
Unveiling the Ionic Diels–Alder Reactions within the Molecular Electron Density Theory




Abstract
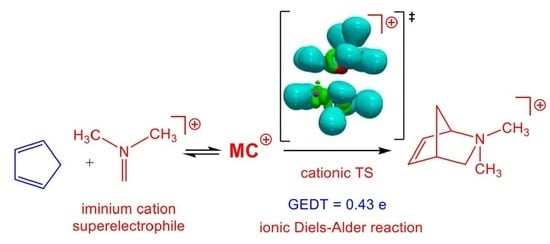
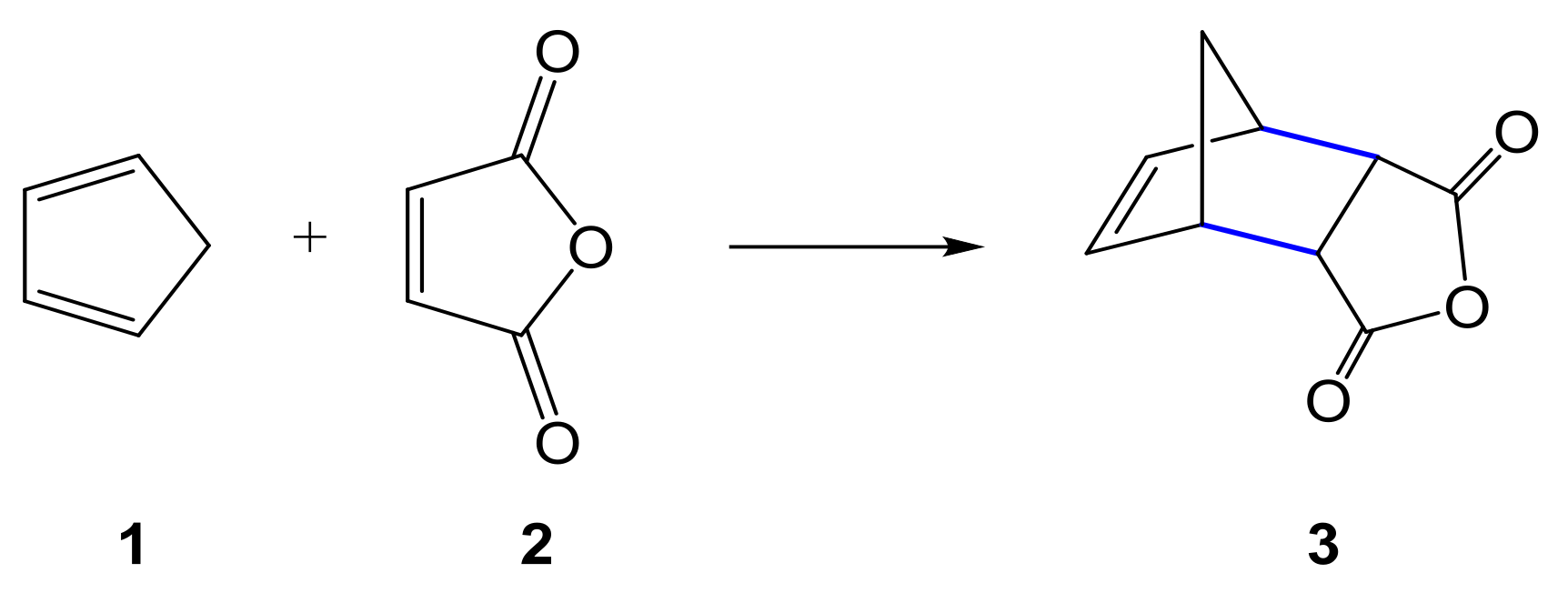
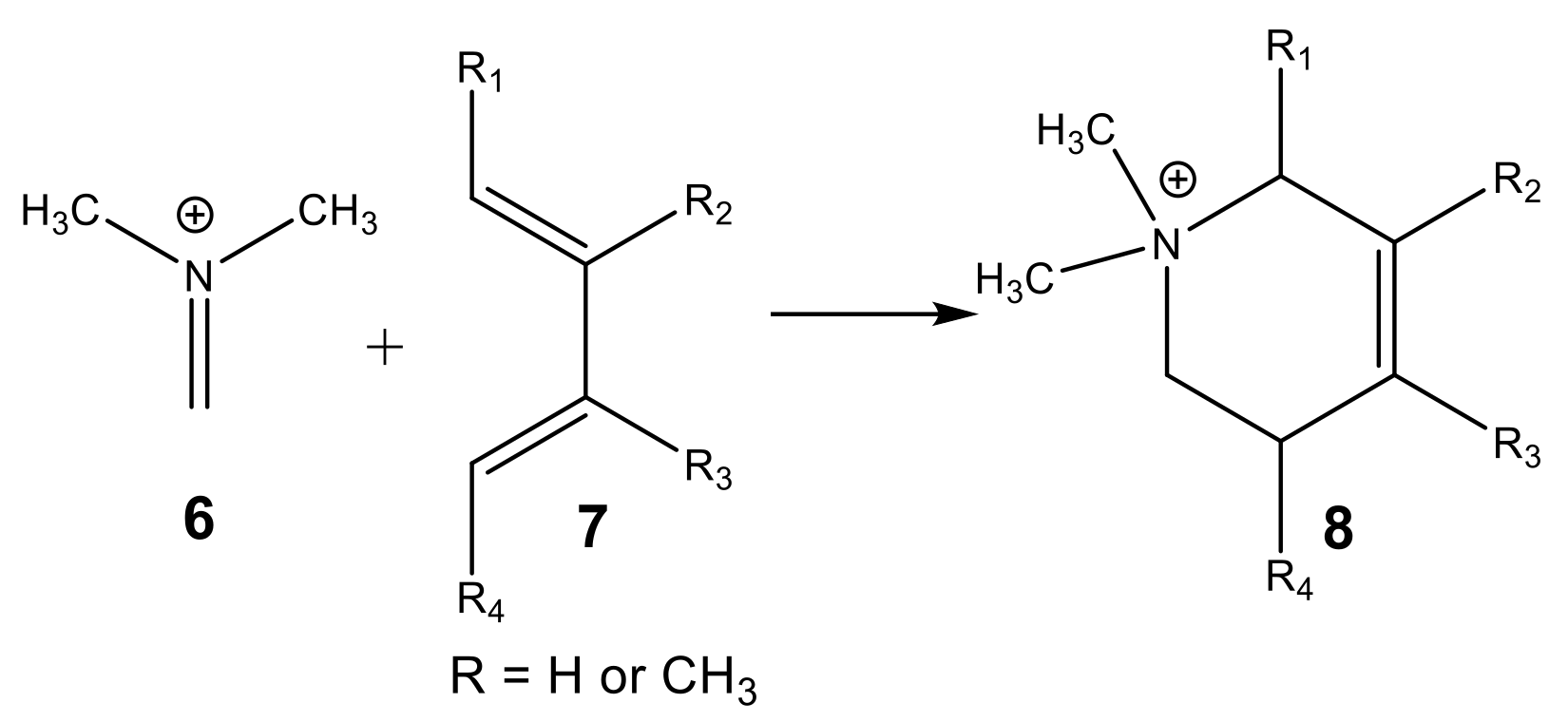
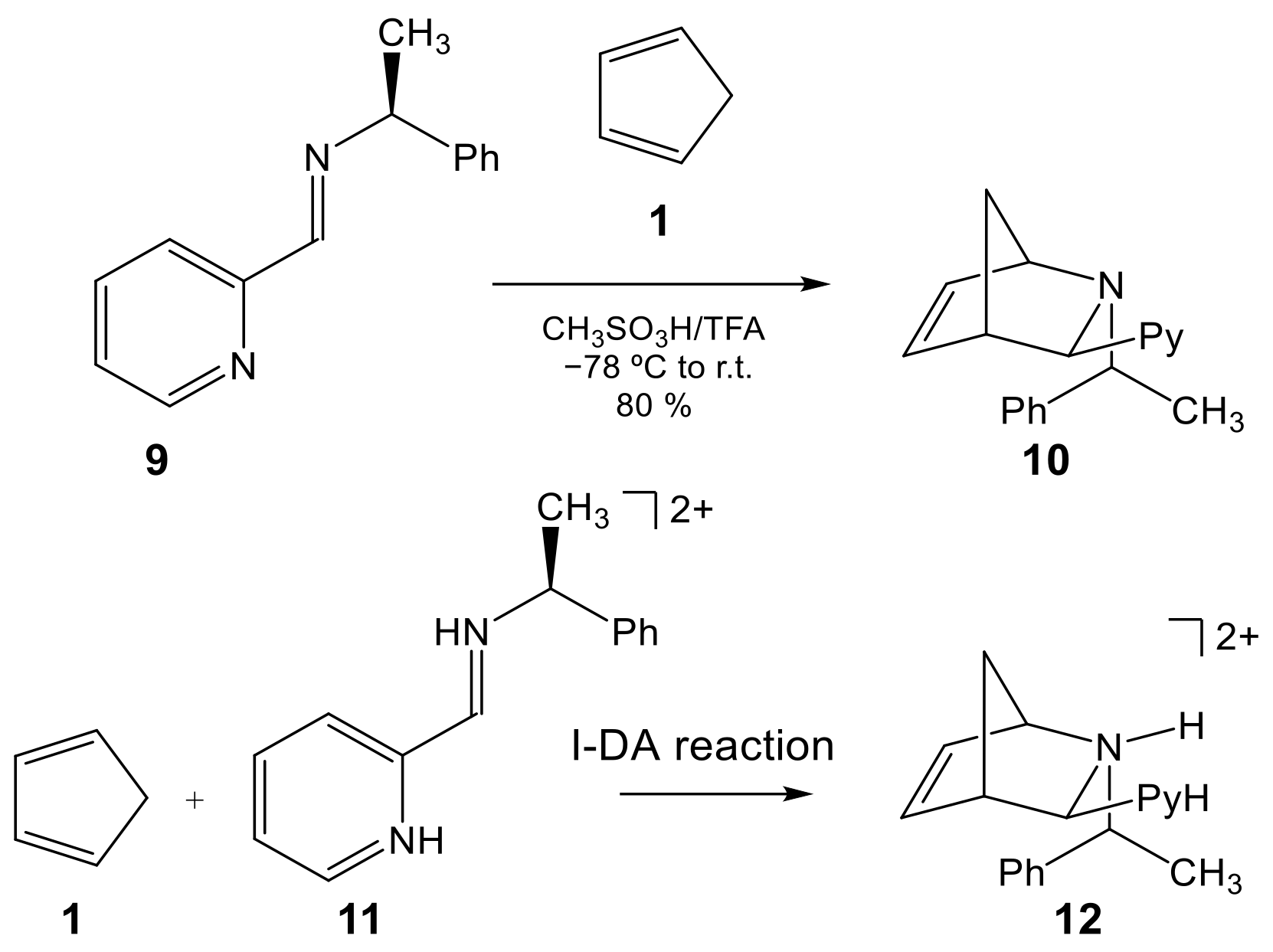
1. Introduction
2. Results and Discussions
2.1. Study of the Electronic Structure and Reactivity at the Ground State of the Reagents
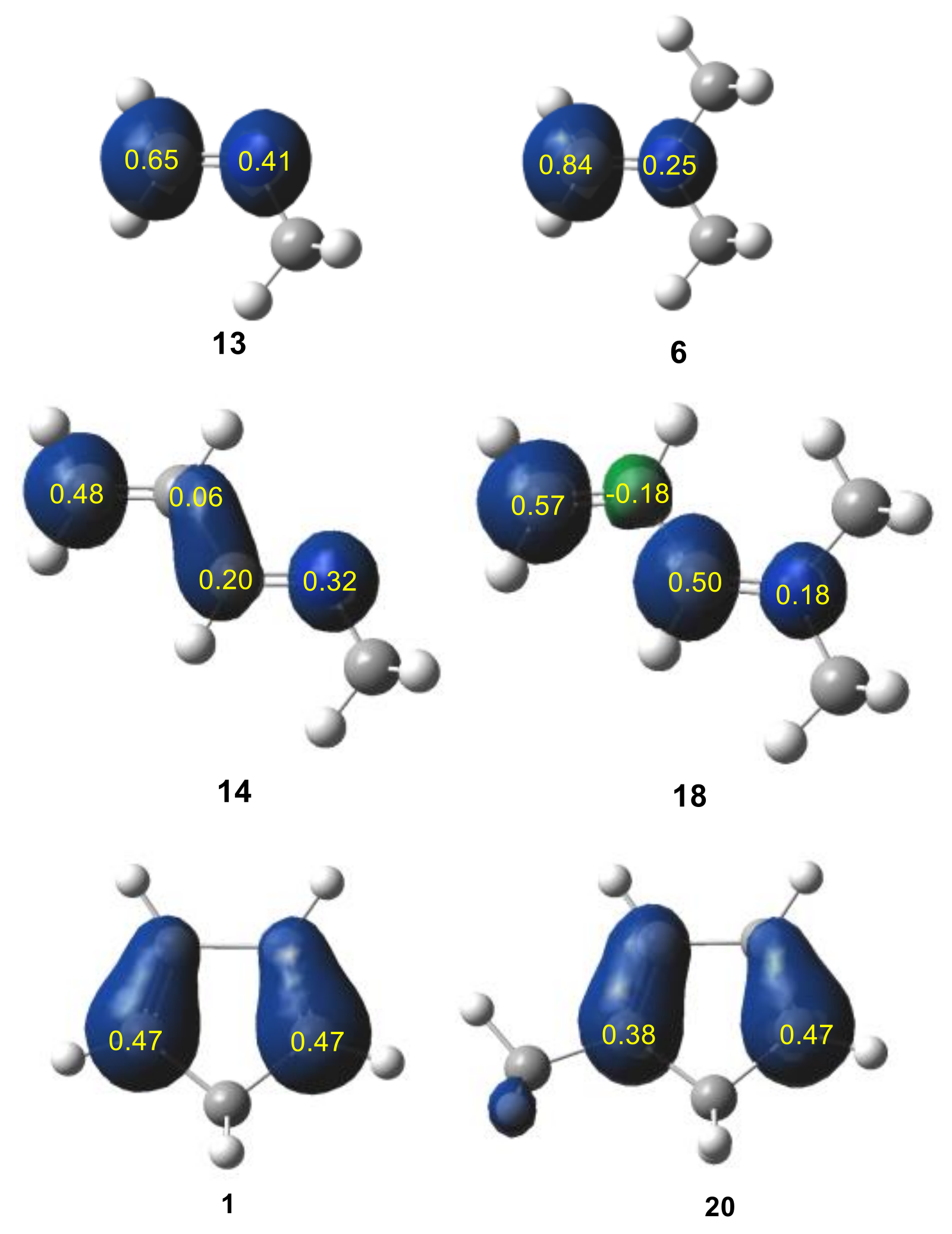
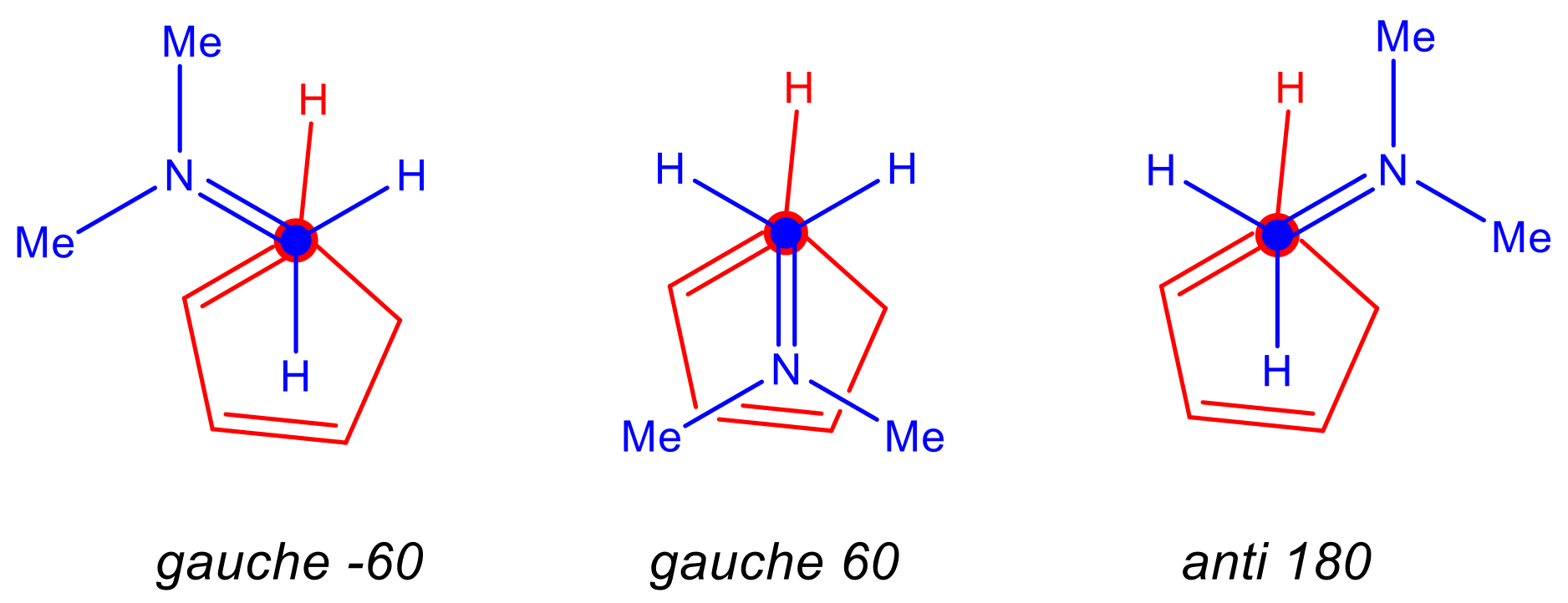
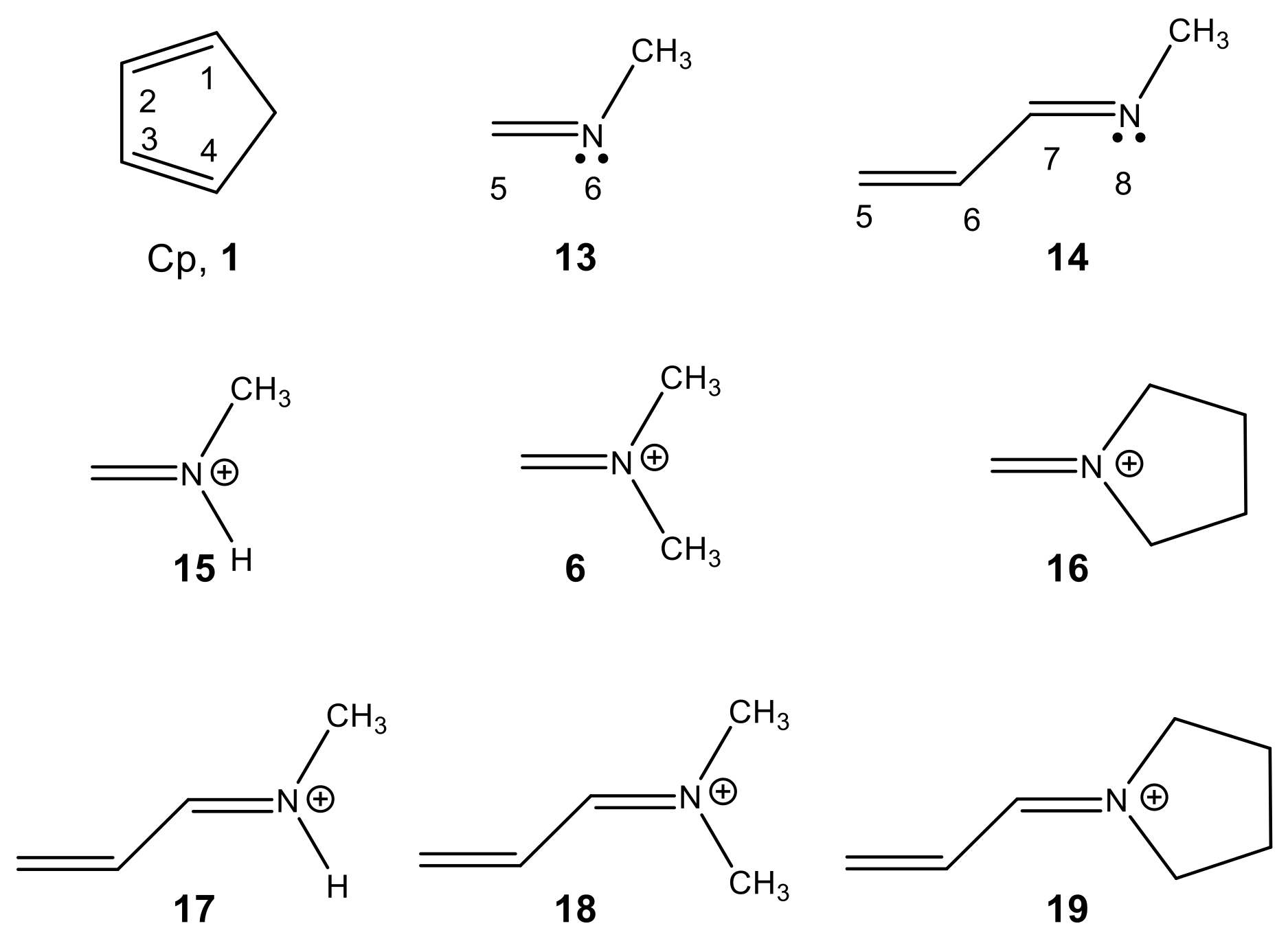
2.1.1. Analysis of the Electronic Structure of Imines and Iminium Cations
2.1.2. Analysis of the CDFT Reactivity Indices at the GS of the Reagents
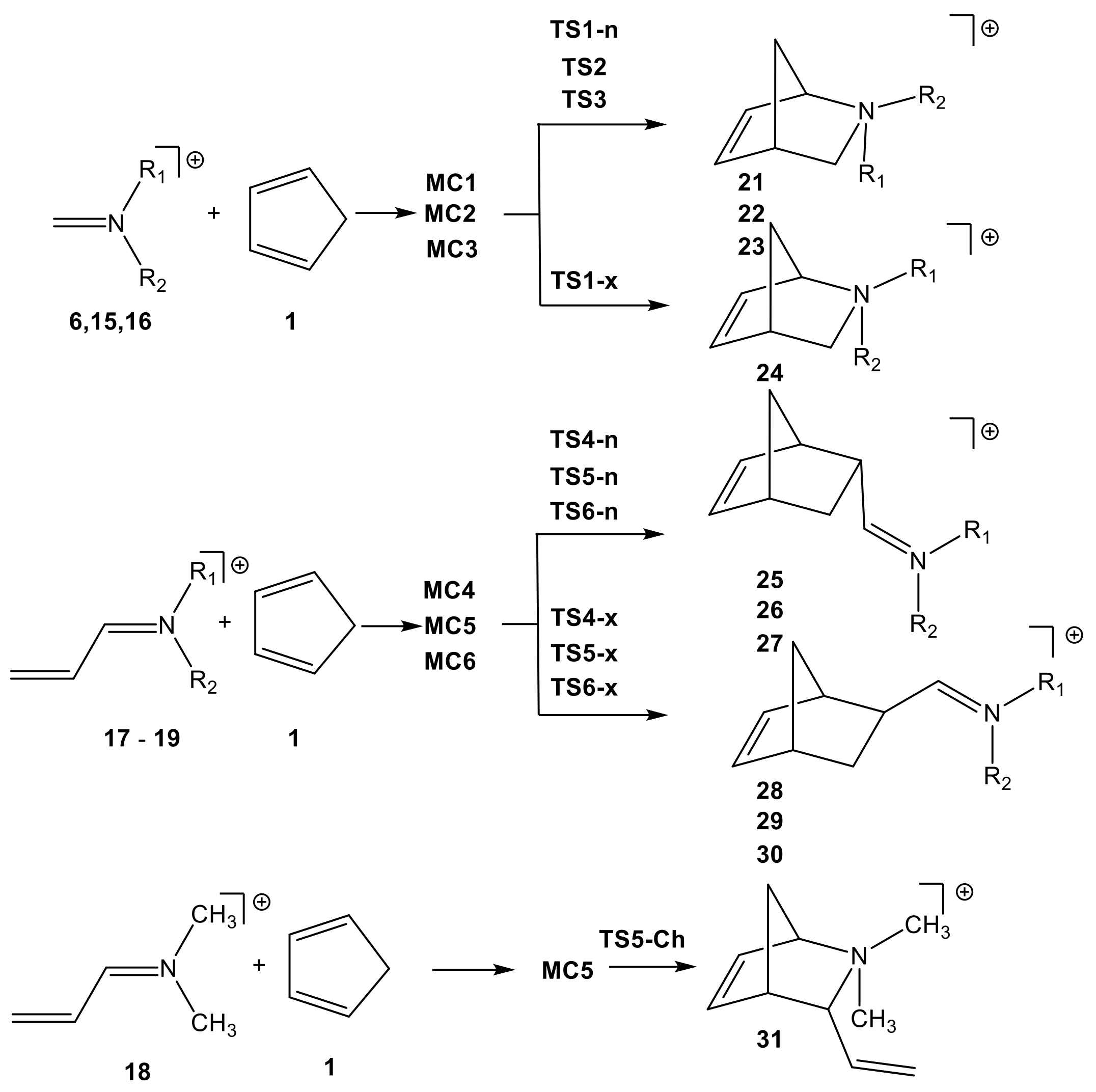
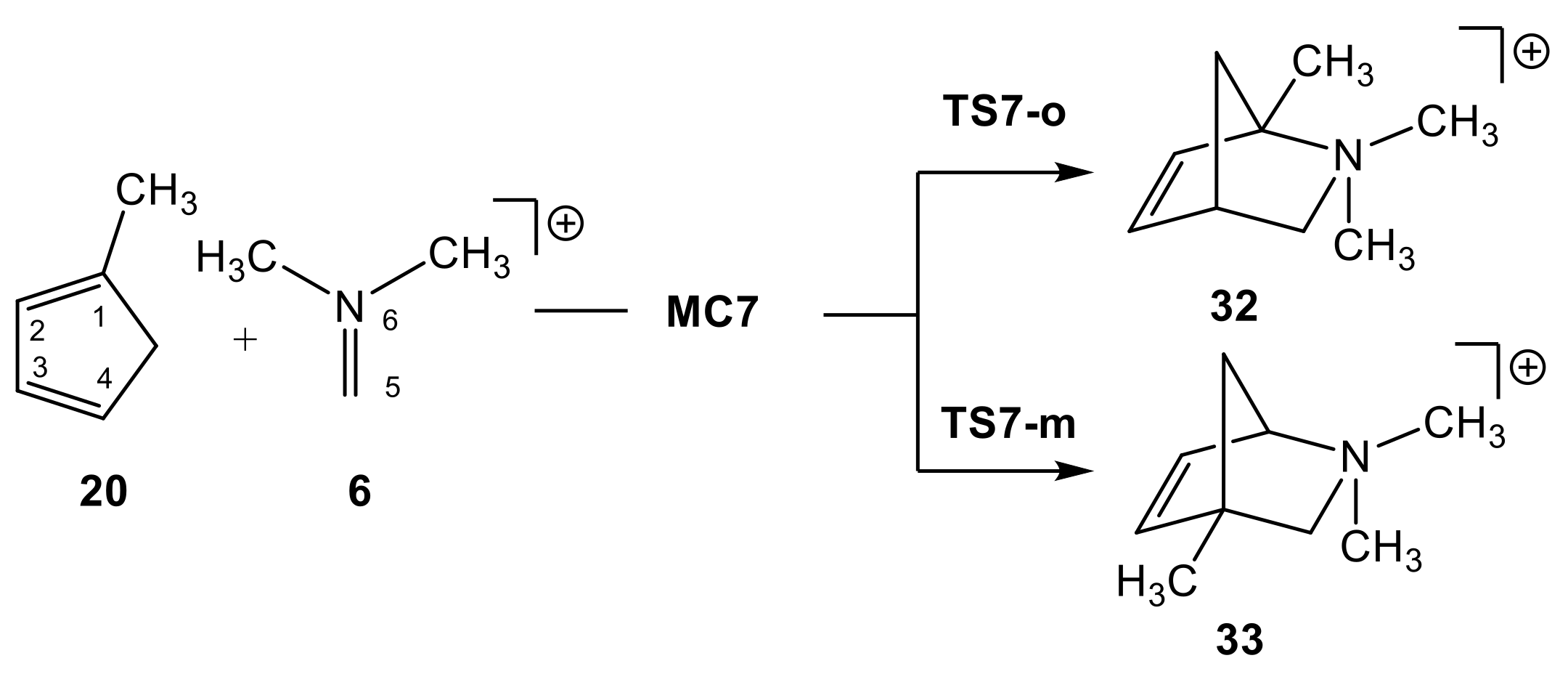
2.2. Study of the Potential Energy Surface of I-DA Reactions
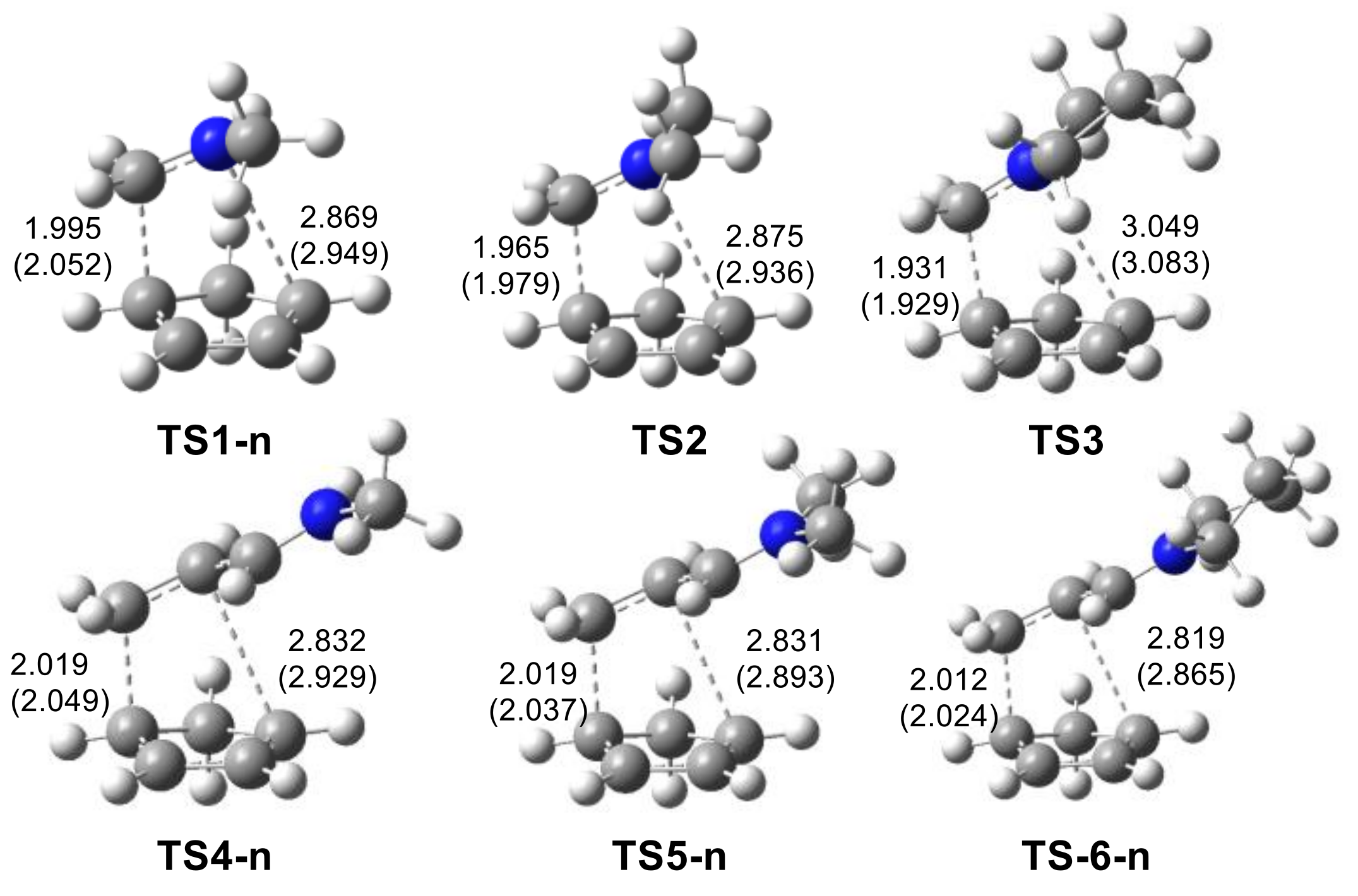
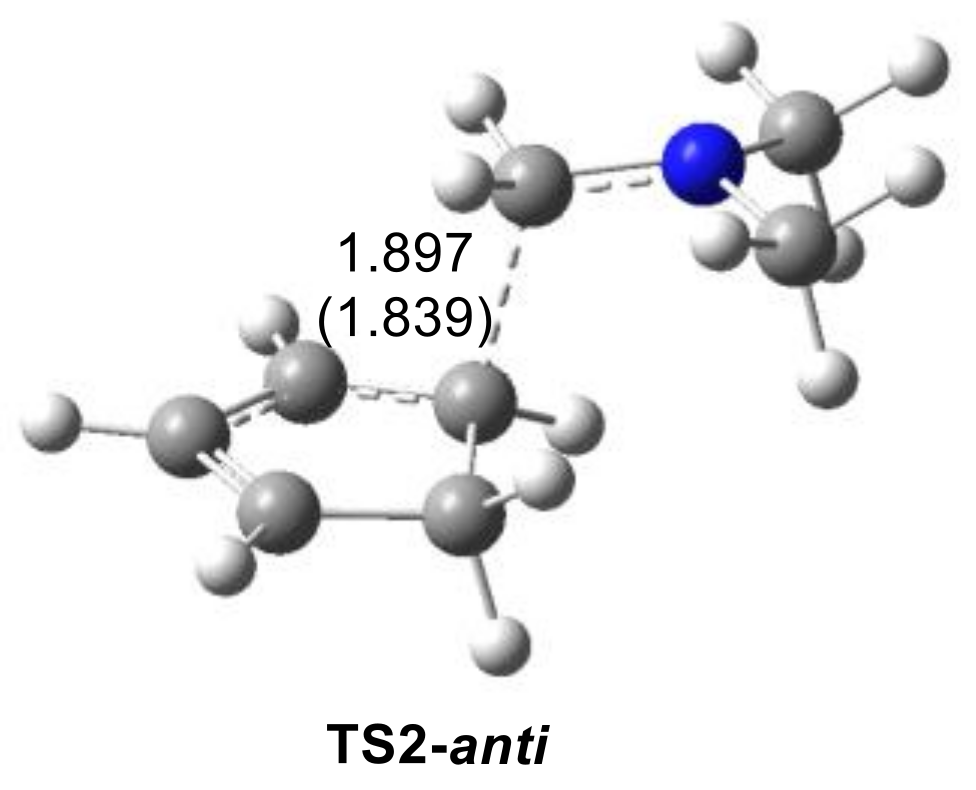
2.2.1. Study of the I-DA Reactions of Iminium Cations 6, 15–19 with Cp 1
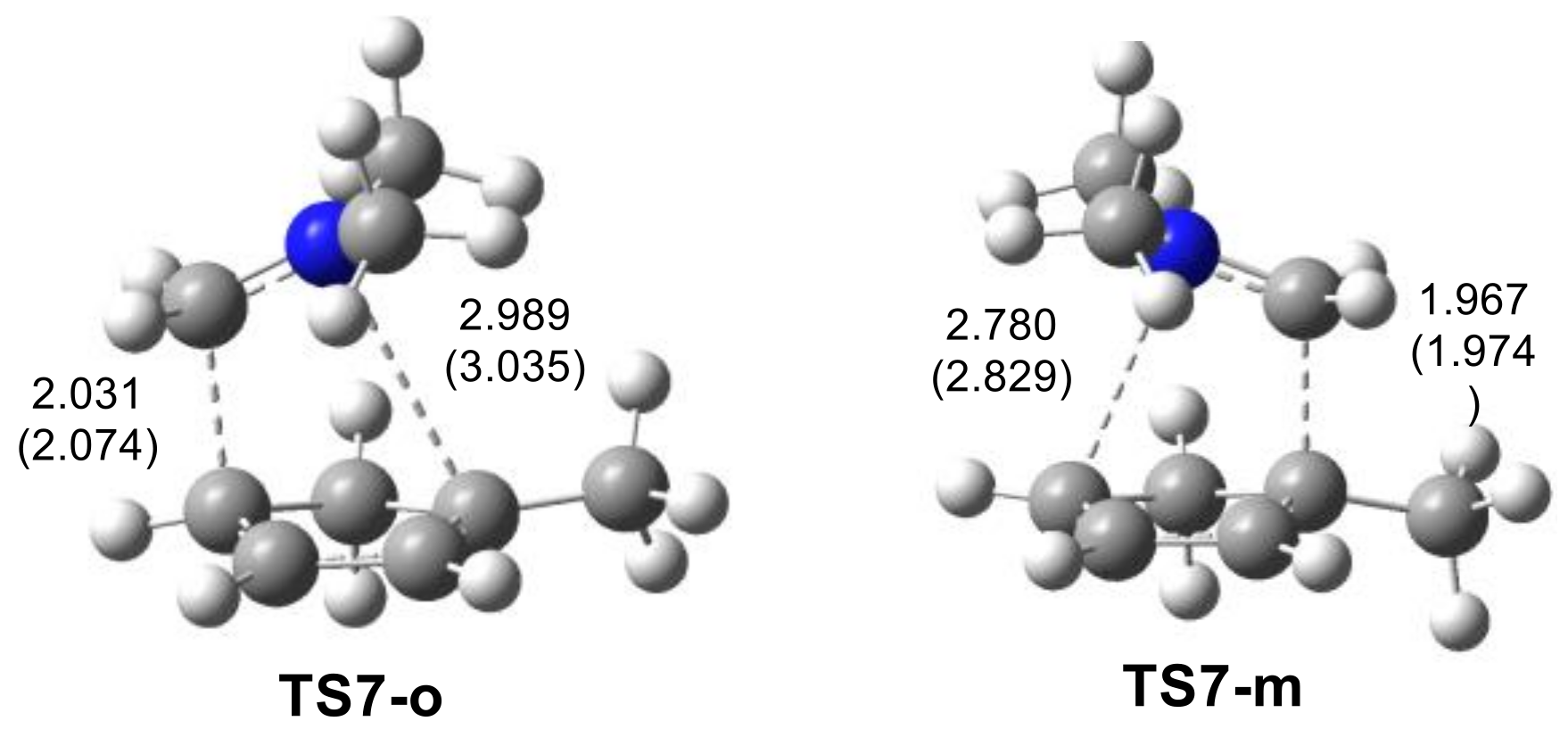
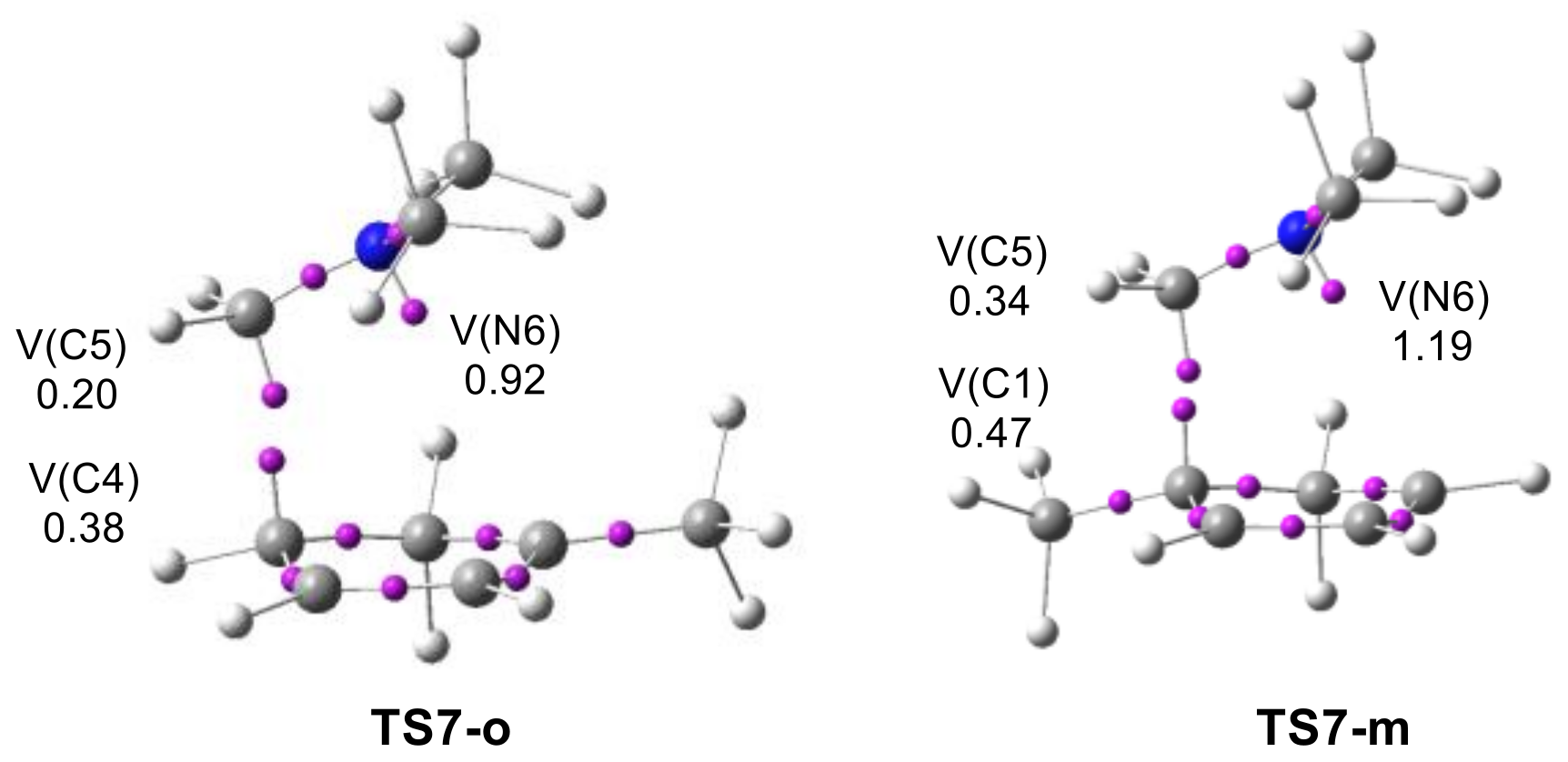
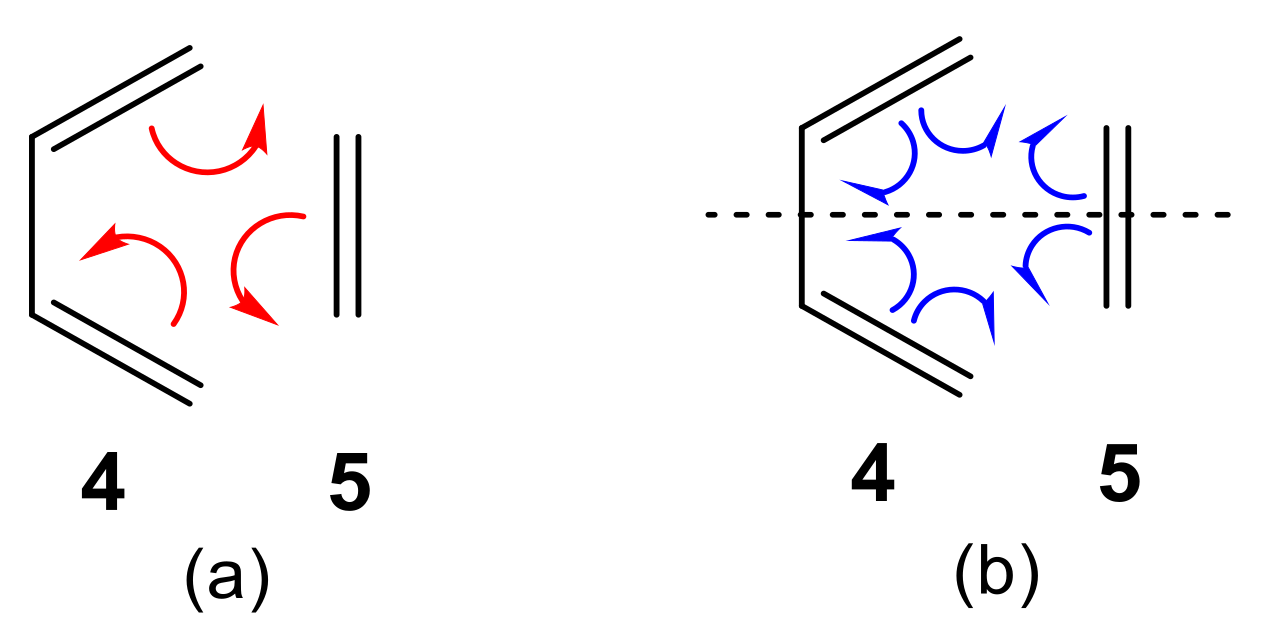
2.2.2. Study of the Regioselectivity in I-DA Reactions
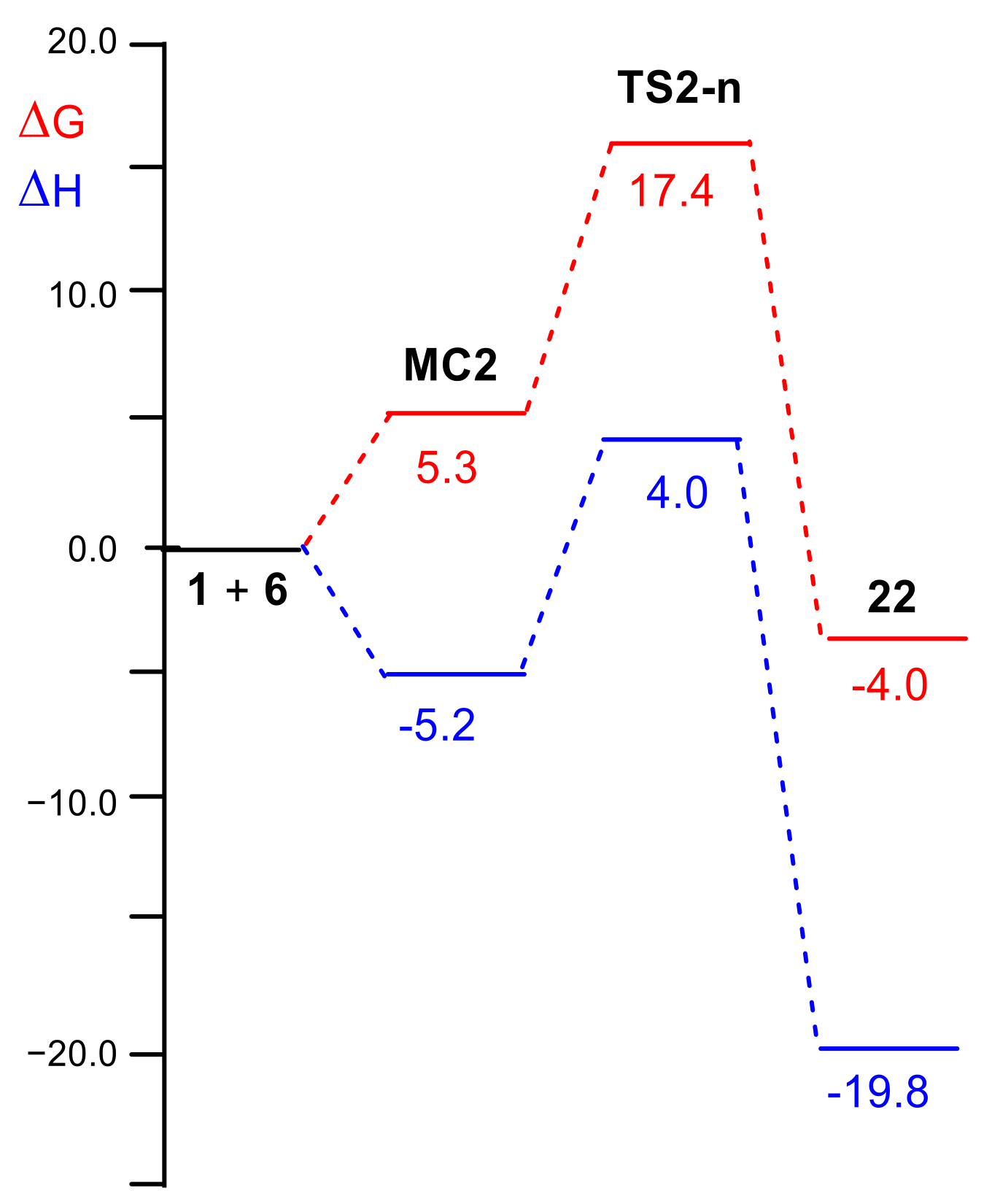
2.2.3. Thermodynamic Analysis of the I-DA Reaction of Iminium Cation 6 with Cp 1
2.3. Topological Analysis of the Bonding Changes along the I-DA Reactions
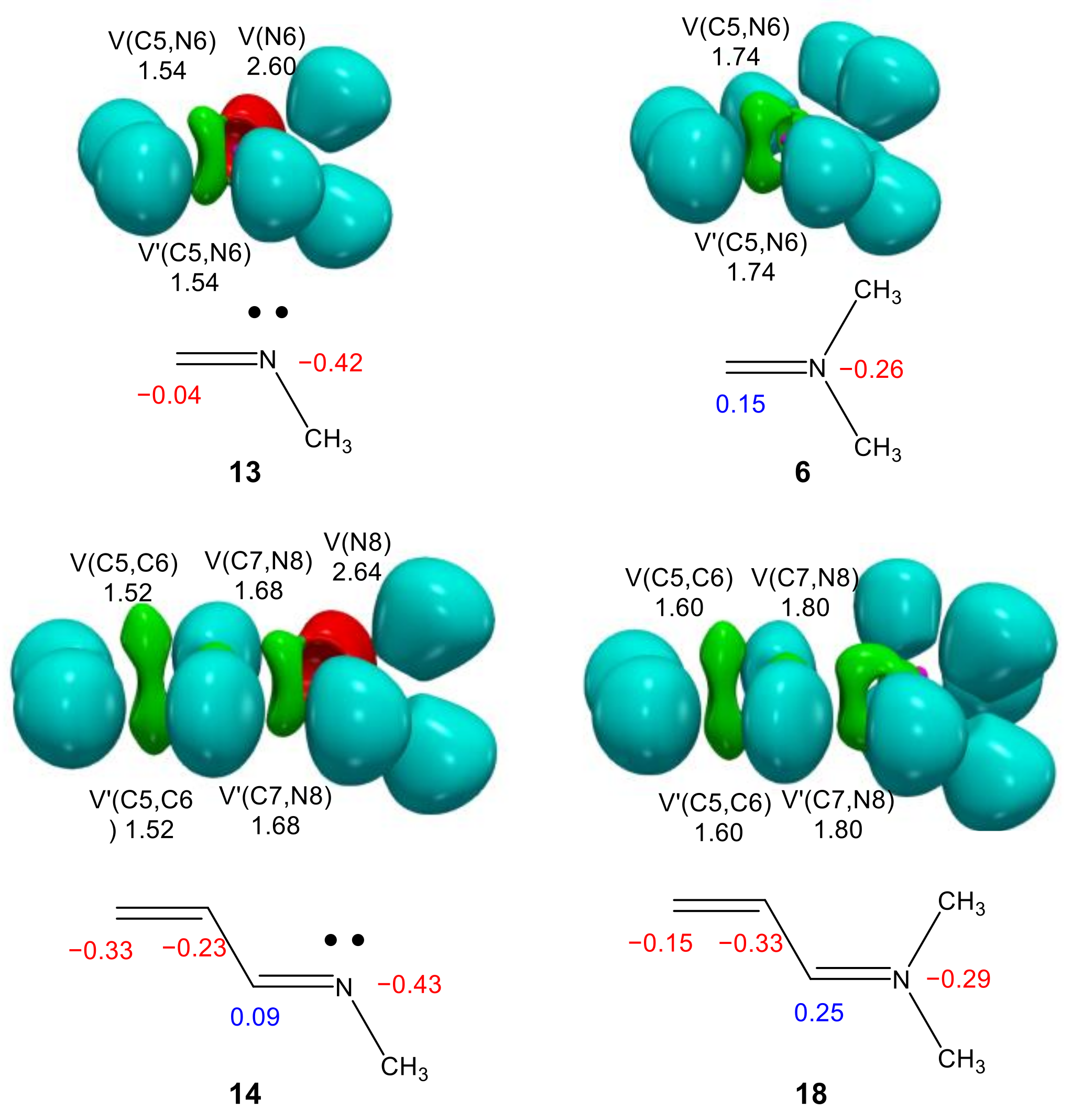
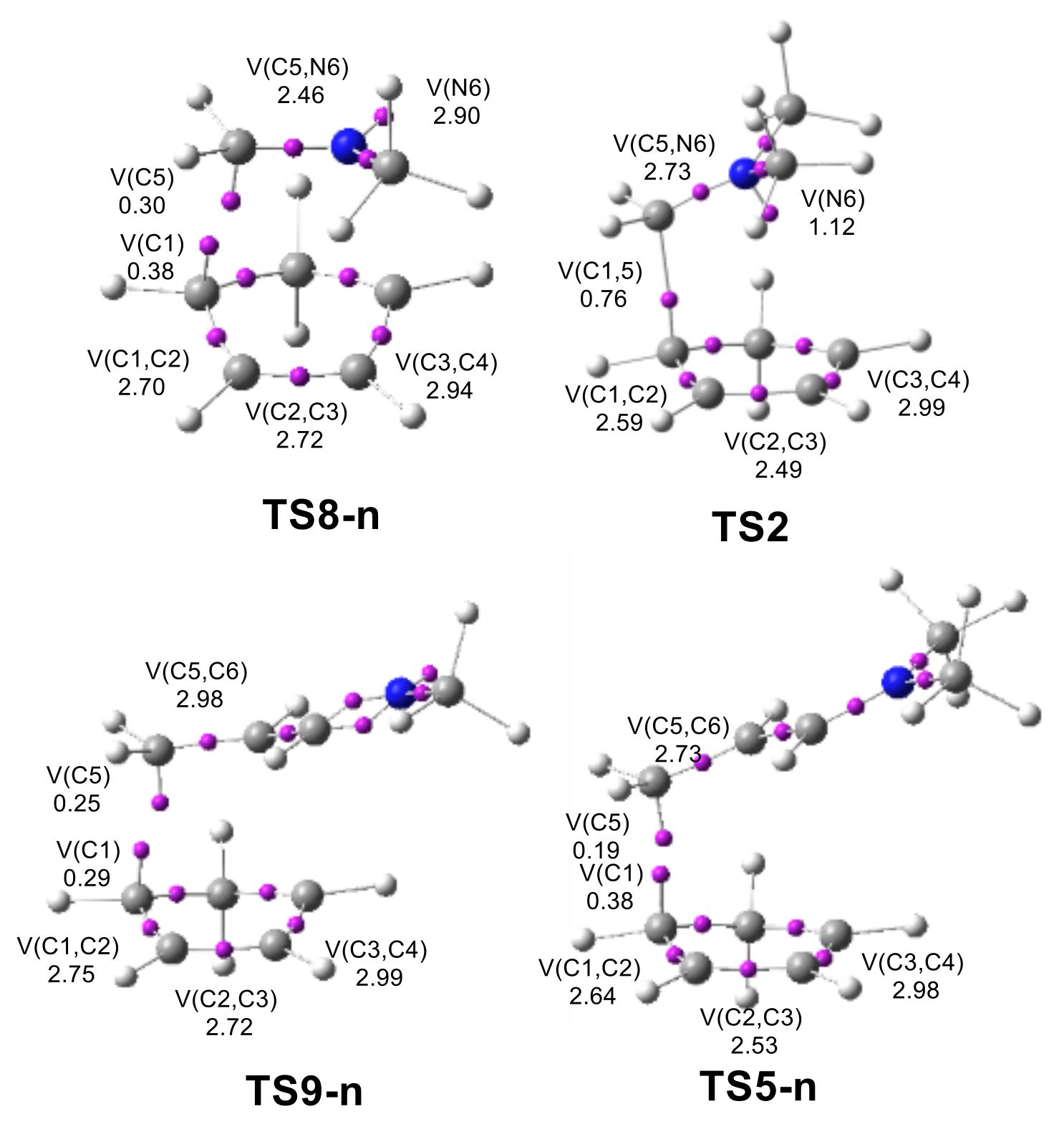
