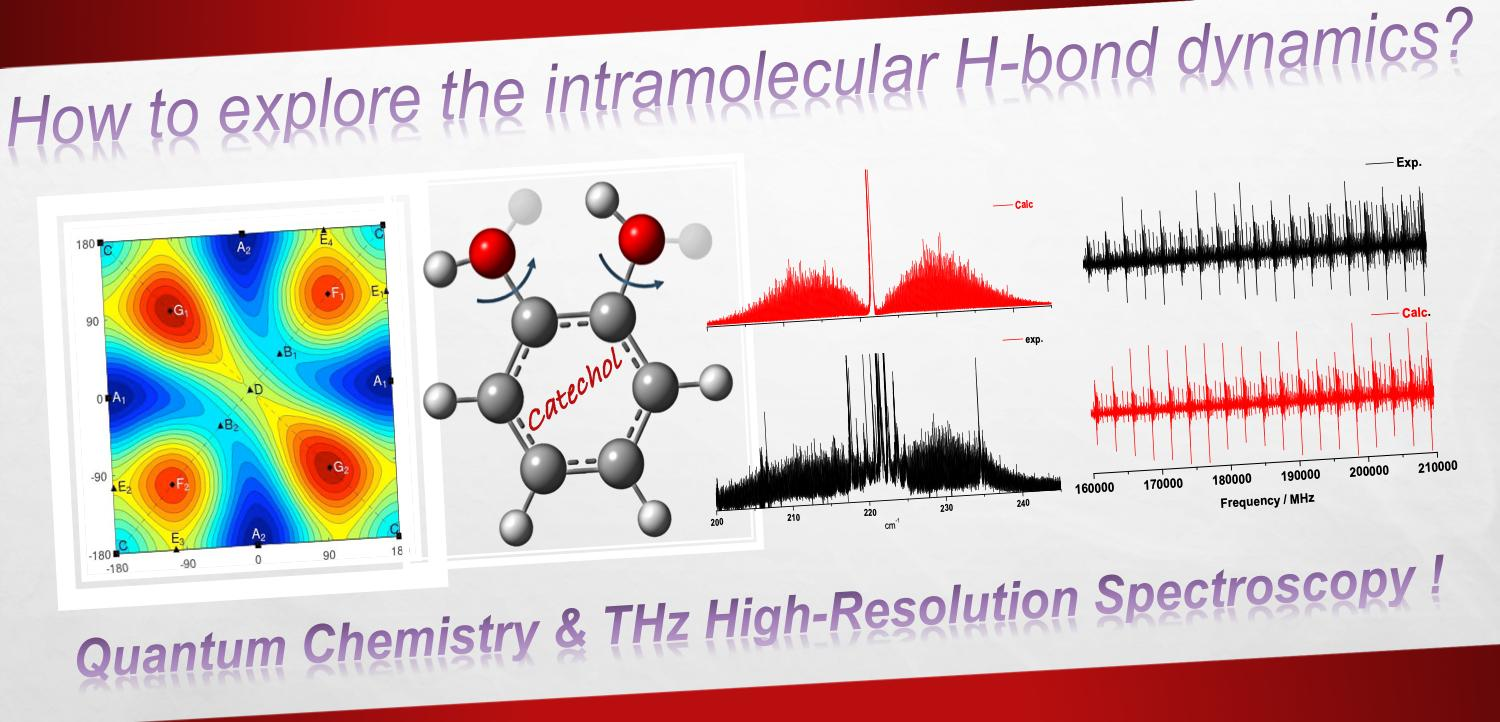
The synchrotron-based FT-far-IR spectrum of catechol, shown in
Figure 3, was measured at high resolution (≃0.001 cm
−1). In this section, we present, first, a vibrational analysis based on the Q-branches observed at medium and high resolutions. Then, we will describe the rovibrational fit of the two intense torsional -OH bands. To the best of our knowledge, it is the first time that a rovibrational assignment was performed on both free and bonded -OH torsional modes involved in the dynamics of the intramolecular HB.
