
DFT Calculations of 1H NMR Chemical Shifts of Geometric Isomers of Conjugated Linolenic Acids, Hexadecatrienyl Pheromones, and Model Triene-Containing Compounds: Structures in Solution and Revision of NMR Assignments



Abstract

1. Introduction
2. Results and Discussion
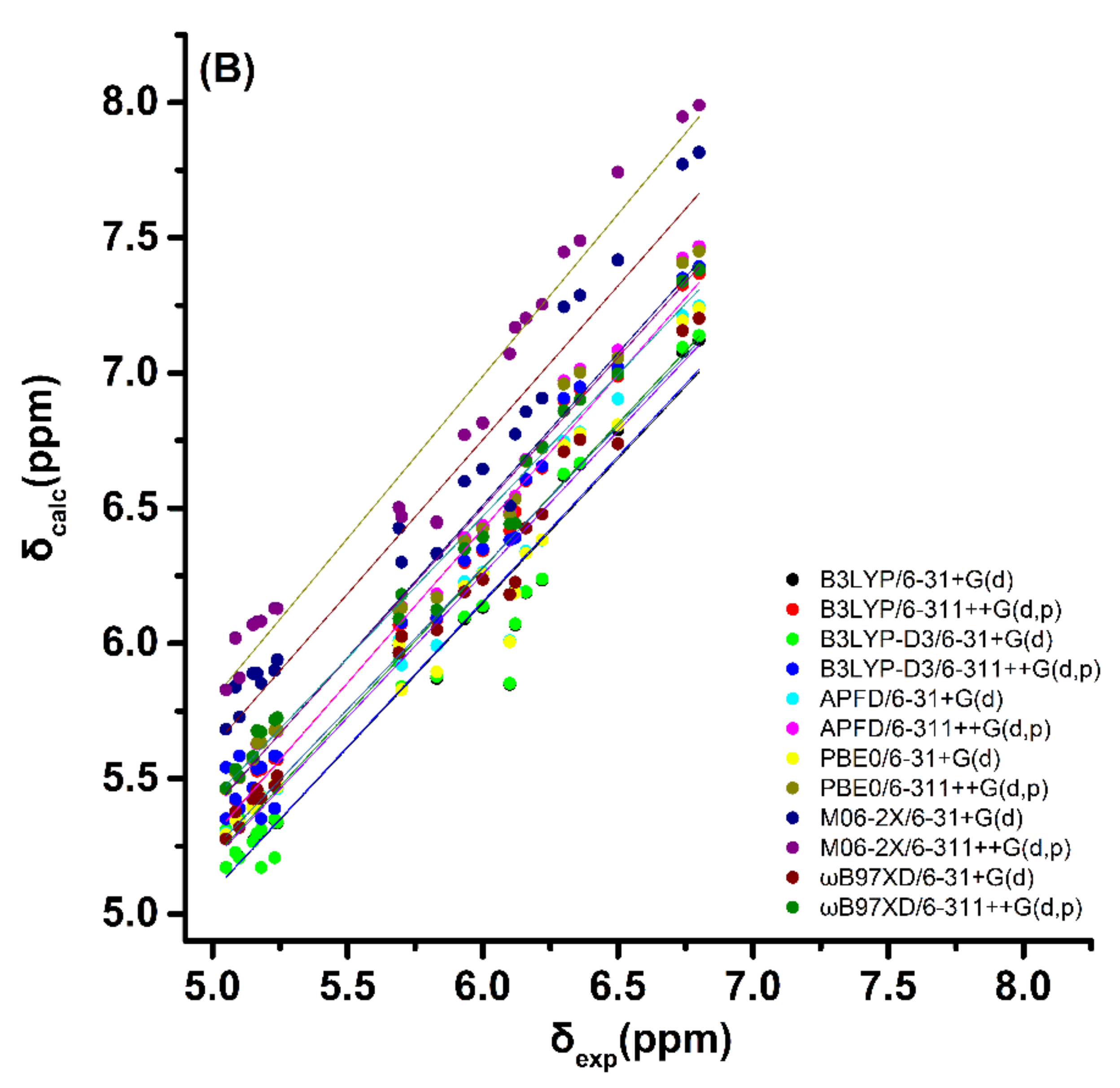
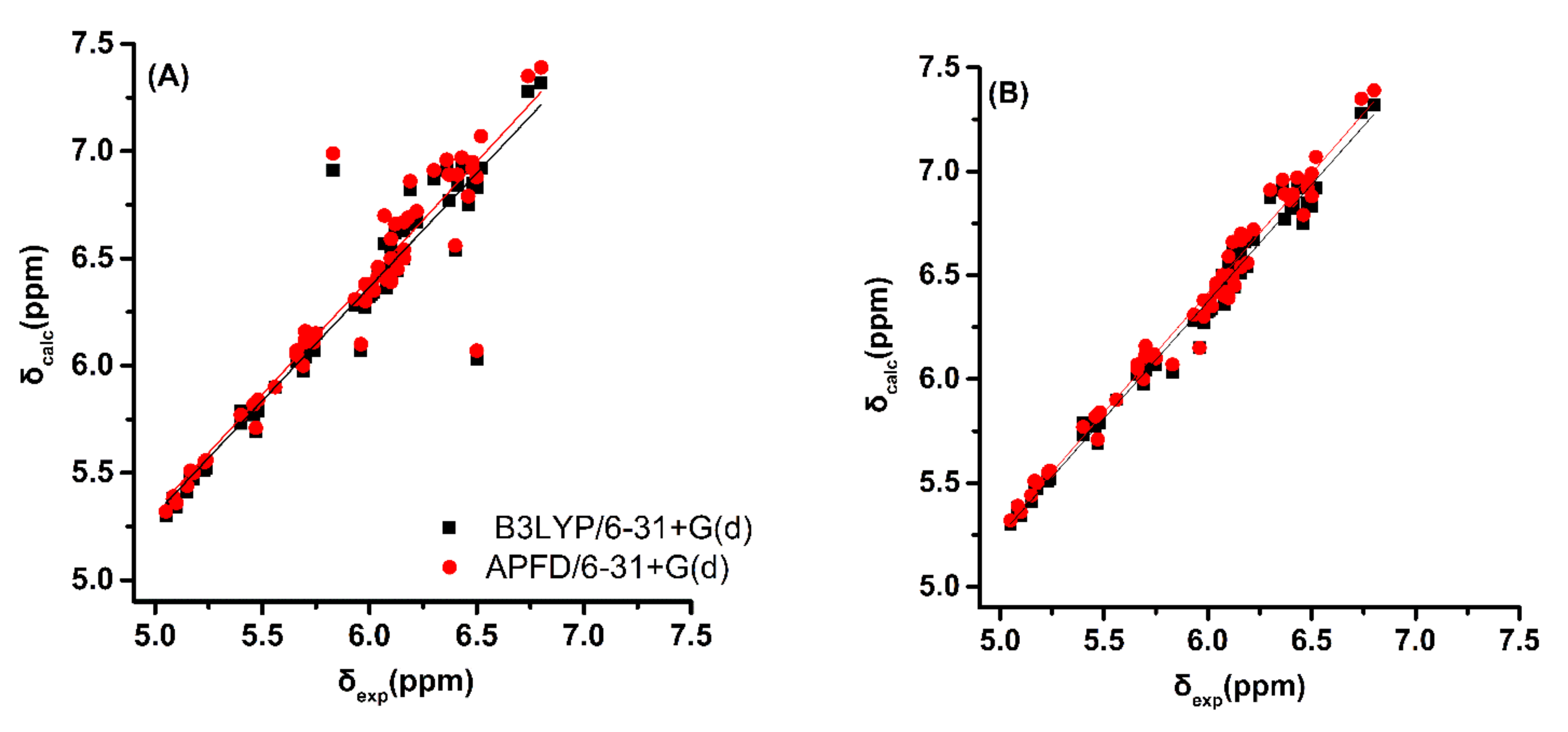
