
Synthesis and Structure of the Bis- and Tris-Polyhedral Hybrid Carboranoclathrochelates with Functionalizing Biorelevant Substituents—The Derivatives of Propargylamine Iron(II) Clathrochelates with Terminal Triple C≡C Bond(s)

 , , and
, , and 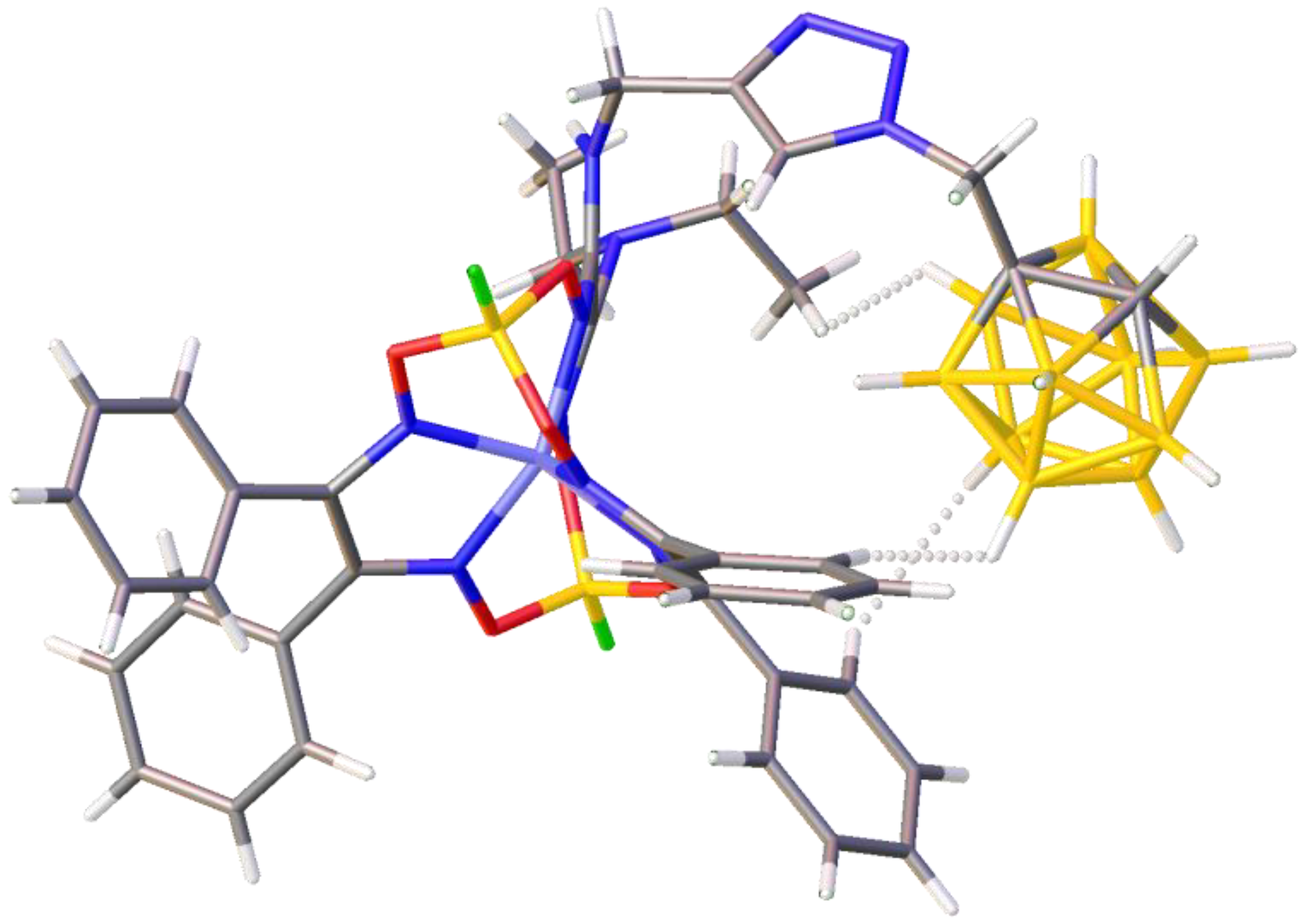{kind=link}
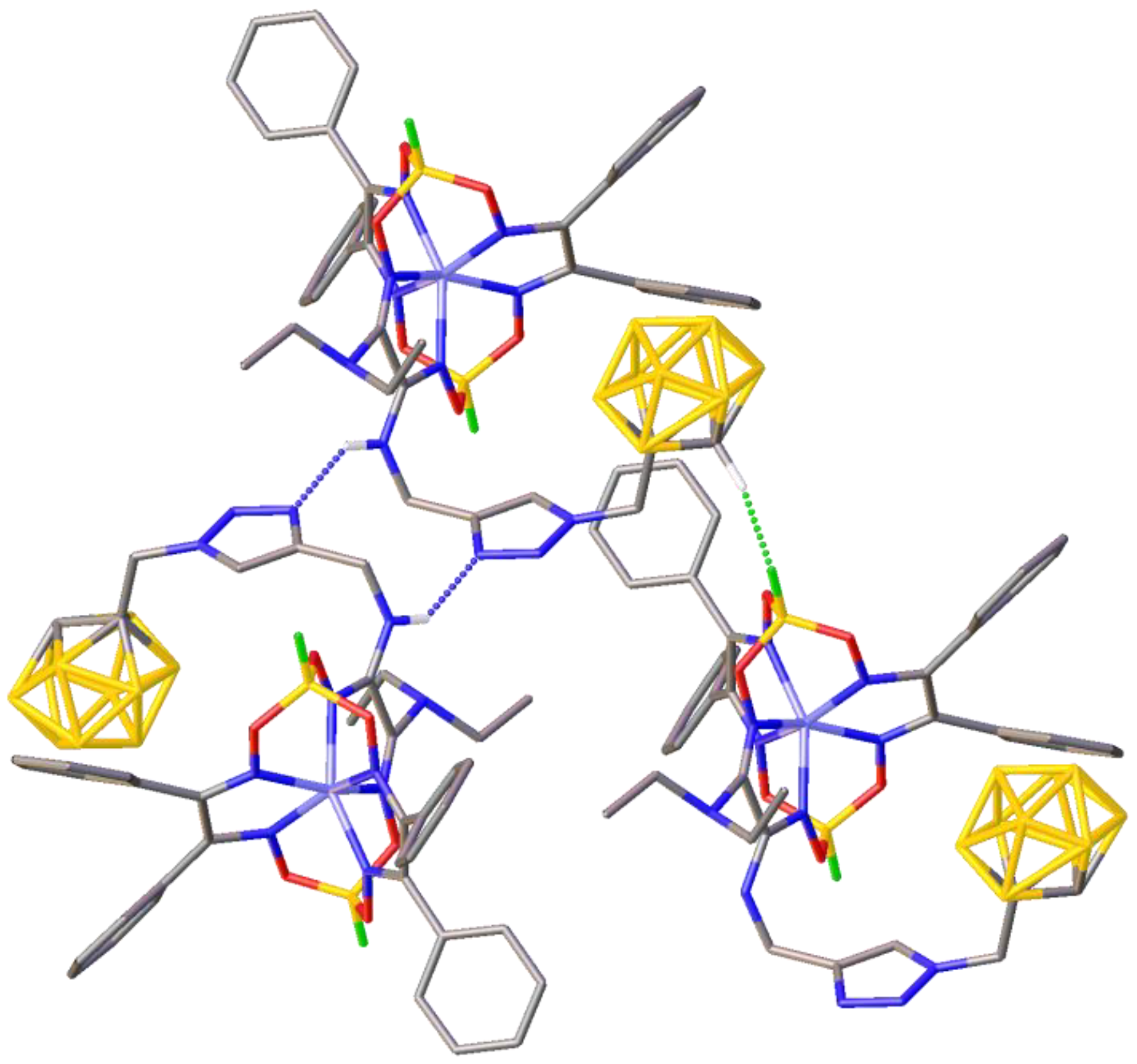{kind=link}
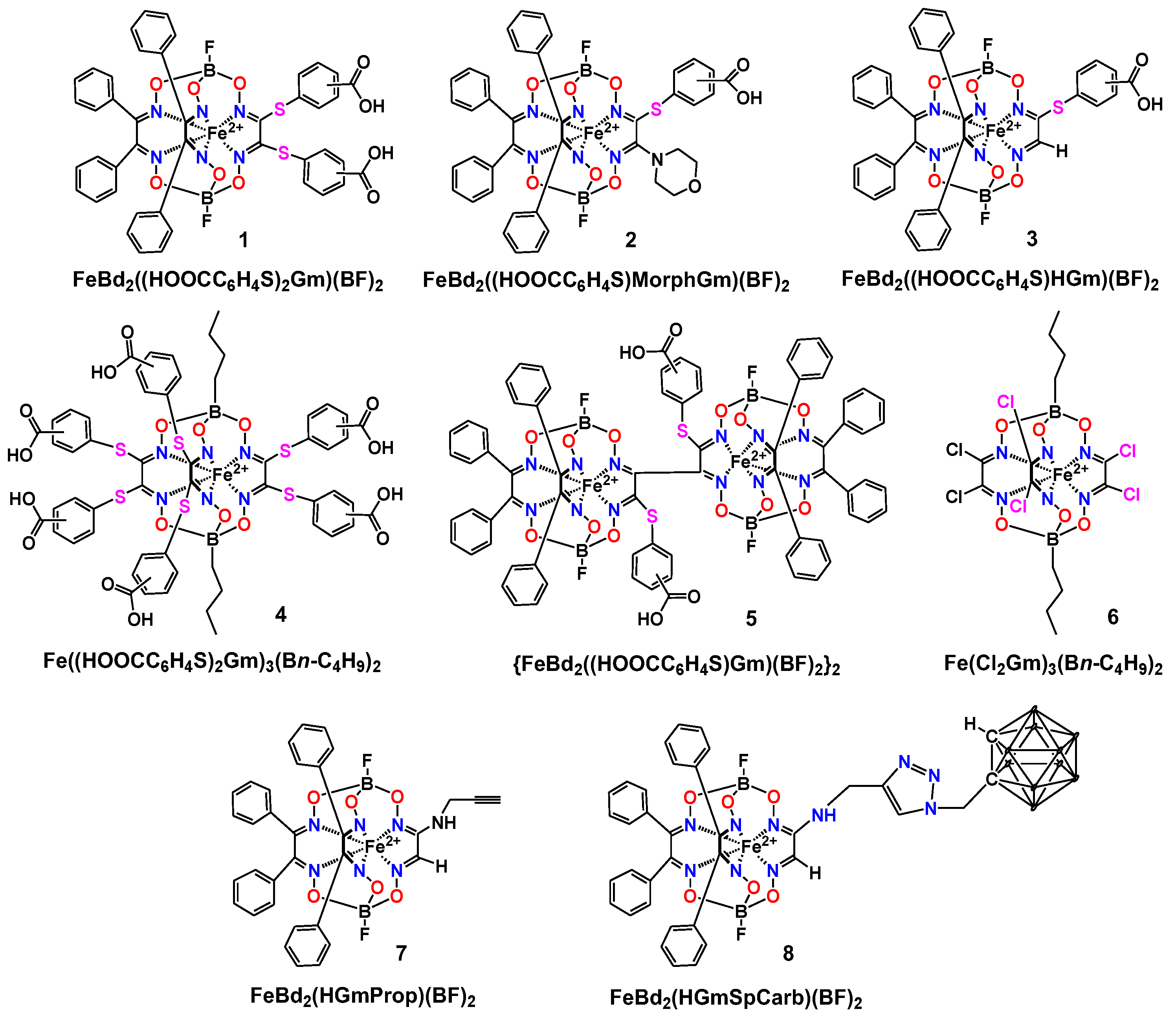{kind=link}
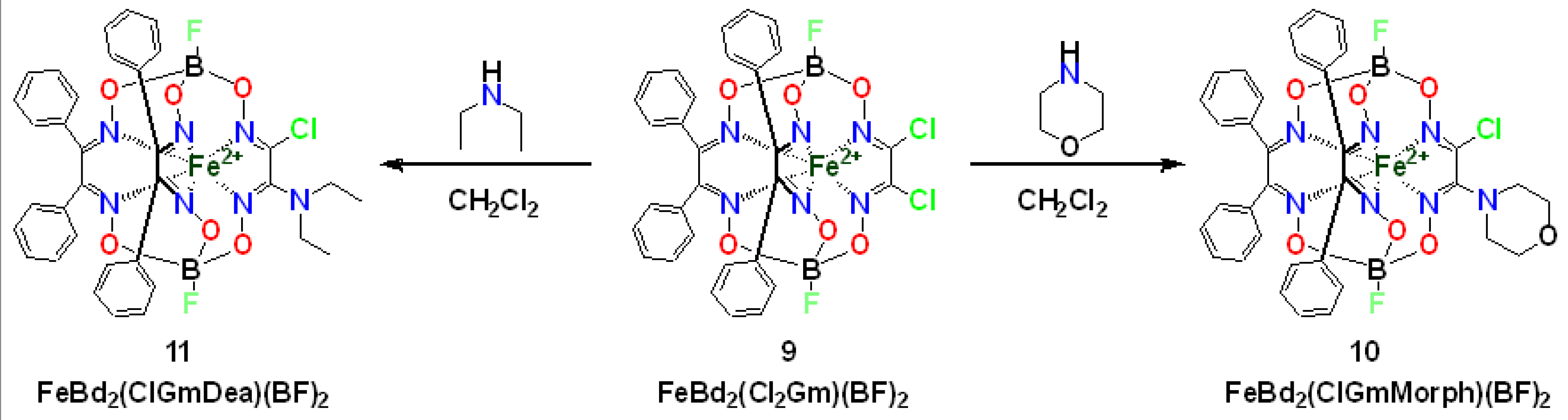{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
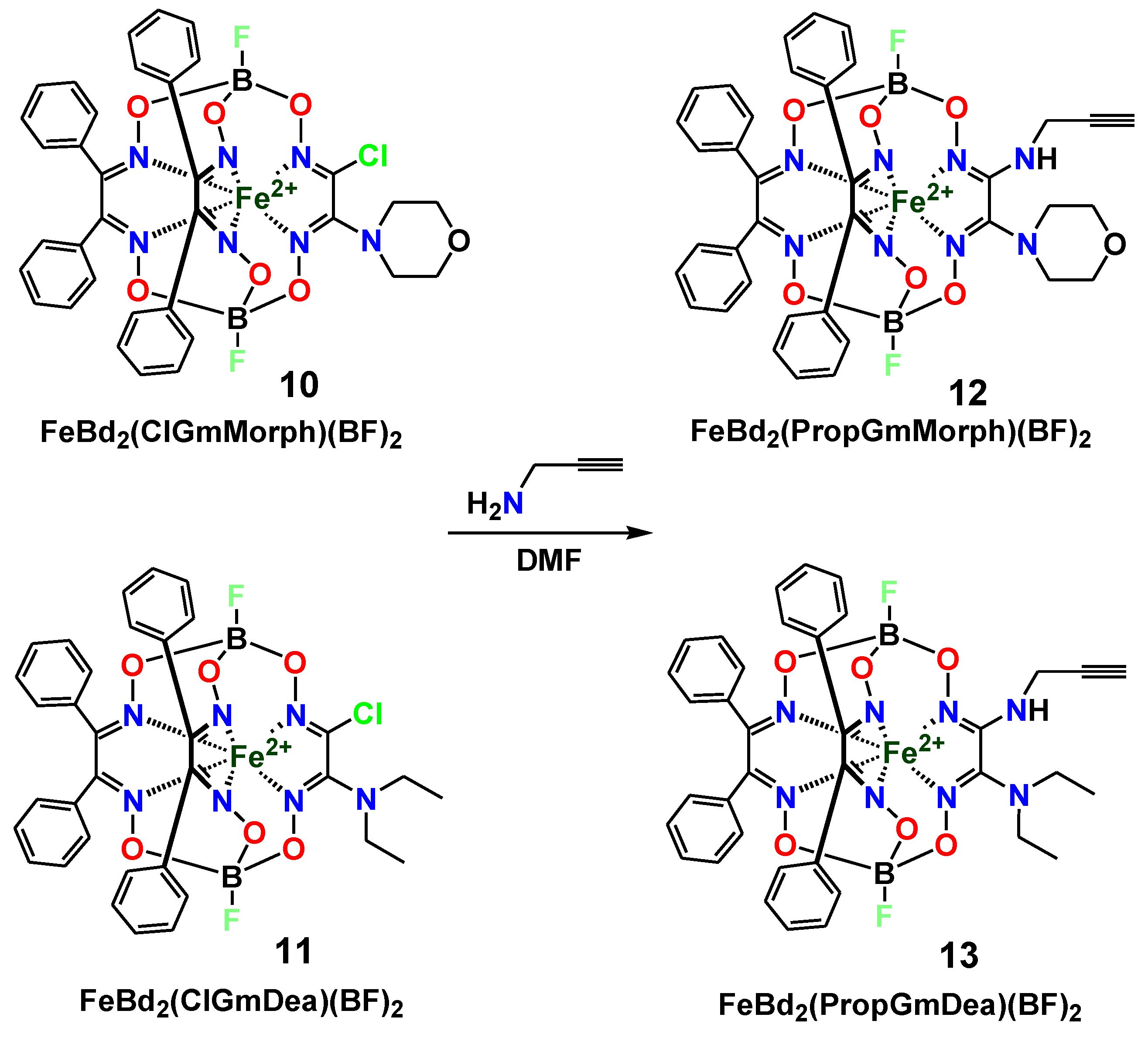
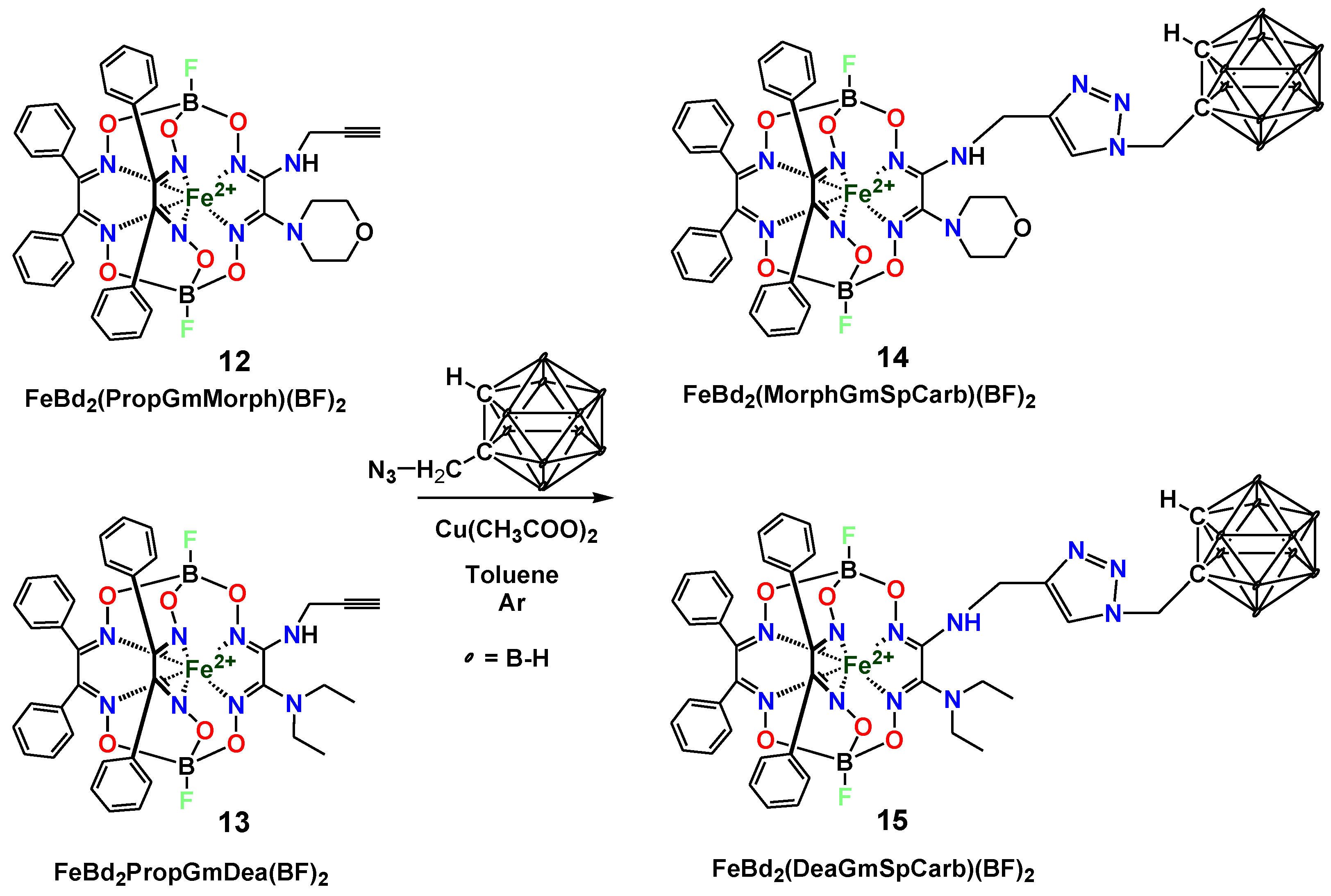
2. Results and Discussion