2.3.1. ELF Analysis of the C–C and N–C Single Bond Formation along the I-DA Reaction of Cp 1 with Iminium Cation 6
2.3.2. Comparative ELF Analysis of the TSs Involved in the P-DA Reaction of Cp 1 with Imines 13 and 14, and Those Involved in the I-DA Reaction of Cp 1 with Iminium Cations 6 and 18
2.3.3. Analysis of the Origin of the Regioselectivity in I-DA Reactions
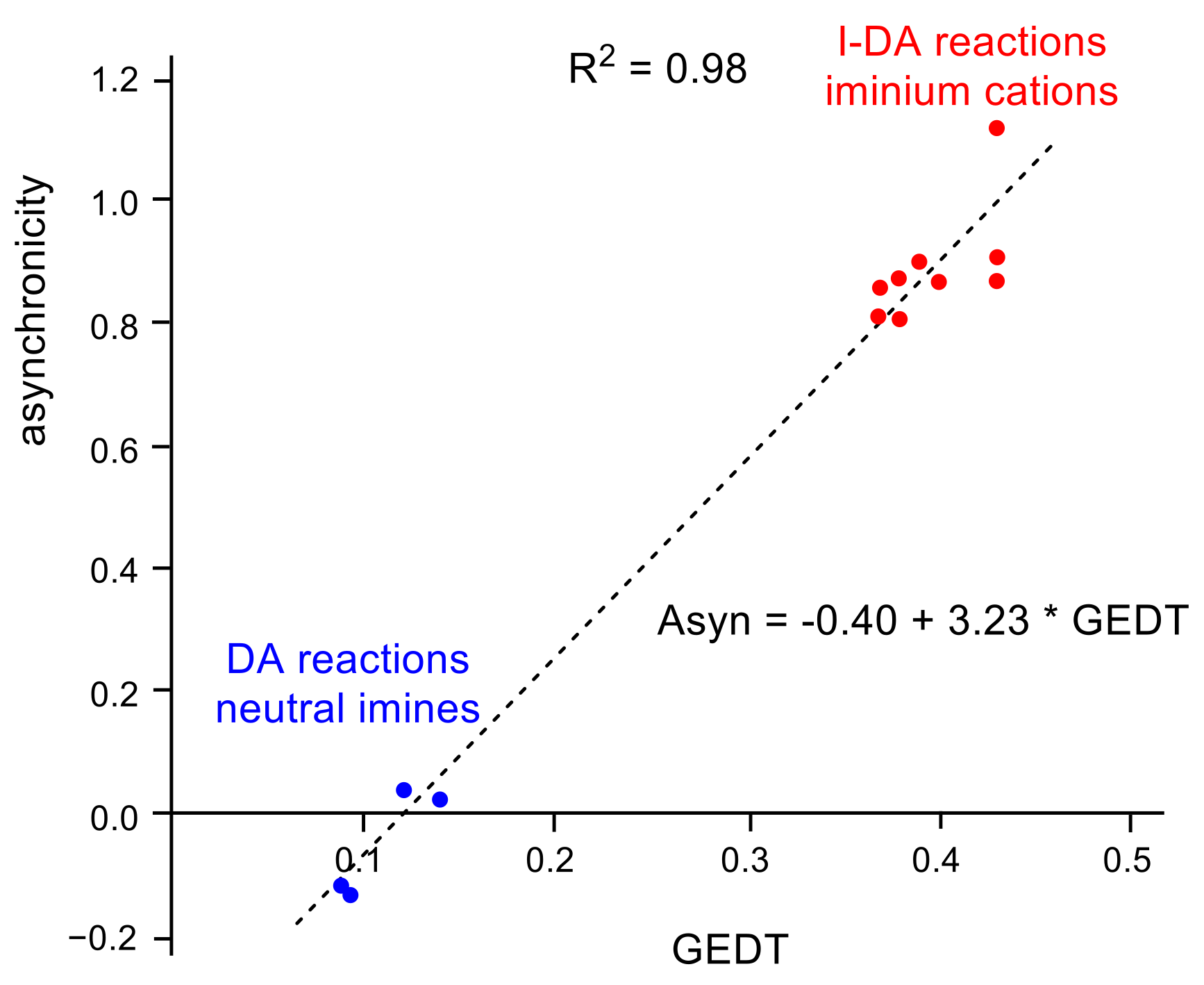
2.4. Origin of the Asynchronicity in I-DA Reactions
3. Conclusions
4. Computational Details
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Carruthers, W. Some Modern Methods of Organic Synthesis, 2nd ed.; Cambridge University Press: Cambridge, UK, 1978. [Google Scholar]
- Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1990. [Google Scholar]
- Woodward, R.B.; Hoffmann, R. The Conservation of the Orbital Symmetry. Angew. Chem. Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Houk, K.N.; Gonzalez, J.; Li, Y. Pericyclic Reaction Transition States: Passions and Punctilios, 1935–1995. Acc. Chem. Res. 1995, 28, 81–90. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of Elementary Chemical Processes by Catastrophe Theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Berski, S.; Andrés, J.; Silvi, B.; Domingo, L.R. The Joint Use of Catastrophe Theory and Electron Localization Function to Characterize Molecular Mechanisms. A Density Functional Study of the Diels−Alder Reaction between Ethylene and 1,3-Butadiene. J. Phys. Chem. A 2003, 107, 6014–6024. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Silvi, B.; Pérez, P. The Mysticism of Pericyclic Reactions: A Contemporary Rationalisation of Organic Reactivity Based on Electron Density Analysis. Eur. J. Org. Chem. 2018, 2018, 1107–1120. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. Molecular Orbitals in Chemistry, Physics, and Biology; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Dewar, M.J.S. Aromaticity and Pericyclic Reaction. Angew. Chem. Int. Ed. 1971, 10, 761–776. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Domingo, L.R.; Arnó, M.; Andrés, J. Influence of Reactant Polarity on the Course of the Inverse-Electron-Demand Diels-Alder Reaction. A DFT Study of Regio- and Stereoselectivity, Presence of Lewis Acid Catalyst, and Inclusion of Solvent Effects in the Reaction between Nitroethene and Substituted Ethenes. J. Org. Chem. 1999, 64, 5867–5875. [Google Scholar]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Contreras, R. Origin of the synchronicity on the transition structures of polar Diels-Alder reactions. Are these reactions [4 + 2] processes? J. Org. Chem. 2003, 68, 3884–3890. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the mechanism of polar Diels–Alder reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Terrier, F.; Sebban, M.; Goumont, R.; Hallé, J.C.; Moutiers, G.; Cangelosi, I.; Buncel, E. Dual