2.1. DFT-Calculated vs. Experimental 1H NMR Chemical Shifts of Model Compounds in Solution: Effects of Various Functionals and Basis Sets
- (i)
- the 1H NMR spectra are recorded in, e.g., a CDCl3 solution at 298 K, and a preliminary assignment is performed using a variety of 1D and 2D NMR experiments;
- (ii)
- the 1H NMR chemical shifts are computed with the CPCM model at the same level of theory as geometry optimization or at the GIAO/B3LYP/6–311+G(2d,p) level, even with less demanding functionals and basis sets;
- (iii)
- a very good linear correlation between experimental NMR chemical shifts, δexp, and calculated shifts, δcalc, provides a strong indication that the assignment procedure is correct.
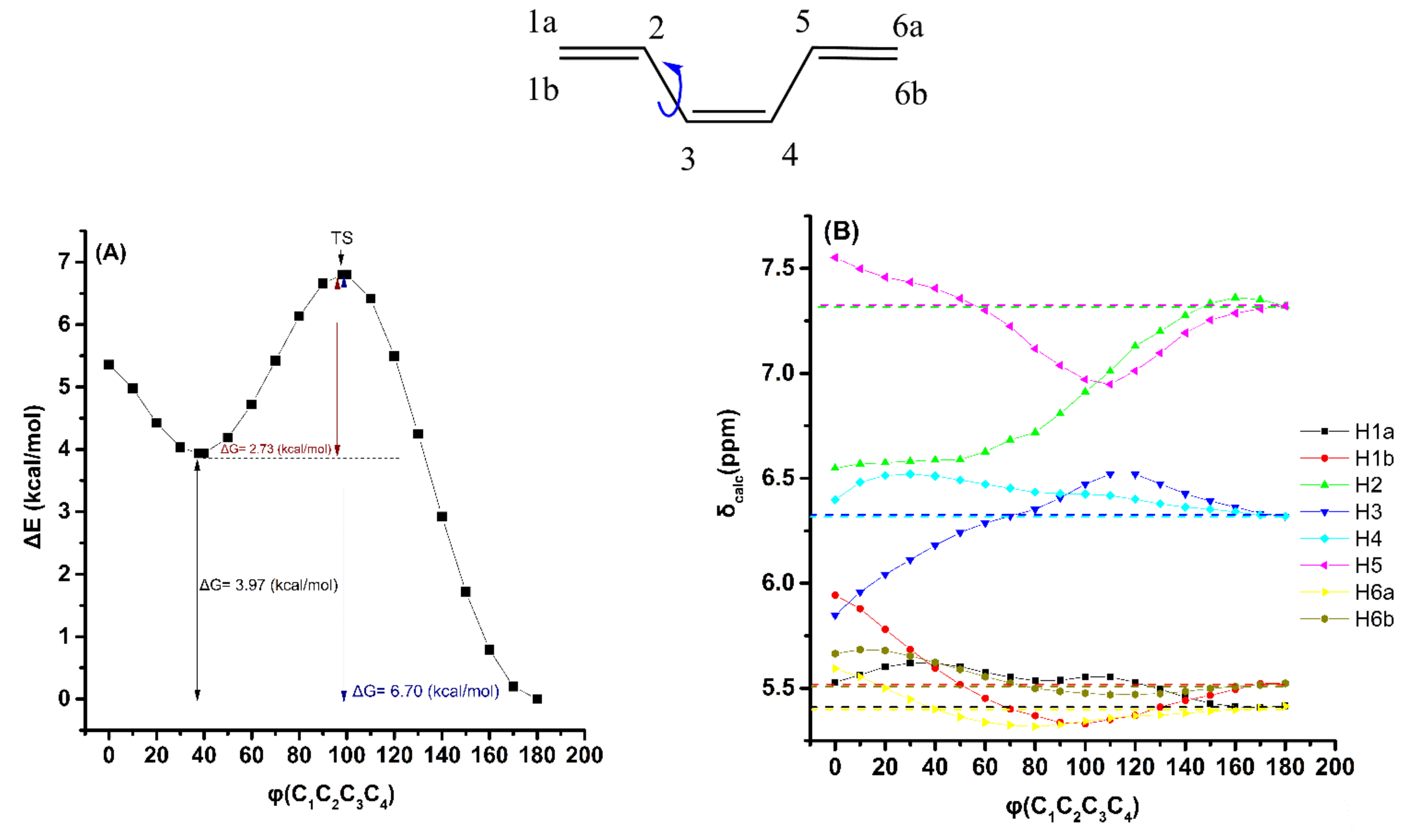
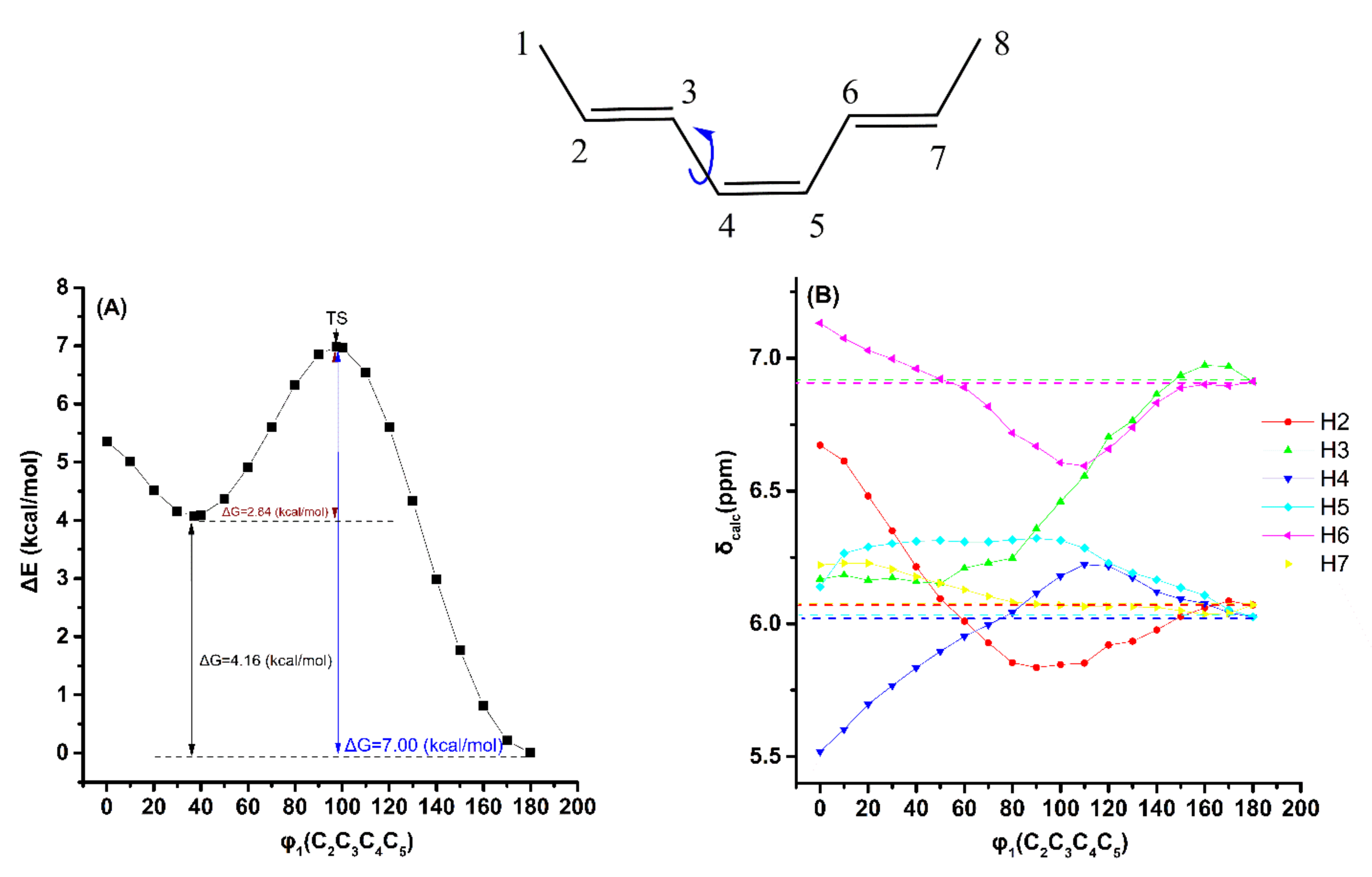
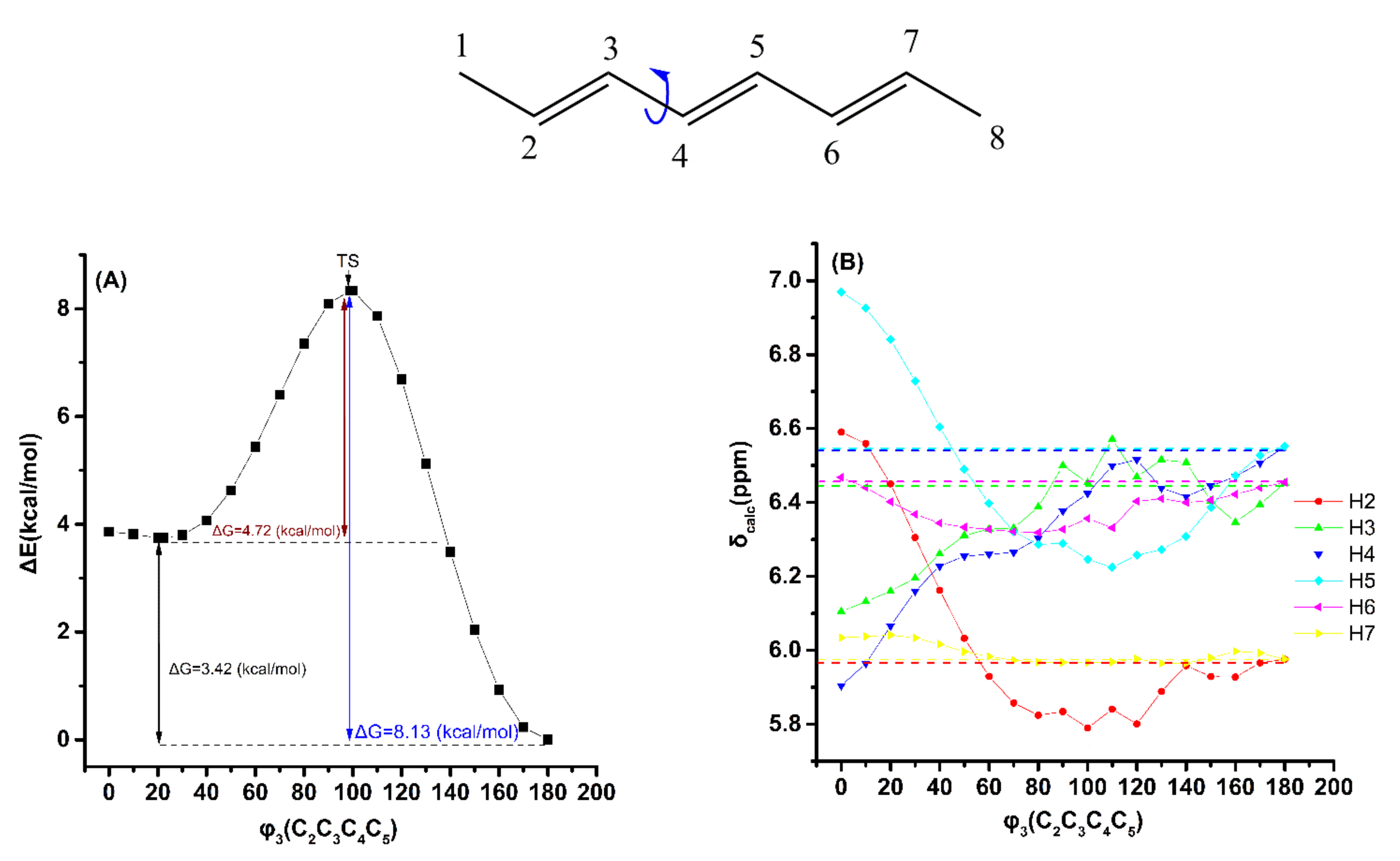
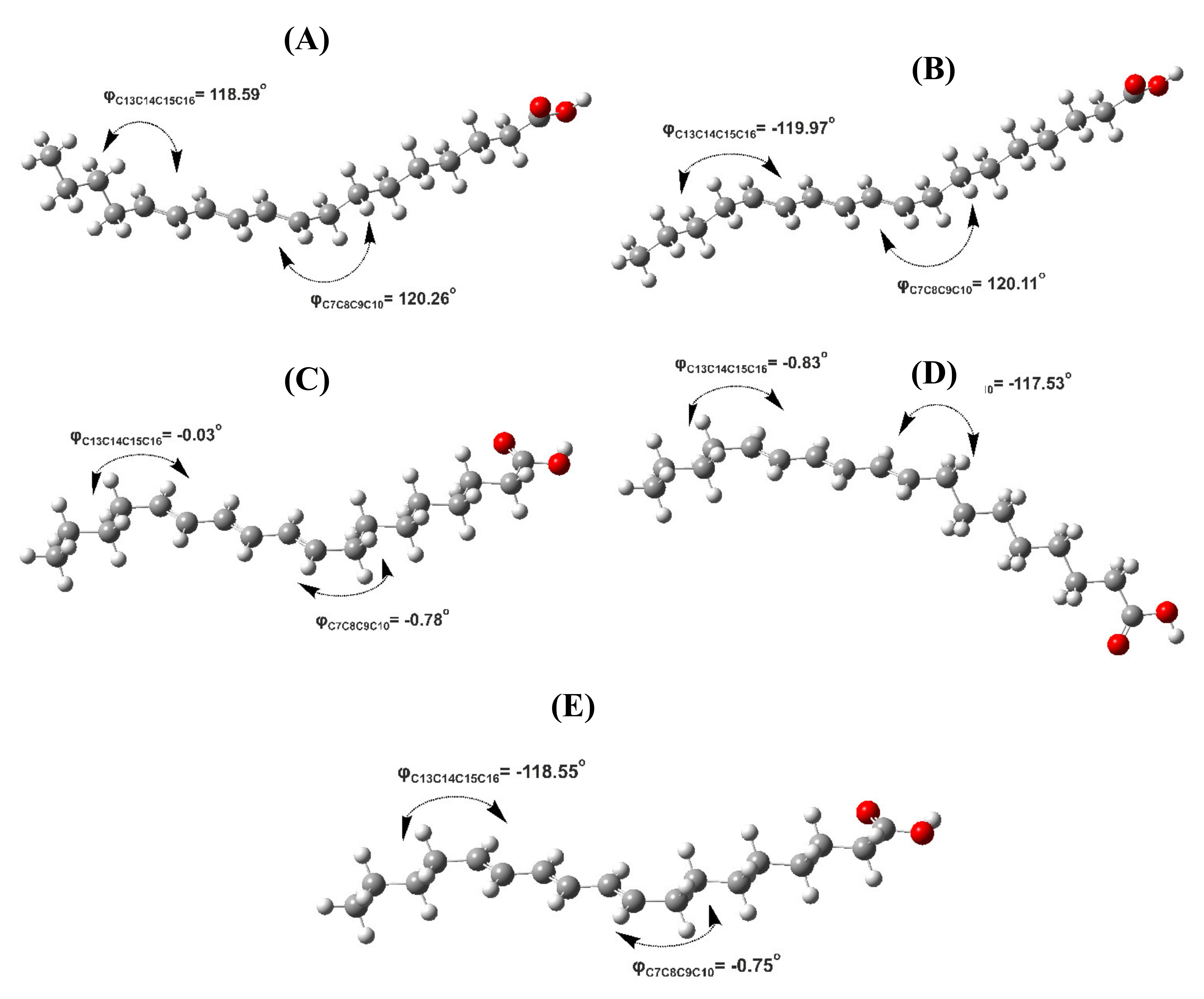
