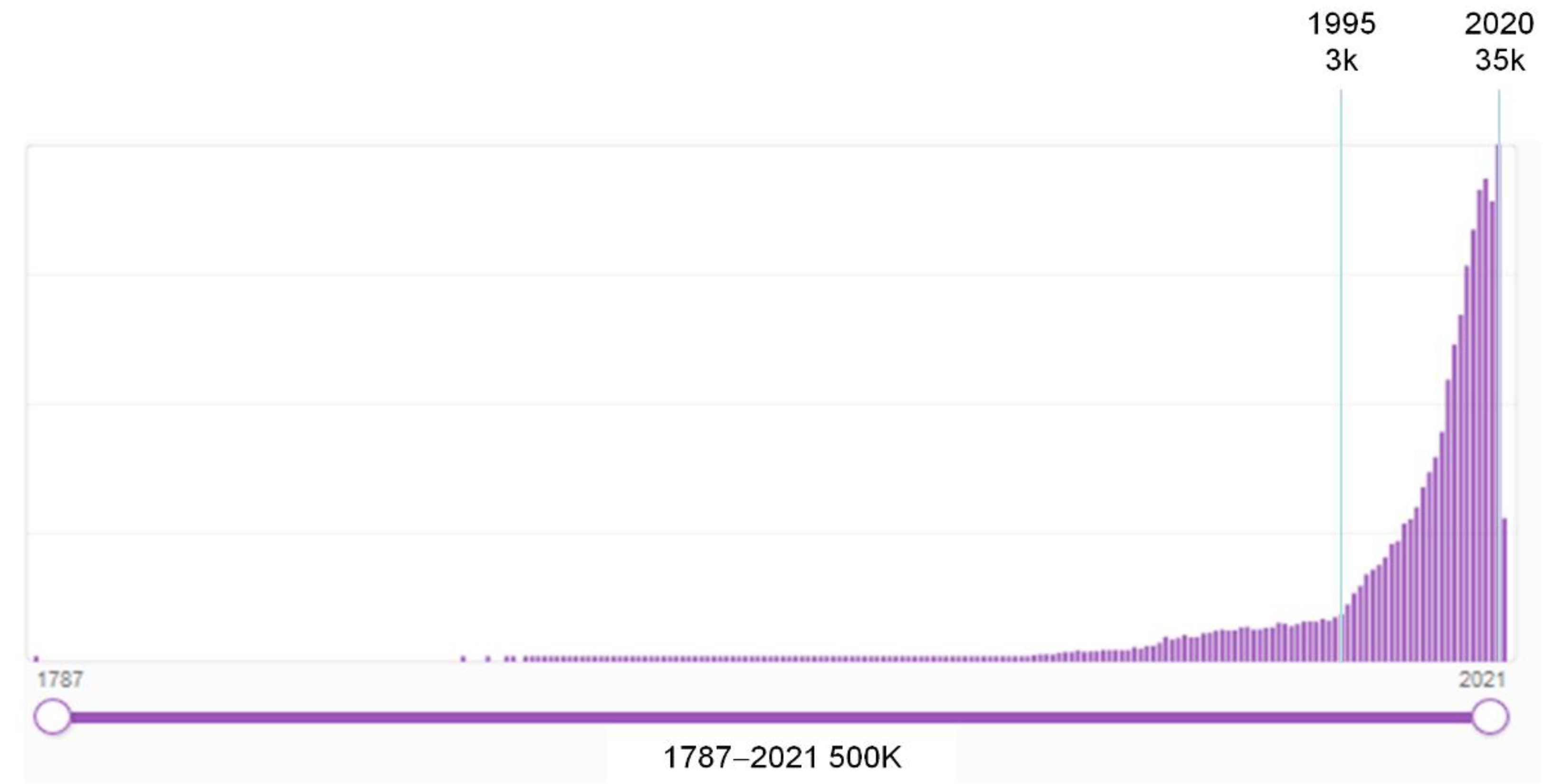
1. Introduction
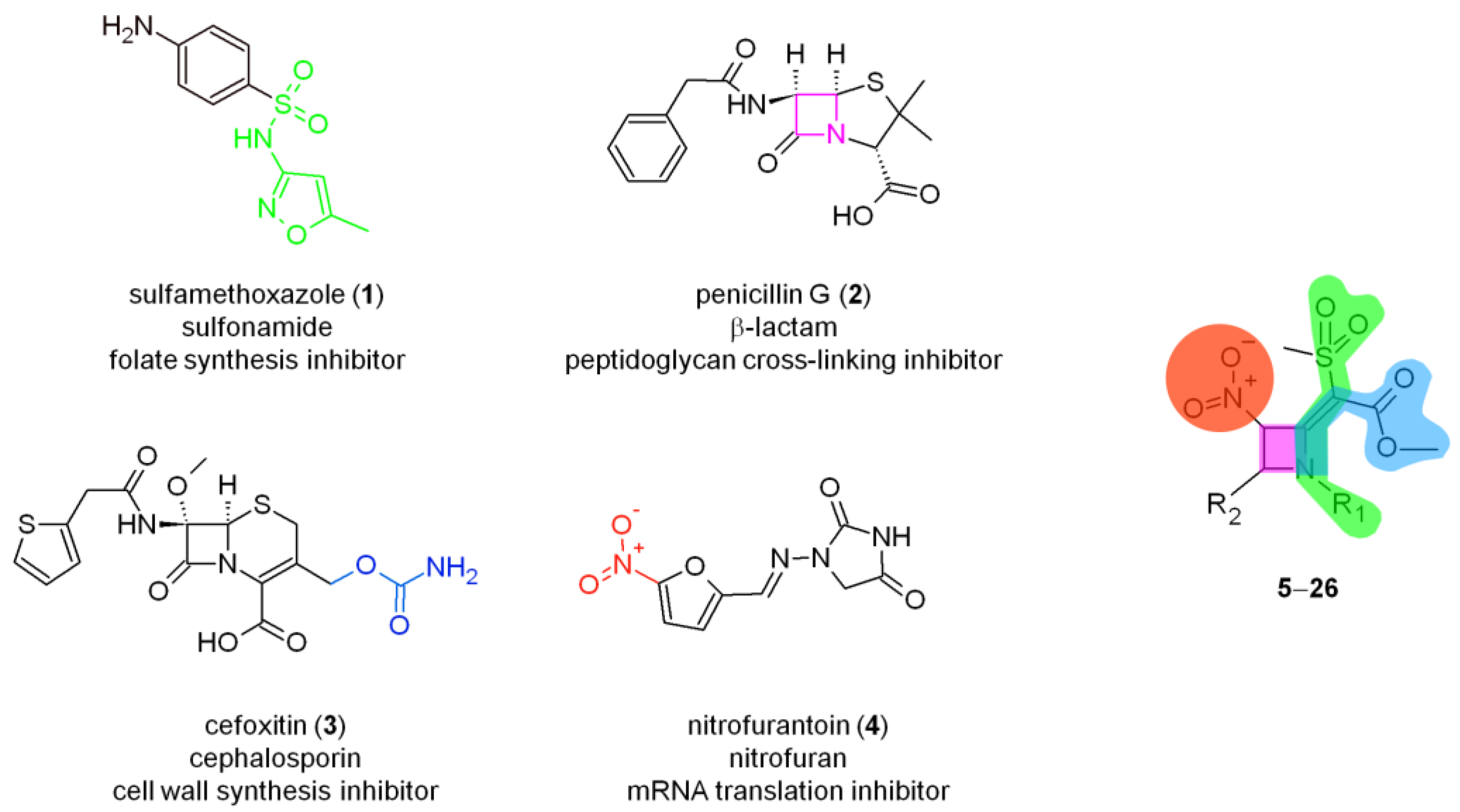
2. Results and Discussion
- mouse (ip): 190 mg/kg
- mouse (po): 2300 mg/kg
- mouse (iv): 56 mg/kg
- mouse (sc): 46 mg/kg
- rat (ip): 51 mg/kg
- rat (po): 2400 mg/kg.