Behavior of 4-Aza-6-nitrobenzofuroxan. A Powerful Electrophile in Hydration and σ-Complex Formation and a Potential Dienophile or Heterodiene in Diels−Alder Type Reactions. J. Org. Chem. 2000, 65, 7391–7398. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P. The Lithium Cation Catalysed Benzene Diels-Alder reaction. Insights on the Molecular Mechanism within the Molecular Electron Density Theory. J. Org. Chem. 2020, 85, 13121–13132. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R.; Sauer, J.; Schmied, B. [2+ + 4] Cycloadditions of Iminium Ions-Concerted or Stepwise Mechanism of Aza Diels-Alder Reactions? Eur. J. Org. Chem. 2000, 2000, 2013–2020. [Google Scholar] [CrossRef]
- Hedberg, C.; Pinho, P.; Roth, P.; Andersson, P.G. Diels−Alder Reaction of Heterocyclic Imine Dienophiles. J. Org. Chem. 2000, 65, 2810–2812. [Google Scholar] [CrossRef]
- Domingo, L.R. A Theoretical Study of the Molecular Mechanism of the Reaction between N,N-Dimethylmethyleneammonium Cation and Cyclopentadiene. J. Org. Chem. 2001, 66, 3211–3214. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the Participation of Quadricyclane as Nucleophile in Polar [2σ + 2σ + 2π] Cycloadditions toward Electrophilic π Molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