3. Experimental
3.1. General Information
3.2. Synthesis
3.2.1. Preparation of the Clathrochelate Precursor FeBd2(HGmProp)(BF)2 (7)
3.2.2. “Click” Reaction of the Clathrochelate Precursor FeBd2(HGmProp)(BF)2 (7)
3.3. X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Voloshin, Y.Z.; Kostromina, N.A.; Krämer, R. Clathrochelates: Synthesis, Structure and Properties; Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Belaya, I.G.; Krämer, R. Cage Metal Complexes: Clathrochelates Revisited; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Novikov, V.V.; Nelyubina, Y.V. Recent advances in biological applications of cage metal complexes. RSC Adv. 2015, 5, 72621–72637. [Google Scholar] [CrossRef]
- Kovalska, V.B.; Vakarov, S.V.; Kuperman, M.V.; Losytskyy, M.Y.; Gumienna–Kontecka, E.; Voloshin, Y.Z.; Varzatskii, O.A. Induced chirality of cage metal complexes switched by their supramolecular and covalent binding. Dalton Trans. 2018, 47, 1036–1052. [Google Scholar] [CrossRef] [PubMed]
- Kovalska, V.; Vakarov, S.; Losytskyy, M.; Kuperman, M.; Chornenka, N.; Toporivska, Y.; Gumienna-Kontecka, E.; Voloshin, Y.; Varzatskii, O.; Mokhir, A. Dicarboxyl-terminated iron(II) clathrochelates as ICD-reporters for globular proteins. RSC Adv. 2019, 9, 24218–24230. [Google Scholar] [CrossRef]