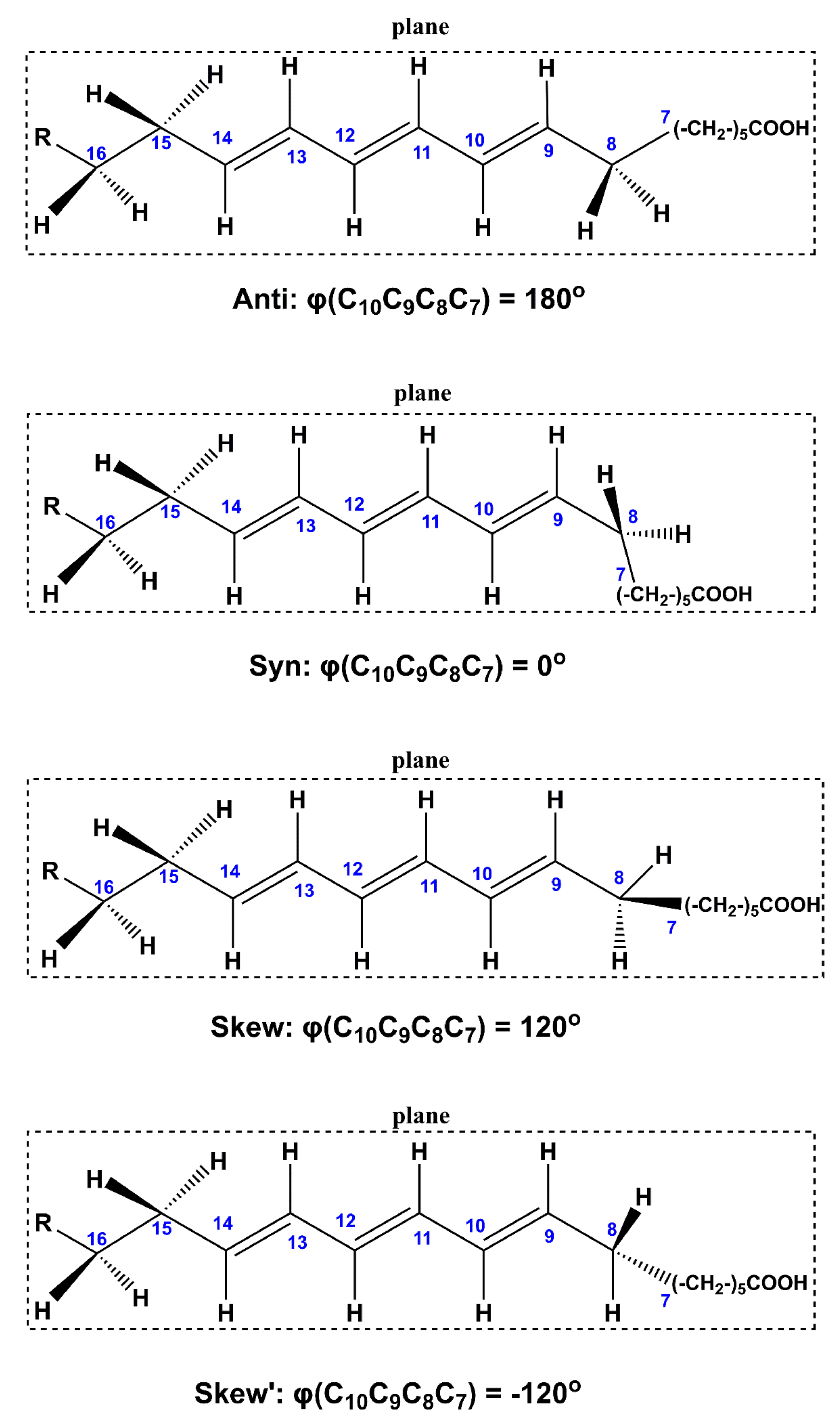
2.2. Out-of-Plane Deformation of the Trienyl System and Steric Effects in Model Compounds
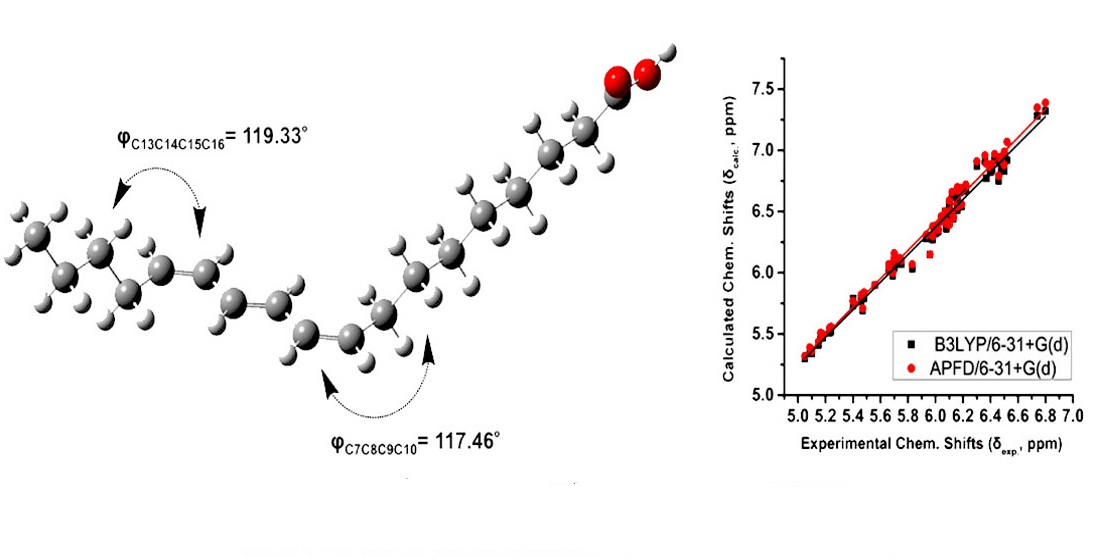
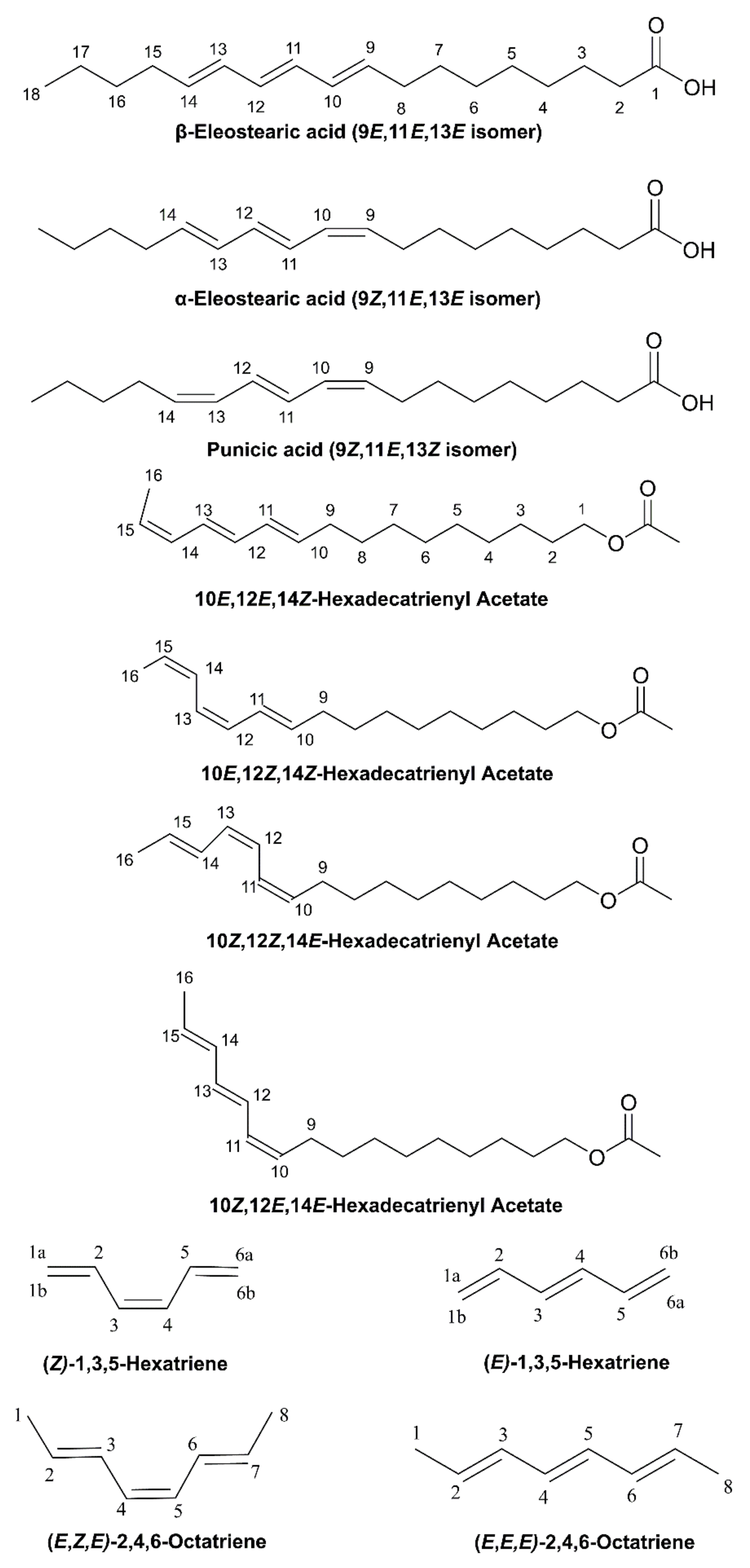
2.3. DFT-Calculated vs. Experimental 1H NMR Chemical Shifts: Structure Elucidation in Solution of Geometric Isomers of 18:3 Conjugated Linolenic Acids and Hexadecatrienyl Pheromones
3. Computational Methods
4. Conclusions
- (a)
- Very good linear correlations can be obtained between DFT-calculated and experimental 1H NMR chemical shifts of the olefinic protons of the lowest-energy DFT-optimized single conformer using standard functionals (B3LYP and PBE0) as well as corrections for dispersion interactions (B3LYP-D3, APFD, M06–2X and ωB97XD). The ωB97XD performs slightly better, but again the accuracy of the functionals used was rather similar.
- (b)
- (c)
- The accuracy of computational 1H NMR chemical shifts can facilitate (i) the unequivocal assignment of the geometric isomerism in conjugated trienyl systems of biological systems, such as CLnAs and hexadecatrienyl pheromones, and especially in the case of problematic resonance assignment due to extensive signal overlap, and (ii) structure elucidation in solution [28,36].