- Domingo, L.R.; Oliva, M.; Andrés, J. A theoretical study of the reaction between cyclopentadiene and protonated imine derivatives: A shift from a concerted to a stepwise molecular mechanism. J. Org. Chem. 2001, 66, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Peréz, P. A Quantum Chemical Topological Analysis of the C-C Bond Formation in Organic Reactions Involving Cationic Species. Phys. Chem. Chem. Phys. 2014, 16, 14108–14115. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P.; Sáez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hamlin, T.A.; Fernandez, I.; Bickelhaupt, F.M. Origin of rate enhancement and asynchronicity in iminium catalyzed Diels–Alder reactions. Chem. Sci. 2020, 11, 8105–8112. [Google Scholar] [CrossRef]
- Van Zeist, W.-J.; Bickelhaupt, F.M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M. Understanding reactivity with Kohn–Sham molecular orbital theory: E2–SN2 mechanistic spectrum and other concepts. J. Comput. Chem. 1999, 20, 114–128. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.J.; Perez, P.; Sáez, J.A. Understanding the Origin of the Asynchronicity in Bond-Formation in Polar Cycloaddition Reactions. A DFT Study of the 1,3-Dipolar Cycloaddition Reaction of Carbonyl Ylides with 1,2-Benzoquinones. RSC Adv. 2012, 2, 1334–1342. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular-systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aizman, A.; Contreras, R. The Electrophilicity Index in Organic Chemistry. In Theoretical Aspects of Chemical Reactivity; Toro-Labbe, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 9, pp. 139–201. [Google Scholar]