3. Materials and Methods
3.1. Chemistry
3.2. Microbiology
3.2.1. Antibacterial Assay
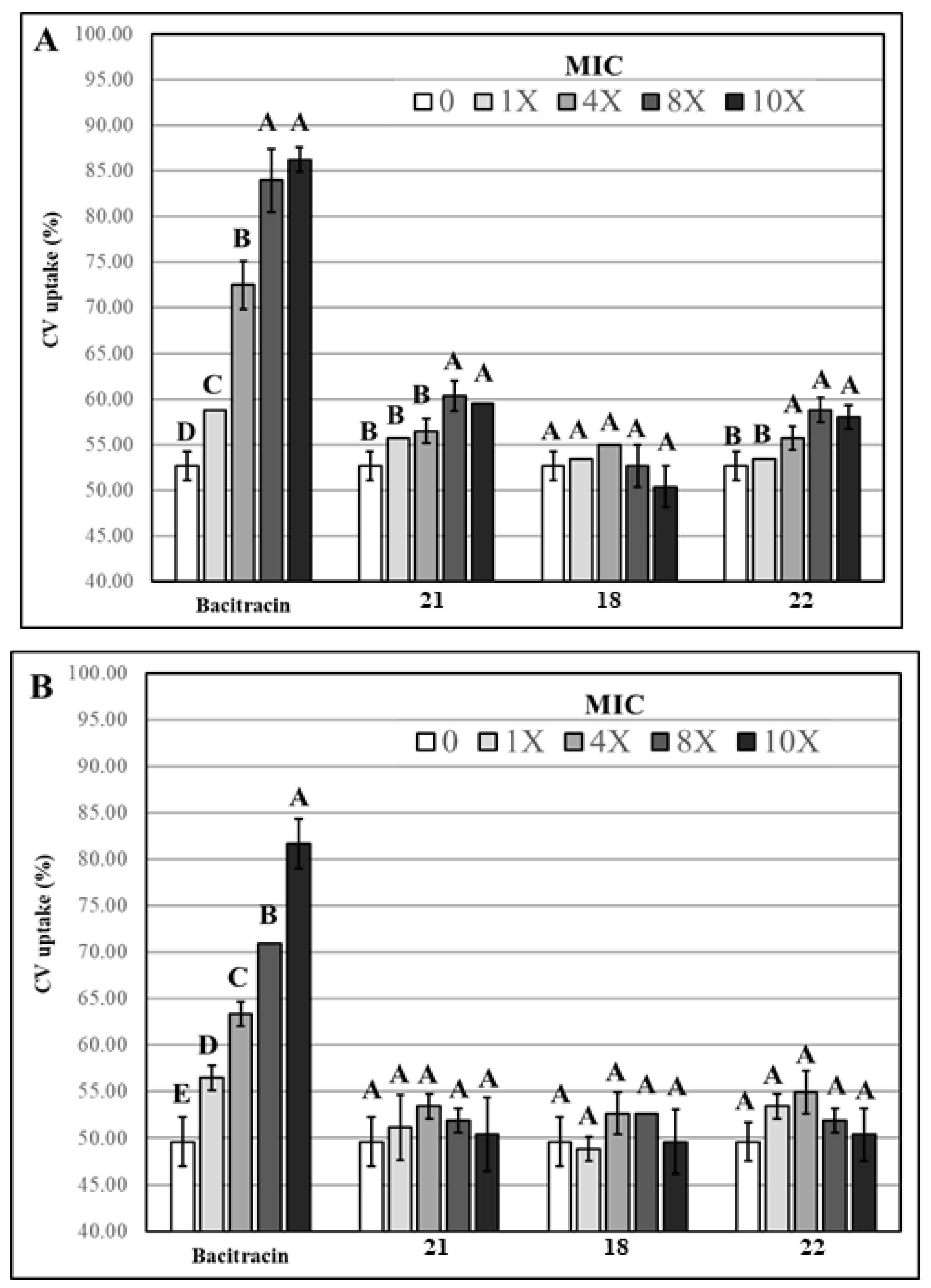
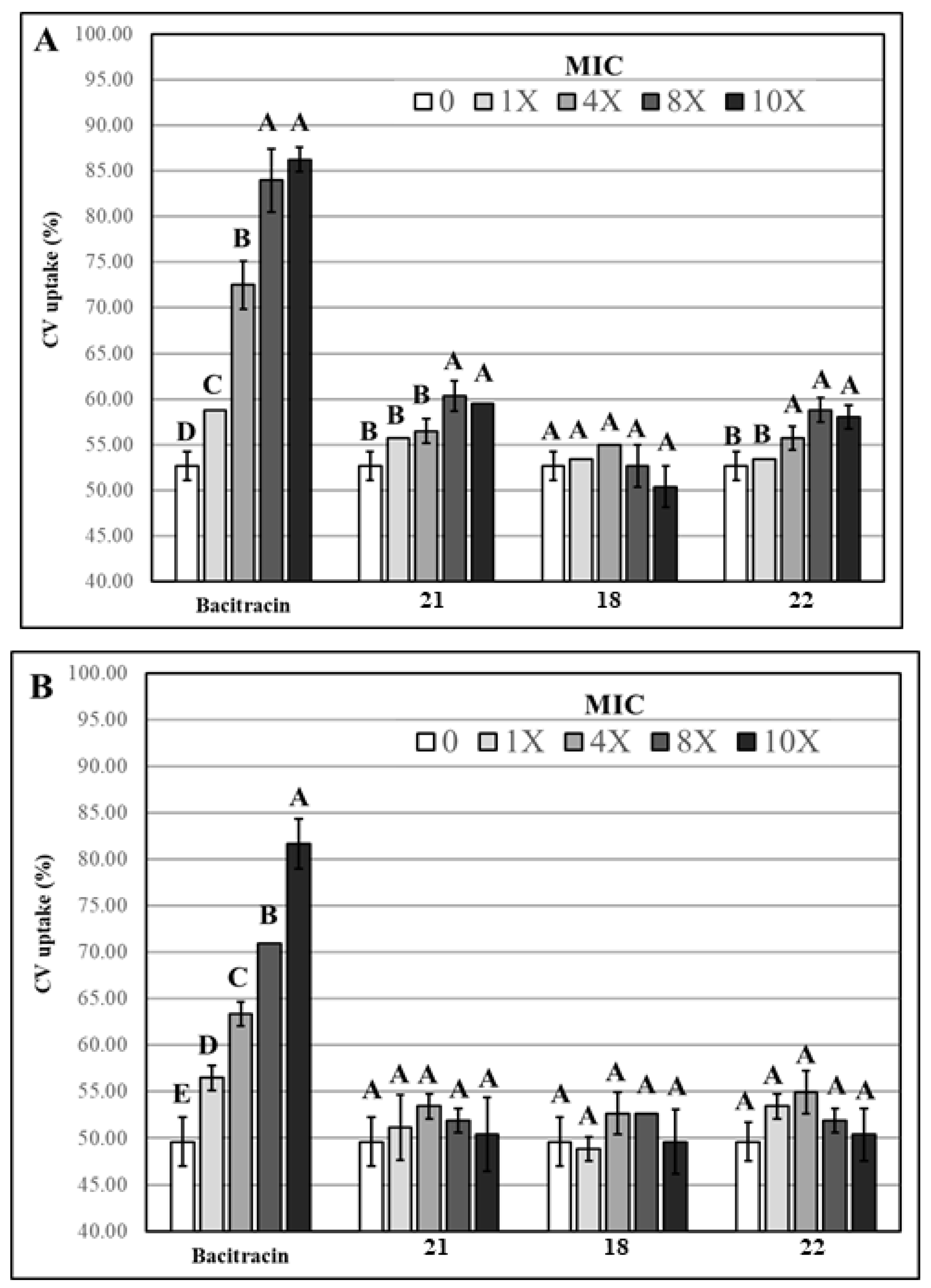
3.2.2. Evaluation of Membrane Permeability
3.2.3. Statistical Analysis
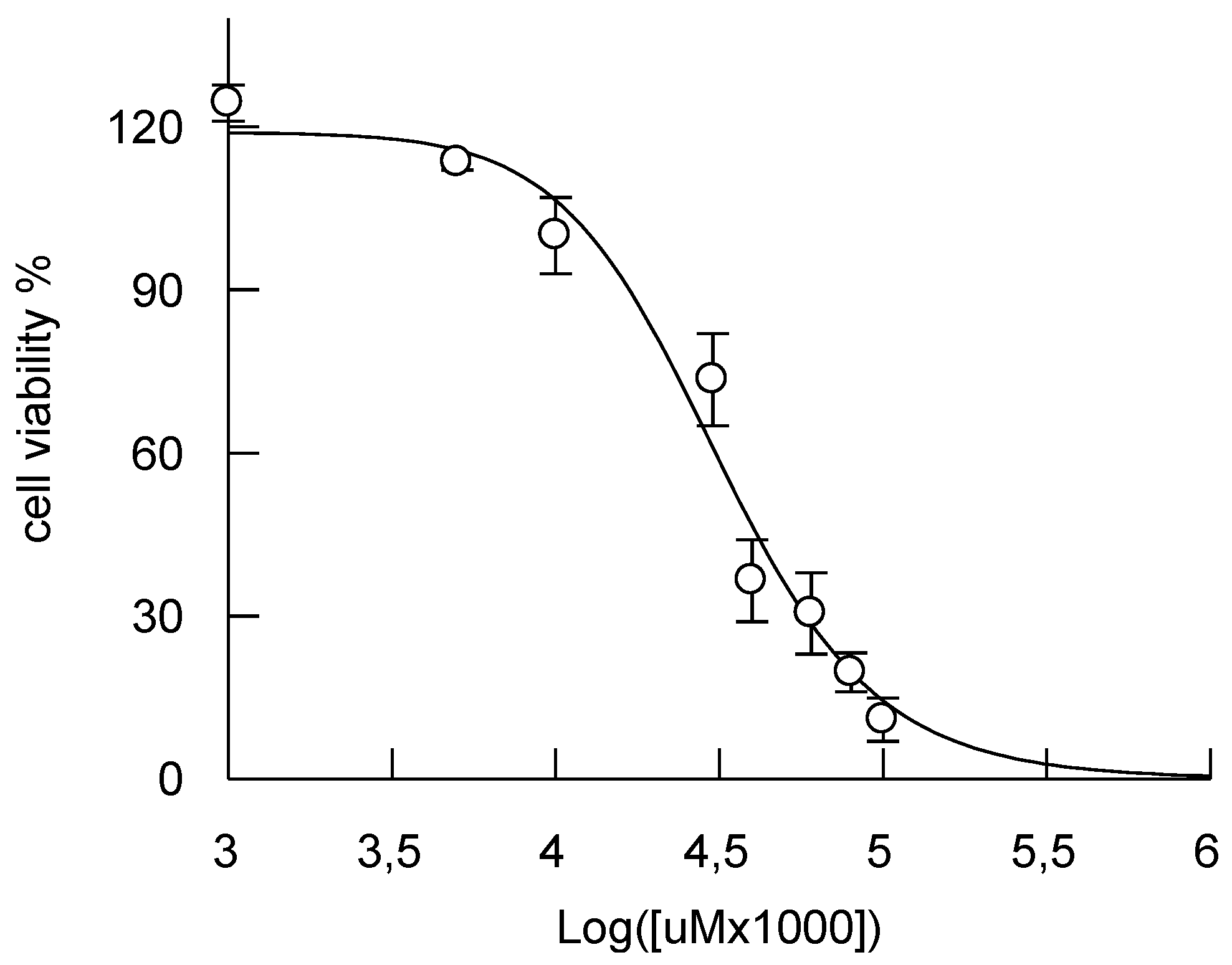
3.3. Cytotoxicity Assay
3.4. Quantum Mechanical Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Levy, S.B. The Antibiotic Paradox: How Miracle Drugs Are Destroying the Miracle; Plenum Publishers: New York, NY, USA, 1992. [Google Scholar]
- Navidinia, M. The clinical importance of emerging ESKAPE pathogens in nosocomial infections. J. Paramed. Sci. 2016, 7, 2008–4978. [Google Scholar]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- SciFindern Home Page. Available online: https://scifinder-n.cas.org (accessed on 13 April 2021).