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Yuan, G.F.; Chena, X.E.; Li, D. Conjugated linolenic acids and their bioactivities: A review. Food Funct. 2014, 5, 1360–1368. [Google Scholar] [CrossRef]
- Dubey, K.K.D.; Sharma, G.; Kumar, A. Conjugated linolenic acids: Implication in cancer. J. Agric. Food Chem. 2019, 67, 6091–6101. [Google Scholar] [CrossRef] [PubMed]
- Ιgarashi, M.; Miyazawa, T. Newly recognized cytotoxic effect of conjugated trienoic fatty acids on cultured human tumor cells. Cancer Lett. 2000, 148, 173–179. [Google Scholar] [CrossRef]
- Özgül-Yücel, S. Determination of conjugated linolenic acid content of selected oil seeds grown in Turkey. J. Am. Oil Chem. Soc. 2005, 82, 893–897. [Google Scholar] [CrossRef]
- Yasui, Y.; Hosokawa, M.; Kohno, H. Growth inhibition and apoptosis induction by all-trans-conjugated linolenic acids on human colon cancer cells. Anticancer Res. 2006, 1855–1860. [Google Scholar]
- Grossmann, M.E.; Mizuno, N.K.; Schuster, T.; Cleary, M.P. Punicic acid is an ω-5 fatty acid capable of inhibiting breast cancer proliferation. Int. J. Oncol. 2009, 36, 421–426. [Google Scholar]
- Lansky, E.P.; Newman, R.A. Punica granatum (Pomegranate) and its potential for prevention and treatment of inflammation and cancer. J. Ethnopharmacol. 2007, 109, 177–206. [Google Scholar] [CrossRef]
- Aruna, P.; Venkataramanamma, D.; Singh, A.K.; Singh, R.P. Health benefits of punicic acid: A review. Compr. Rev. Food Sci. Food Saf. 2016, 15, 16–27. [Google Scholar] [CrossRef]
- Suzuki, R.; Noguchi, R.; Ota, T.; Abe, M.; Miyashita, K.; Kawada, T. Cytotoxic effect of conjugated trienoic fatty acids on mouse tumor and human monocytic leukemia cells. Lipids 2001, 36, 477–482. [Google Scholar] [CrossRef]
- Seol, K.Y.; Honda, H.; Usui, K.; Ando, T.; Matsumoto, Y. 10,12,14-Hexadecatrienyl acetate: Sex pheromone of the mulberry pyralid, Glyphodes pyloalis walker (lepidoptera: Pyralidae). Agric. Biol. Chem. 1987, 51, 2285–2287. [Google Scholar] [CrossRef]
- Gries, R.; Reckziegel, A.; Bogenschütz, H.; Kontzog, H.-G.; Schlegel, C.; Francke, W.; Millar, J.G.; Gries, G. (Z,Z)-11,13-Hexadecadienyl acetate and (Z,E)-11,13,15-hexadecatrienyl acetate: Synergistic sex pheromone components of oak processionary moth, Thaumetopoea processionea (Lepidoptera: Thaumetopoeidae). Chemoecology 2004, 14, 95–100. [Google Scholar] [CrossRef]
- Millar, J.G.; McElfresh, J.S.; Romero, C.; Vila, M.; Mari-Mena, N.; Lopez-Vaamonde, C. Identification of the sex pheromone of a protected species, the Spanish moon moth Graellsia isabellae. J. Chem. Ecol. 2010, 36, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Ogura, Y.; Koyama, M.; Kurane, M.; Uchiyama, M.; Yaul Seol, K.Y. Syntheses and NMR analyses of eight geometrical isomers of 10,12,14-hexadecatrienyl acetate, sex pheromone candidates of the mulberry pyralid. Agric. Biol. Chem. 1988, 52, 2459–2468. [Google Scholar]
- Doolittle, R.E.; Brabham, A.; Tumlinson, J.H. Sex phermonex of Manduca Sexta (L) stereoselective synthesis of (10E, 12E, 14Z)-10,12,14-hexadecatrienal and isomers. J. Chem. Ecol. 1990, 16, 1131–1153. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Ohsawa, H. Sex pheromone candidates with a conjugated triene system: Synthesis and chemical characterization. J. Chem. Ecol. 1993, 19, 119–132. [Google Scholar] [CrossRef]
- Tulloch, A.P.; Bergter, L. Analysis of the conjugated trienoic acid containing oil from fevillea trilobata by 13C nuclear magnetic resonance spectroscopy. Lipids 1979, 14, 996–1002. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, H.-L.; Chen, J.-N.; Chen, Z.-Y.; Yang, L. Identification and characterization of conjugated linolenic acid isomers by Ag+-HPLC and NMR. J. Agric. Food. Chem. 2006, 54, 9004–9009. [Google Scholar] [CrossRef]
- Cao, Y.; Yang, L.; Gao, H.-L.; Chen, J.-N.; Chen, Z.-Y.; Ren, Q.-S. Re-characterization of three conjugated linolenic acid isomers by GC–MS and NMR. Chem. Phys. Lipids 2007, 145, 128–133. [Google Scholar] [CrossRef]
- Sassano, G.; Sanderson, P.; Franx, J.; Groot, P. van Straalen, J.; Bassaganya-Riera, J. Analysis of pomegranate seed oil for the presence of jacaric acid. J. Sci. Food Agric. 2009, 89, 1046–1052. [Google Scholar] [CrossRef]
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.; Tsiafoulis, C.; Gerothanassis, I.P. High resolution NMR spectroscopy as a structural and analytical tool for unsaturated lipids in solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef]
- Albriktsen, P.; Harris, R.K. NMR Studies of conjugated linear trienes. Acta Chem. Scand. 1973, 27, 1875–1882. [Google Scholar] [CrossRef][Green Version]
- Brouwer, A.M.; Bezemer, L.; Jacobs, H.J.C. Steric perturbation of the conjugated triene chromophore. Conformational analysis of (E)- and (2)-3-methyI-1,3,5-hexatriene and (2)-3-tert-butyl- 1,3,5-hexatriene. Red. Trav. Chim. Pays-Bas 1992, 111, 138–143. [Google Scholar] [CrossRef]
- Brouwer, A.M.; Bezemer, L.; Jacobs, H.J.C. Kinetics of thermal interconversion between cis, cis -1,3,5-octatriene, cis, cis, cis -2,4,6-octatriene, and cis, cis, trans-2,4,6-octatriene. Red. Trav. Chim. Pays-Bas 1998, 53, 1132–1137. [Google Scholar]
- Townsend, E.M.; Schrock, R.R.; Hoveyda, A.H. Z-Selective metathesis homocoupling of 1,3-dienes by molybdenum and tungsten monoaryloxide pyrrolide (MAP) complexes. J. Am. Chem. Soc. 2012, 134, 11334–11337. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Goodman, J.M. Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculations. J. Org. Chem. 2009, 74, 4597–4607. [Google Scholar] [CrossRef] [PubMed]