- Kovalska, V.; Kuperman, M.; Losytskyy, M.; Vakarov, S.; Potocki, S.; Yarmoluk, S.; Voloshin, Y.; Varzatskii, O.; Gumienna-Kontecka, E. Induced CD of iron(II) clathrochelates: Sensing of the structural and conformational alterations of serum albumins. Metallomics. 2019, 11, 338–348. [Google Scholar] [CrossRef]
- Novikov, V.V.; Varzatskii, O.A.; Negrutska, V.V.; Bubnov, Y.N.; Palchykovska, L.G.; Dubey, I.Y.; Voloshin, Y.Z. Size matters, so does shape: Inhibition of transcription of T7 RNA polymerase by iron(II) clathrochelates. J. Inorg. Biochem. 2013, 124, 42–45. [Google Scholar] [CrossRef]
- Varzatskii, O.A.; Novikov, V.V.; Shulga, S.V.; Belov, A.S.; Vologzhanina, A.V.; Negrutska, V.V.; Dubey, I.Y.; Bubnov, Y.N.; Voloshin, Y.Z. Copper-promoted reductive homocoupling of quasi-aromatic iron(II) clathrochelates: Boosting the inhibitory activity in a transcription assay. Chem. Commun. 2014, 50, 3166–3168. [Google Scholar] [CrossRef] [PubMed]
- Varzatskii, O.A.; Vologzhanina, A.V.; Novikov, V.V.; Vakarova, S.V.; Oblap, R.V.; Voloshin, Y.Z. Inhibition of DNA synthesis in the transcription system of Taq DNA polymerase by various iron and cobalt(II) tris-dioximate clathrochelates: In vitro study and X-ray structure of leader inhibitors, the carboxyl-terminated macrobicyclic complexes. Inorg. Chim. Acta 2018, 482, 90–98. [Google Scholar] [CrossRef]
- Kovalska, V.B.; Losytskyy, M.Y.; Varzatskii, O.A.; Cherepanov, V.V.; Voloshin, Y.Z.; Mokhir, A.A.; Yarmoluk, S.M.; Volkov, S.V. Study of anti-fibrillogenic activity of iron(II) clathrochelates. Bioorg. Med. Chem. 2014, 22, 1883–1888. [Google Scholar] [CrossRef]
- Mokhir, A.A. (Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany). Private communication, 2019.