- Parkes, A.L. Antibacterial medicinal chemistry–what can we design for? Expert Opin. Drug Discov. 2020, 15, 1005–1013. [Google Scholar] [CrossRef]
- Singh, S.B.; Young, K.; Miesel, L. Screening strategies for discovery of antibacterial natural products. Expert Rev. Anti-Infe. 2011, 9, 589–613. [Google Scholar]
- Lazzara, P.R.; Moore, T.W. Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. RSC Med. Chem. 2020, 11, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Fuson, R.C. The Principle of Vinylogy. Chem. Rev. 1935, 16, 1–27. [Google Scholar] [CrossRef]
- Sharma, R.; Samadhiya, P.; Srivastava, S.D.; Srivastava, S.K. Synthesis and biological activity of 2-oxoazetidine derivatives of phenothiazine. Org Commun. 2011, 4, 42–51. [Google Scholar]
- Patel, R.; Bhandari, A. Synthesis and Antimicrobial Screening of Some Azetidine Derivatives. Am. J. Adv. Drug Deliv. 2014, 2, 104–109. [Google Scholar]
- Tavani, C.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G. Densely Functionalized 2-Methylideneazetidines from Nitrodienic Building Blocks. Eur. J. Org. Chem. 2018, 126–136. [Google Scholar] [CrossRef]
- Sana, S.; Datta, S.; Biswas, D.; Sengupta, D. Assessment of synergistic antibacterial activity of combined biosurfactants revealed by bacterial cell envelop damage. BBA Biomembr. 2018, 1860, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Luo, M.; Fu, Y.J.; Zu, Y.G.; Wang, W.; Zhang, L.; Yao, L.P.; Zhao, C.J.; Sun, Y. Effect of corilagin on membrane permeability of Escherichia coli, Staphylococcus aureus and Candida albicans. Phytother. Res. 2013, 27, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz Nizer, W.S.; Ferraz, A.C.; Moraes, T.D.F.S.; Lima, W.G.; Dos Santos, J.P.; Duarte, L.P.; Ferreira, J.M.S.; de Brito Magalhães, C.L.; Vieira-Filho, S.A.; Andrade, A.C.S.P.; et al. Pristimerin isolated from Salacia crassifolia (Mart. Ex. Schult.) G. Don. (Celastraceae) roots as a potential antibacterial agent against Staphylococcus aureus. J. Ethnopharmacol. 2021, 266, 113423. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. BBA Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef] [Green Version]
- The Bacterial Diversity Metadatabase. Available online: https://bacdive.dsmz.de/ (accessed on 11 June 2021).
- Revell, K.D.; Heldreth, B.; Long, T.E.; Jang, S.; Turos, E. N-thiolated β-lactams: Studies on the mode of action and identification of a primary cellular target in Staphylococcus aureus. Bioorg. Med. Chem. 2007, 15, 2453–2467. [Google Scholar] [CrossRef] [Green Version]
- Lanne, A.; Cui, Y.; Browne, E.; Craven, P.G.E.; Cundy, N.J.; Coltman, N.J.; Dale, K.; Feula, A.; Frampton, J.; Goff, A. Azetidines kill Mycobacterium tuberculosis without detectable resistance by blocking mycolate assembly. bioRxiv 2020. [Google Scholar] [CrossRef]