- Domingo, L.R.; Pérez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Aurell, M.J.; Domingo, L.R.; Perez, P.; Contreras, R. A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 2004, 60, 11503–11509. [Google Scholar] [CrossRef]
- Hammond, G.S. A Correlation of Reaction Rates. J. Am. Chem. Soc. 1955, 77, 334–338. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A Molecular Electron Density Theory Study of the Reactivity of Tetrazines in Aza-Diels-Alder Reactions. RSC Adv. 2020, 10, 15394–15405. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Unveiling the Lewis Acid Catalysed Diels–Alder Reactions Through the Molecular Electron Density Theory. Molecules 2020, 25, 2535. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Domingo, L.R.; Jones, R.A.; Picher, M.T.; Sepulveda-Arques, J. Theoretical study of the reaction of dimethyl acetylenedicarboxilate with 1-methyl-2-(1-substituted vinyl) pyrroles. Tetrahedron 1995, 51, 8739–8748. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Bickelhaupt, F.M.; Fernández, I. The Pauli Repulsion-Lowering Concept in Catalysis. Acc. Chem. Res. 2021, 54, 1972–1981. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hehre, M.J.; Radom, L.; Schleyer, P.V.R.; Pople, J. Ab initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Schlegel, H.B. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific Publishing: Singapore, 1994. [Google Scholar]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- González, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: And overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions–Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Cances, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M.; Tomasi, J. Geometry optimization of molecular structures in solution by the polarizable continuum model. J. Comput. Chem. 1998, 19, 404–417. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization, Visualization Handbook; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar] [CrossRef]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware: Clifton Park, NY, USA, 2015; ISBN 978-1930934306. [Google Scholar]




















{kind=link}
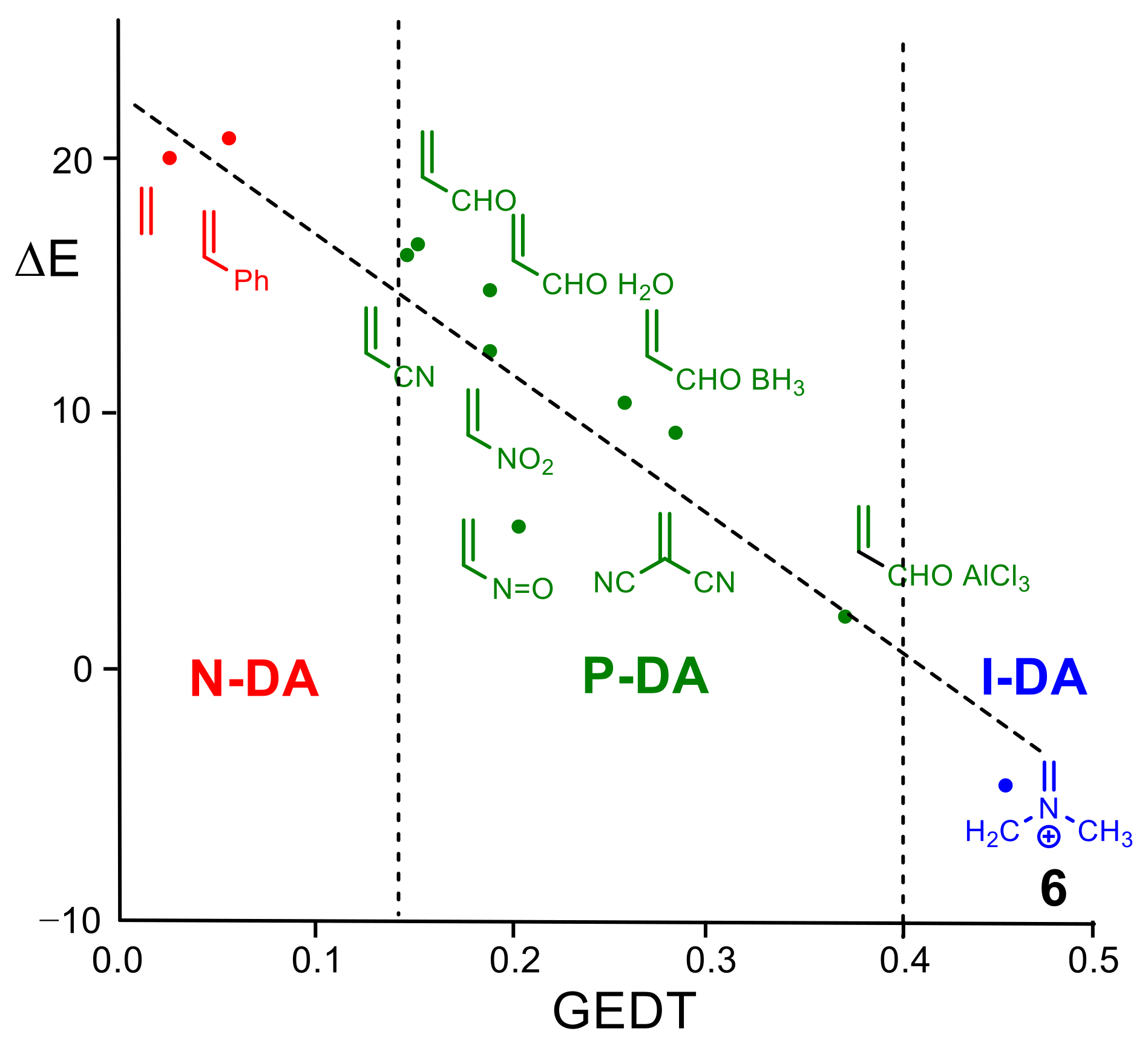{kind=link}
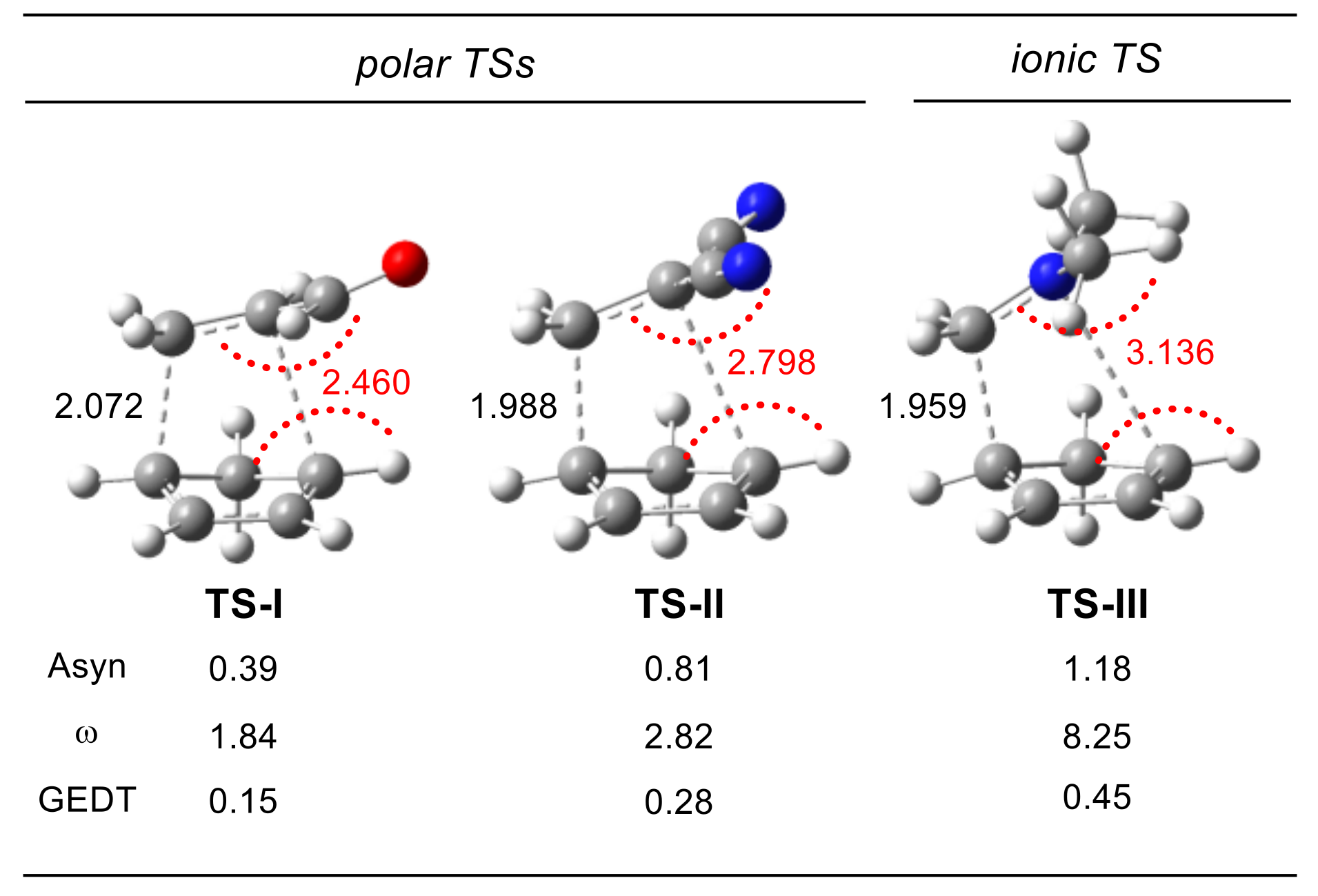{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| μ | η | ω | N | |
|---|---|---|---|---|
| 17 | −10.11 | 5.36 | 9.53 | −3.67 |
| 15 | −11.83 | 7.80 | 8.97 | −6.61 |
| 19 | −9.76 | 5.32 | 8.95 | −3.30 |
| 18 | −9.53 | 5.22 | 8.71 | −3.02 |
| 6 | −11.18 | 7.56 | 8.26 | −5.84 |
| 16 | −10.51 | 6.73 | 8.20 | −4.75 |
| 14 | −3.91 | 5.84 | 1.31 | 2.30 |
| 13 | −3.46 | 6.80 | 0.88 | 2.26 |
| Cp 1 | −3.01 | 5.48 | 0.83 | 3.37 |
| CpMe 20 | −2.84 | 5.31 | 0.76 | 3.62 |
| 15 | 6 | 16 | |||