2.2.1. Vibrational Analysis
The catechol molecule belongs to the
symmetry group with 36 normal modes: 25 A’ and 11 A” modes. In this study, we chose to keep the vibrational labelling of Gaussian output files with a classification based on the
symmetry instead of the labelling based on the
symmetry of monosubstituted benzenes [
23] used in previous catechol studies [
16,
17].
Table 3 presents the low-frequency vibrational modes lying in the far-IR spectral range shown in
Figure 3. Ten fundamental vibrations are located below
: the antisymmetric
and symmetric
out-of-plane O-C-C-O twisting and wagging modes; the antisymmetric
and symmetric
in-plane O-C-C-O rocking and scissoring modes; the free
and the bonded
-OH torsion; and four low-frequency ring deformations,
,
,
, and
.
Considering the theoretical calculations, the two modes associated to the free and bonded -OH torsion have strong intensities (
) compared to the other low-frequency modes (
). Indeed the FT-far-IR spectrum exhibits two intense rovibrational bands around
and
, respectively, for the
free and the
bonded -OH torsion. These two bands are pure
c-type, and their rotational structures are resolved in the P- and R-branches allowing the rovibrational analysis of catechol (see
Section 2.2).
The measured vibrational band centres agree with the REMPI gas phase measurements of [
4]. If we consider now the calculated harmonic and anharmonic IR frequencies and intensities at the B3LYP–D3/aug–cc–pVTZ level of theory, which provides us the best global agreement with the experimental ones, we note that the computational method fails to determine the anharmonic contribution of the -OH torsional modes. This could be explained by the large amplitude motions in these out-of-plane (oop) modes involved in the intramolecular HB, which cannot be treated with perturbation methods. These two torsional modes may be better described in a variational calculation with a Hamiltonian model of reduced dimensionality using our 2D PES [
24].
Except for the two -OH torsional modes, all low-frequency fundamental vibrations of catechol exhibited poor IR activities with calculated intensities below
. Yet, numerous other intense Q-branches were observed on both sides of the
and
band centres (see
Figure 3). Considering the calculated frequencies and intensities, these Q-branches were not assigned to fundamental bands but to hot bands of the -OH torsional modes. None of the other modes have been measured yet in a gas phase far-IR spectrum except the
mode, involving the intramolecular HB stretching, observed in the REMPI experiment of Bakker et al. [
4] at
in good agreement with our calculated values.
In our FT-far-IR spectra, no vibrational signature was visible in this region (see
Figure 3). Compared to the
c-type bands of the torsional modes, this A’ symmetry mode does not induce an out-of-plane variation of the dipole moment leading to a sharp Q-branch, which is easy to identify in the spectrum. The other O-C-C-O bending modes were observed only by solid state Raman measurements [
16], and the measured frequencies are in the range defined by our B3LYP–D3 harmonic and anharmonic calculations. Finally, the overtones
and
and the combination band
were predicted in the
region by the anharmonic B3LYP–D3/aug–cc–pVTZ calculation. These bands are too weak to be observed and may be masked by the intense
bonded -OH torsional band.
As mentioned previously, the numerous Q-branches, observed in the room temperature FT-far-IR spectra revealed complex vibrational patterns of hot bands on both sides of the and Q-branches. Their strong intensities reflect the strong c-type transition dipole between the GS and the two -OH torsional oop states. Some of these hot bands are localised far from the and fundamental vibrational band centres, especially in the blue side of . This experimental observation assesses the strong anharmonicity of some modes confirmed by the anharmonic DFT calculations.
In
Figure 4, we propose a tentative assignment of the hot bands around the
free -OH torsional band taking into account the four lowest energy vibrational states
,
,
, and
that we know to be thermally excited due to the mm-wave analysis described in
Section 2.3. First, we attempted to identify the four hot band sequences
(with
), labelled
in
Figure 3, starting from the
vibrational centre at
.
The assignment of the i mode to the sequence was performed in two steps: (i) according to the Boltzmann factor, the fit of the exponential decay of integrated intensities between successive components of the hot band sequence provides an estimation of the energy difference between consecutive levels; (ii) anharmonic coefficients were deduced from the previous step and are compared with those obtained directly with the frequency progression of the hot band sequence.