- Boyd, D.B. β-Lactam Antibacterial Agents: Computational Chemistry Investigations. In The Amide Linkage; Greenberg, A., Breneman, C.M., Liebman, J.F., Eds.; Wiley: New York, NY, USA, 2000; pp. 337–375. [Google Scholar]
- Cooper, R.D.G.; De Marco, P.V.; Cheng, J.C.; Jones, N.D. Structural Studies on Penicillin Derivatives. J. Am. Chem. Soc. 1969, 91, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.D.; van der Veen, J.M. Conformations of penicillin G: Crystal structure of procaine penicillin G monohydrate and a refinement of the structure of potassium penicillin G. J. Chem. Soc. Perkin I. 1978, 185–190. [Google Scholar] [CrossRef]
- Carocci, A.; Roselli, M.; Budriesi, R.; Micucci, M.; Desaphy, J.F.; Altamura, C.; Cavalluzzi, M.M.; Toma, M.; Passeri, G.I.; Milani, G. Synthesis and Evaluation of Voltage-Gated Sodium Channel Blocking Pyrroline Derivatives Endowed with Both Antiarrhythmic and Antioxidant Activities. Chem. Med. Chem. 2021, 16, 578–588. [Google Scholar] [CrossRef]
- Gnocchi, D.; Cavalluzzi, M.M.; Mangiatordi, G.F.; Rizzi, R.; Tortorella, C.; Spennacchio, M.; L28ntini, G.; Altomare, A.; Sabbà, C.; Mazzocca, A. Xanthenylacetic Acid Derivatives Effectively Target Lysophosphatidic Acid Receptor 6 to Inhibit Hepatocellular Carcinoma Cell Growth. Chem. Med. Chem. 2021, 16, 1–10. [Google Scholar]
- Smith, D.J.; Anderson, R.C. Toxicity and Metabolism of Nitroalkanes and Substituted Nitroalkanes. Agric. Food Chem. 2013, 61, 763–779. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Mangiatordi, G.F.; Nicolotti, O.; Lentini, G. Ligand Efficiency Metrics in Drug Discovery: The Pros and Cons from a Practical Perspective. Expert Opin. Drug Discov. 2017, 12, 1087–1104. [Google Scholar] [CrossRef] [PubMed]
- Roselli, M.; Carocci, A.; Budriesi, R.; Micucci, M.; Toma, M.; Di Cesare Mannelli, L.; Lovece, A.; Catalano, A.; Cavalluzzi, M.M.; Bruno, C.; et al. Synthesis, Antiarrhythmic Activity, and Toxicological Evaluation of Mexiletine Analogues. Eur. J. Med. Chem. 2016, 121, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comp. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity Index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Benzi, A.; Bianchi, L.; Giorgi, G.; Maccagno, M.; Petrillo, G.; Tavani, C. 2-Aryl-3-Vinyl Substituted Imidazo [1,2-a] pyridines and Fluorescent Electrocyclization Derivatives therefrom. ChemistrySelect 2020, 5, 4552–4558. [Google Scholar] [CrossRef]
- Cavalluzzi, M.M.; Budriesi, R.; De Salvia, M.A.; Quintieri, L.; Piarulli, M.; Milani, G.; Gualdani, R.; Micucci, M.; Corazza, I.; Rosato, A.; et al. Lubeluzole: From Anti-Ischemic Drug to Preclinical Antidiarrheal Studies. Pharmacol. Rep. 2021, 73, 172–184. [Google Scholar] [CrossRef]
- Berridge, M.V.; Tan, A.S. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): Subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 1993, 303, 474–482. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. I.; Basis, Form, Scope, Parameterization, and Performance of Mmff94. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Davidson, E.R.; Feller, D. Basis Set Selection for Molecular Calculations. Chem. Rev. 1986, 86, 681–696. [Google Scholar] [CrossRef]