- Blechinger, J.; Varzackii, O.A.; Kovalska, V.; Zelinskii, G.E.; Voloshin, Y.Z.; Kinski, E.; Mokhir, A. Cytotoxicity of electrophilic iron(II)–clathrochelates in human promyelocytic leukemia cell line. Bioorg. Med. Chem. Lett. 2016, 26, 626–629. [Google Scholar] [CrossRef]
- Hosmane, N.S.; Eagling, R. Handbook of Boron Science: With Applications in Organometallics, Catalysis, Materials and Vedicine; World Scientific Publishing: Singapore, 2018. [Google Scholar]
- Grimes, R.N. Carboranes; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Nakamura, H.; Kirihata, M. Boron Compounds: New Candidates for Boron Carriers in BNCT. In Neutron Capture Therapy: Principles and Applications; Sauerwein, W., Wittig, A., Moss, R., Nakagawa, Y., Eds.; Springer: Heidelberg, Germany, 2012; pp. 99–116. [Google Scholar]
- Omarova, E.O.; Nazarov, P.A.; Firsov, A.M.; Strakhovskaya, M.G.; Arkhipova, A.Y.; Moisenovich, M.M.; Agapov, I.I.; Ol’shevskaya, V.A.; Zaitsev, A.V.; Kalinin, V.N.; et al. Carboranyl-chlorin e6 as a potent antimicrobial photosensitizer. Plos ONE 2015, 10, e0141990. [Google Scholar] [CrossRef]
- Lesnikowski, Z.L. Challenges and opportunities for the application of boron clusters in drug design. J. Med. Chem. 2016, 59, 7738–7758. [Google Scholar] [CrossRef] [PubMed]
- Ol’shevskaya, V.A.; Makarenkov, A.V.; Kononova, E.G.; Petrovskii, P.V.; Verbitskiy, E.V.; Rusinov, G.L.; Charushin, V.N.; Hey-Hawkins, E.; Kalinin, V.N. Novel bis [(1,2,3-triazolyl)methyl] carborane derivatives via regiospecific copper-catalyzed 1,3-dipolar cycloaddition. Polyhedron 2012, 42, 302–306. [Google Scholar] [CrossRef]
- Rokitskaya, T.I.; Khailova, L.S.; Makarenkov, A.V.; Shunaev, A.V.; Tatarskiy, V.V.; Shtil, A.A.; Ol’shevskaya, V.A.; Antonenko, Y.N. Carborane derivatives of 1,2,3-triazole depolarize mitochondria by transferring protons through the lipid part of membranes. BBA – Biomembranes 2019, 1861, 573–583. [Google Scholar] [CrossRef]
- Couto, M.; Alamón, C.; Fernanda García, M.; Kovacs, M.; Trias, E.; Nievas, S.; Pozzi, E.; Curotto, P.; Thorp, S.; Dagrosa, M.A.; et al. Closo-carboranyl- and metallacarboranyl [1,2,3] triazolyl-decorated lapatinib-scaffold for cancer therapy combining tyrosine kinase inhibition and boron neutron capture therapy. Cells 2020, 9, 1408. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Zhang, Z.; Liu, T. A realistic appraisal of boron neutron capture therapy as a cancer treatment modality. Cancer Commun. 2018, 38, 36. [Google Scholar] [CrossRef]
- Hua, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.H.; Zhang, R.M. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Xuan, S.; Vicente, M.G.H. Recent advances in boron delivery agents for boron neutron capture therapy (BNCT). In Boron-Based Compounds: Potential and Emerging Applications in Biomedicine; Hey-Hawkins, E., Vinas, C., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2018; pp. 298–342. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgencycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew.Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Brase, A.S.; Gil, C.; Kneppei, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Singh, B.M.S.; Chowdhury, S.; Koley, S. Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click chemistry: 1,2,3-triazoles as pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef]
- Zelinskii, G.E.; Belov, A.S.; Vologzhanina, A.V.; Limarev, I.P.; Pavlov, A.A.; Olshevskaya, V.A.; Makarenkov, A.V.; Dorovatovskii, P.V.; Lebed, E.G.; Voloshin, Y.Z. Iron(II) clathrochelate with terminal triple C≡C bond and its carboranoclathrochelate derivative with a flexible linker between the polyhedral cages: Synthesis and X-ray structure. Chem. Sel. 2019, 4, 11572–11577. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Zavodnik, V.E.; Varzatskii, O.A.; Belsky, V.K.; Vorontsov, I.I.; Antipin, M.Y. Reactions of chloride iron(II) clathrochelates with aliphatic amines: An unexpected influence of the nature of the amine and solvent on the reaction products. Inorg. Chim. Acta 2001, 321, 116–134. [Google Scholar] [CrossRef]