Our hot band assignment suggested a large negative diagonal constant of about cm−1, which constitutes an additional signature of the strong anharmonicity of the free -OH torsion. The off-diagonal anharmonic constants deduced from the three other hot band sequences were smaller in absolute value with , and estimated, respectively, about , , and . As was underlined previously, the anharmonic DFT calculation failed to evaluate the anharmonicity of the -OH torsions, which prevents a reliable comparison between the experimental and computed constants.
Afterwards, we noticed that the pattern of these hot band sequences observed around the
band centre was repeating in each component of the strongly blue-shifted
sequence. Furthermore, we observed hot band sequences starting from combination levels. An example is given in the red panel of
Figure 4 with two sequences, one blue-shifted (
) and one red-shifted (
) from the
origin.
Finally, for a complete hot band assignment, including
, a specific study should be performed in a future article in order to confirm and finalize our present work. As was done for thermally excited molecules, such as benzene [
25] or naphthalene [
26], a suitable treatment based on an anharmonic force field would have to be carried out for the hot bands rovibrational assignment. In the case of catechol with a high number of low-energy states, the possibility of resonances between close-lying states should be considered, and, in addition to the anharmonic force field calculation, the multi-state interaction problem should be solved by considering numerous Coriolis/Fermi/Darling–Dennison couplings to have a better prediction of the hot band sequences.
2.2.2. Rovibrational Analysis
In our rovibrational analysis, we treated the
and
as isolated vibrational states, i.e., we considered no explicit Coriolis interactions and no vibrational resonances with other close lying states. This turned out to be sufficient considering the accuracy of our data and the range of the observed
J values. The fitted parameters in the far-IR analysis of the
and
bands are presented in
Table 4, and the simulated far-IR spectra with these parameters are compared to the experimental ones in
Figure 5.
The two experimental rovibrational patterns shown on a spectral range of 50 cm
−1 (
Figure 5, black curves) exhibit several hot bands, which overlap the P, Q, and R branches of the fundamentals. In this work, the rovibrational lines belonging to hot bands were not included in the fit (see
Figure 5, red curves) and numerous rovibrational lines remain unassigned (see
Figure 5, zoomed parts). Nevertheless, more than 11,000 rovibrational
c-type transitions were assigned in the FT-far-IR spectrum (6937 for
and 4131 for
) highlighting the highly congested far-IR spectrum of catechol.
Numerous rovibrational lines are blended, especially between quasi-degenerate transitions with or . Therefore, the number of fitted transitions is larger than the number of fitted frequencies. A development of the centrifugal distortion up to quartic terms in the effective Hamiltonian was sufficient to reproduce the experimental transition frequencies of the two bands and at the experimental accuracy with an unitless RMS close to 1.1.
All the molecular parameters for the two torsional bands were determined with a high degree of accuracy. In particular, the vibrational band centres of
and
were fitted with an accuracy better than 10
−4 cm
−1, four orders of magnitude better than those obtained with the REMPI technique in [
4,
7]. The complete set of rotational and centrifugal distortion constants were perfectly determined, and the detailed fit is given in the
Supplementary Materials.
From the results of
Table 4, we determined the differences between the GS rotational constants and those in the
and
torsionally excited states (ES) according to the relation
with
and similarly for the other rotational constants
B and
C. The vibration-rotation constants
allow for estimation of the influence of the two torsional modes on the overall rotation of the catechol molecule.
From the fitted values of
Table 4 for the free -OH torsional mode, we obtained
,
, and
. Similarly, for the bonded -OH torsional modes, the values were
, and
. While the rotation of catechol in the ring plane around the c-axis is almost not affected by the -OH torsional modes, we observed significant decreasing of the
,
, and
rotational constants.
With a bonded -OH almost parallel to the b-axis (see
Figure 1, right part), the largest vibration-rotation constant
was obtained for a rotation around the a-axis (
). The free -OH torsion
also perturbed the overall rotation around the a- and b-axes but with a lesser influence compared to
(
). Here, we observe that the free and the bonded -OH torsions differently affected the overall rotation of the catechol molecule.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}