- Lange, A.W.; Herbert, J.M.A. Smooth, Nonsingular, and Faithful Discretization Scheme for Polarizable Continuum Models: The Switching/Gaussian Approach. J. Chem. Phys. 2010, 133, 244111. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Herbert, J.M. Symmetric versus Asymmetric Discretization of the Integral Equations in Polarizable Continuum Solvation Models. Chem. Phys. Lett. 2011, 509, 77–87. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, D.L. “Collateral Damage” from Cephalosporin or Quinolone Antibiotic Therapy. Clin. Infect. Dis. 2004, 38, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wijdeveld, M.; Nieuwdorp, M.; IJzerman, R. The Interaction between Microbiome and Host Central Nervous System: The Gut-Brain Axis as a Potential New Therapeutic Target in the Treatment of Obesity and Cardiometabolic Disease. Expert Opin. Ther. Tar. 2020, 24, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Chu, H.; Duan, Y.; Schnabl, B. Gut Microbiota in Liver Disease: Too Much Is Harmful, Nothing at All Is Not Helpful Either. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G563–G573. [Google Scholar] [CrossRef]
- Zhou, A.; Tang, L.; Zeng, S.; Lei, Y.; Yang, S.; Tang, B. Gut Microbiota: A New Piece in Understanding Hepatocarcinogenesis. Cancer Lett. 2020, 474, 15–22. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
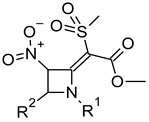
| Compound | R1 | R2 | S. Aureus ATCC 29,213 (G) | S. Aureus ATCC 6538 P (G +) | S. Aureus ATCC 6538 (G) | E. Faecalis ATCC 29,212 (G) | cLog Pb |
| 5 | Me | p-tolyl | >512 (>1000) | >512 (>1000) | 512 (>1000) | >512 (>1000) | 0.42 |
| 6 | Et | p-tolyl | >512 (>1000) | 256 (696) | 256 (696) | >512 (>1000) | 0.75 |
| 7 | C6H11 | p-tolyl | >512 (>1000) | >512 (>1000) | 512 (>1000) | >512 (>1000) | 1.96 |
| 8 | n-Bu | Ph | >512 (>1000) | >512 (>1000) | >512 (>1000) | >512 (>1000) | 1.17 |
| 9 | n-Bu | 3,5-(CF3)2Ph | >512 (>988) | >512 (>988) | >512 (>988) | >512 (>988) | 3.01 |
| 10 | n-Bu | p-MeSO2Ph | 256 (556) | 128 (278) | 128 (278) | 256 (556) | 0.40 |
| 11 | Bn | C6H11 | >512 (>1000) | >512 (>1000) | 256 (607) | >512 (>1000) | 1.91 |
| 12 | n-Bu | 2-thienyl | 64 (165) | 32 (82) | 32 (82) | >512 (>1000) | −0.14 |
| 13 | Ph | p-tolyl | 256 (615) | 256 (615) | 256 (615) | 256 (615) | 2.49 |
| 14 | Bn | p-tolyl | 32 (74) | 16 (37) | 16 (37) | >512 (>1000) | 2.10 |
| 15 | Bn | 2-thienyl | 16 (38) | 16 (38) | 16 (38) | 32 (76) | 0.35 |
| 16 | o-ClBn | 2-thienyl | 64 (140) | 32 (70) | 128 (280) | 32 (70) | 0.91 |
| 17 | m-ClBn | 2-thienyl | 32 (70) | 16 (35) | 128 (280) | 64 (140) | 0.91 |
| 18 | p-ClBn | 2-thienyl | 16 (35) | 8 (17.5) | 16 (35) | 8 (17.5) | 0.91 |