| MC1 | −6.6 | MC2 | −6.6 | MC3 | −7.2 |
| TS1-n | −0.6 | TS2 | 2.9 | TS3 | 2.8 |
| TS1-x | 0.6 | ||||
| 21 | −28.4 | 22 | −24.0 | 23 | −24.6 |
| 24 | −28.1 | ||||
| 17 | 18 | 19 | |||
| MC4 | −5.6 | MC5 | −5.6 | MC6 | −5.8 |
| TS4-n | 3.7 | TS5-n | 3.1 | TS6-n | 3.7 |
| TS4-x | 4.4 | TS5-x | 3.6 | TS6-x | 4.1 |
| TS5-Ch | 10.8 | ||||
| 25 | −28.3 | 26 | −28.3 | 27 | 27.0 |
| 28 | −27.7 | 29 | −27.7 | 30 | −27.7 |
| 31 | −15.3 |
| MC7 | −7.5 |
| TS7-o | −0.8 |
| TS7-m | 2.7 |
| 32 | −24.3 |
| 33 | −24.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domingo, L.R.; Ríos-Gutiérrez, M.; Aurell, M.J. Unveiling the Ionic Diels–Alder Reactions within the Molecular Electron Density Theory. Molecules 2021, 26, 3638. https://doi.org/10.3390/molecules26123638
Domingo LR, Ríos-Gutiérrez M, Aurell MJ. Unveiling the Ionic Diels–Alder Reactions within the Molecular Electron Density Theory. Molecules. 2021; 26(12):3638. https://doi.org/10.3390/molecules26123638
Chicago/Turabian StyleDomingo, Luis R., Mar Ríos-Gutiérrez, and María José Aurell. 2021. "Unveiling the Ionic Diels–Alder Reactions within the Molecular Electron Density Theory" Molecules 26, no. 12: 3638. https://doi.org/10.3390/molecules26123638
APA StyleDomingo, L. R., Ríos-Gutiérrez, M., & Aurell, M. J. (2021). Unveiling the Ionic Diels–Alder Reactions within the Molecular Electron Density Theory. Molecules, 26(12), 3638. https://doi.org/10.3390/molecules26123638