- Ol’shevskaya, V.A.; Makarenkov, A.V.; Kononova, E.G.; Petrovskii, P.V.; Verbitskii, E.V.; Rusinov, G.L.; Kalinin, V.N.; Charushin, V.N. 1,3-Dipolar cycloaddition of [(o-carboran-1-yl)methyl] azide to alkynes. Dokl. Chem. 2010, 434, 245–248. [Google Scholar] [CrossRef]
- Meng, D.J.; Fokin, V.V.; Finn, M.G. Kinetic resolution by copper-catalyzed azide-alkyne cycloaddition. Tetrahedron Lett. 2005, 46, 4543–4546. [Google Scholar] [CrossRef]
- Reddy, K.R.; Rajgopal, K.; Kantam, M.L. Copper (II)-promoted regioselective synthesis of 1, 4-disubstituted 1, 2, 3-triazoles in water. Synlett 2006, 6, 957–959. [Google Scholar] [CrossRef]
- Kamata, K.; Yamaguchi, S.; Kotani, M.; Yamaguchi, K.; Mizuno, N. Efficient oxidative alkyne homocoupling catalyzed by a monomeric dicopper-substituted silicotungstate. Angew. Chem. Int. Ed. Engl. 2008, 47, 2407–2410. [Google Scholar] [CrossRef]
- Belov, A.S.; Prikhod’ko, A.I.; Novikov, V.V.; Vologzhanina, A.V.; Bubnov, Y.N.; Voloshin, Y.Z. First “click” synthesis of the ribbed-functionalized metal clathrochelates: Cycloaddition of benzyl azide to propargylamineiron(II) macrobicycle and the unexpected transformations of the resulting cage complex. Eur. J. Inorg. Chem. 2012, 4507–4514. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Zavodnik, V.E.; Varzatskii, O.A.; Belsky, V.K.; Palchik, A.V.; Strizhakova, N.G.; Vorontsov, I.I.; Antipin, M.Y. Clathrochelatemonoribbed-functionalized iron(II) α-dioximates: Synthetic pathways and structural and electrochemical features. Dalton Trans. 2002, 1193–1202. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Cryst. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Belov, A.S.; Vologzhanina, A.V.; Novikov, V.V.; Negrutska, V.V.; Dubey, I.Y.; Mikhailova, Z.A.; Lebed, E.G.; Voloshin, Y.Z. Synthesis of the first morpholine-containing iron(II) clathrochelates: A new class of efficient functionalized transcription inhibitors. Inorg. Chim. Acta 2014, 421, 300–306. [Google Scholar] [CrossRef]
- Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G.W. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. 2011, D67, 271–281. [Google Scholar] [CrossRef]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. 2006, D62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystalstructure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelinskii, G.E.; Limarev, I.P.; Vologzhanina, A.V.; Olshevskaya, V.A.; Makarenkov, A.V.; Dorovatovskii, P.V.; Chuprin, A.S.; Vershinin, M.A.; Dudkin, S.V.; Voloshin, Y.Z. Synthesis and Structure of the Bis- and Tris-Polyhedral Hybrid Carboranoclathrochelates with Functionalizing Biorelevant Substituents—The Derivatives of Propargylamine Iron(II) Clathrochelates with Terminal Triple C≡C Bond(s). Molecules 2021, 26, 3635. https://doi.org/10.3390/molecules26123635
Zelinskii GE, Limarev IP, Vologzhanina AV, Olshevskaya VA, Makarenkov AV, Dorovatovskii PV, Chuprin AS, Vershinin MA, Dudkin SV, Voloshin YZ. Synthesis and Structure of the Bis- and Tris-Polyhedral Hybrid Carboranoclathrochelates with Functionalizing Biorelevant Substituents—The Derivatives of Propargylamine Iron(II) Clathrochelates with Terminal Triple C≡C Bond(s). Molecules. 2021; 26(12):3635. https://doi.org/10.3390/molecules26123635
Chicago/Turabian StyleZelinskii, Genrikh E., Ilya P. Limarev, Anna V. Vologzhanina, Valentina A. Olshevskaya, Anton V. Makarenkov, Pavel V. Dorovatovskii, Alexander S. Chuprin, Mikhail A. Vershinin, Semyon V. Dudkin, and Yan Z. Voloshin. 2021. "Synthesis and Structure of the Bis- and Tris-Polyhedral Hybrid Carboranoclathrochelates with Functionalizing Biorelevant Substituents—The Derivatives of Propargylamine Iron(II) Clathrochelates with Terminal Triple C≡C Bond(s)" Molecules 26, no. 12: 3635. https://doi.org/10.3390/molecules26123635
APA StyleZelinskii, G. E., Limarev, I. P., Vologzhanina, A. V., Olshevskaya, V. A., Makarenkov, A. V., Dorovatovskii, P. V., Chuprin, A. S., Vershinin, M. A., Dudkin, S. V., & Voloshin, Y. Z. (2021). Synthesis and Structure of the Bis- and Tris-Polyhedral Hybrid Carboranoclathrochelates with Functionalizing Biorelevant Substituents—The Derivatives of Propargylamine Iron(II) Clathrochelates with Terminal Triple C≡C Bond(s). Molecules, 26(12), 3635. https://doi.org/10.3390/molecules26123635