| 19 | 3,4-Cl2Bn | 2-thienyl | 8 (16.2) | 8 (16.2) | 16 (32) | 16 (32) | 1.47 |
| 20 | o-ClBn | p-tolyl | 32 (69) | 16 (34) | 128 (276) | 64 (138) | 2.71 |
| 21 | m-ClBn | p-tolyl | 8 (17.2) | 8 (17.2) | 8 (17.2) | 16 (34) | 2.71 |
| 22 | p-ClBn | p-tolyl | 4 (8.6) | 4 (8.6) | 4 (8.6) | 4 (8.6) | 2.71 |
| 23 | 3,4-Cl2Bn | p-tolyl | 8 (16.0) | 8 (16.0) | 16 (32) | 32 (64) | 3.26 |
| 24 | Bn | p-MeSO2Ph | 256 (518) | 128 (259) | 256 (518) | 256 (518) | 0.89 |
| 25 | Bn | 3,5-(CF3)2Ph | 64 (116) | 8 (14.5) | 64 (116) | 128 (232) | 3.50 |
| 26 | p-ClBn | 3,5-(CF3)2Ph | >512 (>1000) | >512 (>1000) | >512 (>1000) | >512 (>1000) | 4.06 |
| norfloxacin | - | - | 1 (3.1) | 0.50 (1.57) | 0.50 (1.57) | 2 (6.2) | 0.82 |
 | |||
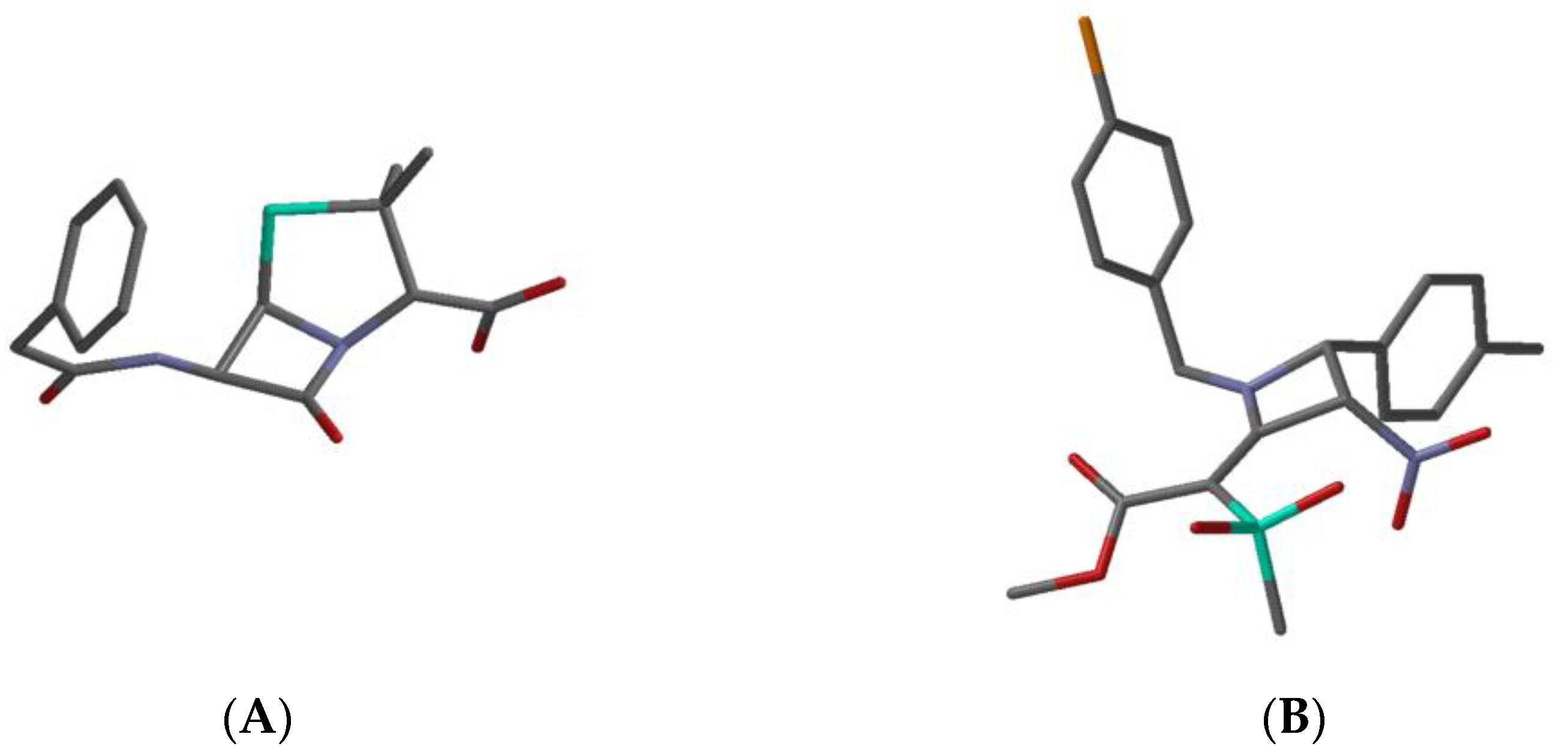
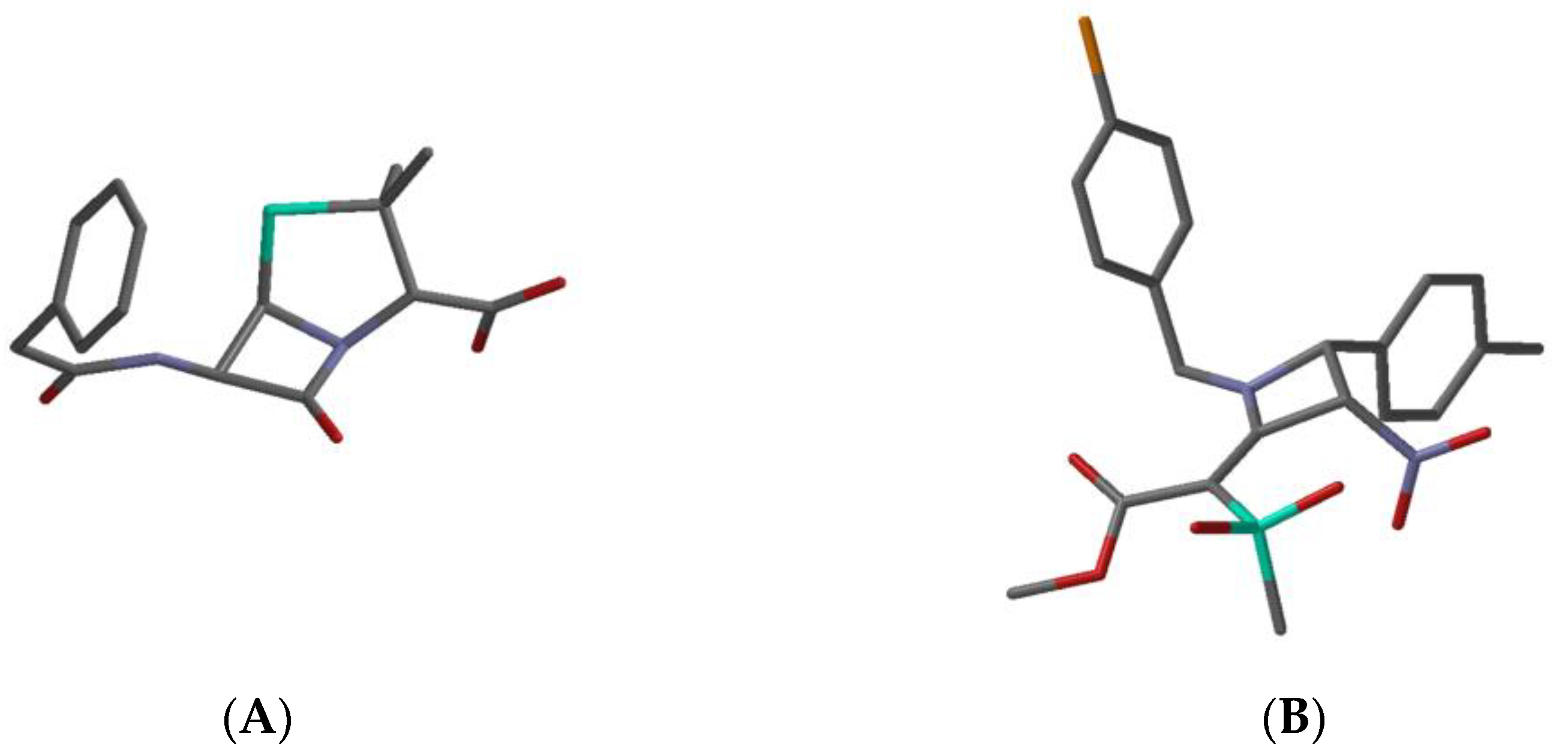
| Penicillin G a | Compd 22 b | Reported qualitative relationship between β-lactam stereoelectronic features and antibacterial activity [ref1]; ↑: improves, ↓: lowers | |
| C=X bond length (Å) | 1.20 | 1.37 | short ↑; long ↓ |
| C-N bond length (Å) | 1.40 | 1.35 | long ↑; short ↓ |
| C=X bond order | high ↑; low ↓ | ||
| Löwdin | 2.22 | 1.55 | |
| Mulliken | 1.92 | 1.53 | |
| C=X stretching frequency (cm−1) c | 1838 | 1624 | high ↑; low ↓ |
| net atomic charge on X (units of electrons) | less negative ↑; more negative ↓ | ||
| electrostatic | −0.45 | −0.67 | |
| Mulliken | −0.46 | −0.40 | |
| natural | −0.56 | −0.57 | |
| Sum of bond angles at nitrogen (°) | 332 | 359 | << 360 ↑; < 360 ↓ |
| Woodward h value d | 0.46 | 0.19 | high ↑; low ↓ |
| electrophilicity index (eV) e | 0.90 | 1.05 | high ↑; low ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrillo, G.; Tavani, C.; Bianchi, L.; Benzi, A.; Cavalluzzi, M.M.; Salvagno, L.; Quintieri, L.; De Palma, A.; Caputo, L.; Rosato, A.; et al. Densely Functionalized 2-Methylideneazetidines: Evaluation as Antibacterials. Molecules 2021, 26, 3891. https://doi.org/10.3390/molecules26133891
Petrillo G, Tavani C, Bianchi L, Benzi A, Cavalluzzi MM, Salvagno L, Quintieri L, De Palma A, Caputo L, Rosato A, et al. Densely Functionalized 2-Methylideneazetidines: Evaluation as Antibacterials. Molecules. 2021; 26(13):3891. https://doi.org/10.3390/molecules26133891
Chicago/Turabian StylePetrillo, Giovanni, Cinzia Tavani, Lara Bianchi, Alice Benzi, Maria Maddalena Cavalluzzi, Lara Salvagno, Laura Quintieri, Annalisa De Palma, Leonardo Caputo, Antonio Rosato, and et al. 2021. "Densely Functionalized 2-Methylideneazetidines: Evaluation as Antibacterials" Molecules 26, no. 13: 3891. https://doi.org/10.3390/molecules26133891
APA StylePetrillo, G., Tavani, C., Bianchi, L., Benzi, A., Cavalluzzi, M. M., Salvagno, L., Quintieri, L., De Palma, A., Caputo, L., Rosato, A., & Lentini, G. (2021). Densely Functionalized 2-Methylideneazetidines: Evaluation as Antibacterials. Molecules, 26(13), 3891. https://doi.org/10.3390/molecules26133891