- Saielli, G.; Nicolaou, K.C.; Ortiz, A.; Zhang, H.; Bagno, A. Addressing the stereochemistry of complex organic molecules by density functional theory-NMR: Vannusal B. in retrospective. J. Am. Chem. Soc. 2011, 133, 6072–6077. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Tantillo, D.J. Walking in the woods with quantum chemistry –applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 2013, 30, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Kontogianni, V.G.; Tsiafoulis, C.G.; Tzakos, A.G.; Gerothanassis, I.P. Investigation of solute–solvent interactions in phenol compounds: Accurate ab initio calculations of solvent effects on 1H NMR chemical shifts. Org. Biomol. Chem. 2013, 11, 7400–7411. [Google Scholar] [CrossRef]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef]
- Jaremko, L.; Jaremko, M.; Buczek, A.; Broda, A.M.; Kupka, T.; Jackowski, K. 1H and 13C shielding measurements in comparison with DFT calculations performed for 2-(acetylamino)-N,N-dimethyl-3-phenylacrylamide isomers. Chem. Phys. Lett. 2015, 627, 1–6. [Google Scholar] [CrossRef]
- Navarro-Vázquez, A. State of the art and perspectives in the application of quantum chemical prediction of 1H and 13C chemical shifts and scalar couplings for structural elucidation of organic compounds. Magn. Reson. Chem. 2017, 55, 2–32. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, G.; Benedit, G.; Fernández, R.; Pérez, M.; Rodriguez, J.; Jiménez, C.; Cuevas, C. Can stereoclusters separated by two methylene groups be related by DFT studies? The case of the cytotoxic meroditerpeneshalioxepines. J. Nat. Prod. 2018, 81, 343–348. [Google Scholar] [CrossRef]
- Krivdin, L.B. Computational protocols for calculating 13C NMR chemical shifts. Progr. NMR Spectrosc. 2019, 112–113, 103–156. [Google Scholar] [CrossRef] [PubMed]
- Krivdin, L.B. Computational 1H NMR: Part 1. Theoretical background. Magn. Reson. Chem. 2019, 57, 897–914. [Google Scholar] [CrossRef]
- Semenov, V.A.; Krivdin, L.B. Computational 1H and 13C NMR of strychnobaillonine: On the way to larger molecules calculated at lower computational costs. Magn. Reson. Chem. 2021, 59, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Siskos, M.G.; Tzakos, A.G.; Gerothanassis, I.P. Accurate ab initio calculations of O–H…O and O–H…O proton chemical shifts: Towards elucidation of the nature of the hydrogen bond and prediction of hydrogen bond distances. Org. Biomol. Chem. 2015, 13, 8852–8868. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Tzakos, A.G.; Gerothanassis, I.P. 1H NMR chemical shift assignment, structure and conformational elucidation of hypericin with the use of DFT calculations–The challenge of accurate positions of labile hydrogens. Tetrahedron 2016, 72, 8287–8293. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen atomic positions of O–H…O hydrogen bonds in solution and in the solid state: The synergy of quantum chemical calculations with 1H-NMR chemical shifts and X-ray diffraction methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Refinement of labile hydrogen positions based on DFT calculations of 1H NMR chemical shifts: Comparison with X-ray and neutron diffraction methods. Org. Biomol. Chem. 2017, 15, 4655–4666. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. DFT-calculated structures based on 1H NMR chemical shifts in solution vs. structures solved by single-crystal X-ray and crystalline-sponge methods: Assessing specific sources of discrepancies. Tetrahedron 2018, 74, 4728–4737. [Google Scholar] [CrossRef]
- Torralba, M.P.; Sanz, D.; Claramunt, R.M.; Alkorta, I.; Dardonville, C.; Elguero, J. The structure of fosfomycin salts in solution and in the solid state by nuclear magnetic resonance spectroscopy and DFT calculations. Tetrahedron 2018, 74, 3937–3942. [Google Scholar] [CrossRef]
- Mari, S.H.; Varras, P.C.; Wahab, A.-t.; Choudhary, I.M.; Siskos, M.G.; Gerothanassis, I.P. Solvent-dependent structures of natural products based on the combined use of DFT calculations and 1H-NMR chemical shifts. Molecules 2019, 24, 2290. [Google Scholar] [CrossRef]
- Siskos, M.G.; Varras, P.C.; Gerothanassis, I.P. DFT calculations of O-H....O 1H NMR chemical shifts in investigating enol–enol tautomeric equilibria: Probing the impacts of intramolecular hydrogen bonding vs. stereoelectronic interactions. Tetrahedron 2020, 76, 130979. [Google Scholar] [CrossRef]
- Ahmed, R.; Varras, P.C.; Siskos, M.G.; Siddiqui, H.; Choudhary, M.I.; Gerothanassis, I.P. NMR and computational studies as analytical and high-resolution structural tool for complex hydroperoxides and endo-hydroperoxides of fatty acids in solution. Molecules 2020, 25, 4902. [Google Scholar] [CrossRef] [PubMed]
- Venianakis, T.; Oikonomaki, C.; Siskos, M.G.; Varras, P.C.; Primikyri, A.; Alexandri, E.; Gerothanassis, I.P. DFT Calculations of 1H- and 13C-NMR chemical shifts of geometric isomers of conjugated linoleic acid (18:2 ω-7) and model compounds in solution. Molecules 2020, 25, 3660. [Google Scholar] [CrossRef] [PubMed]
- Touw, S.I.E.; de Groot, H.J.M.; Buda, F. DFT calculations of the 1H NMR chemical shifts and 13C chemical shifts tensors of retinal isomers. J. Mol. Struct. (Theochem) 2004, 711, 141–147. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Abraham, R.J.; Canton, M.; Grifiths, L. Proton chemical shifts in alkenes and anisotropic and steric effects in double bond. Magn. Reson. Chem. 2001, 39, 421–431. [Google Scholar] [CrossRef]
- Cheney, B.V. Magnetic deshielding of protons due to intramolecular steric interactions with proximate hydrogens. J. Am. Chem. Soc. 1968, 90, 5386–5390. [Google Scholar] [CrossRef]
- Marshall, T.W.; Pople, J.A. Nuclear magnetic shielding of a hydrogen atom in an electric field. Mol. Phys. 1958, 1, 199–202. [Google Scholar] [CrossRef]
- Cobb, T.B.; Memory, J.B. High-resolution NMR spectra of polycyclic hydrocarbons. II. Pentacyclic compounds. J. Phys. Chem. 1967, 47, 2020–2025. [Google Scholar] [CrossRef]
- Grimme, S.; Diedrich, C.; Korth, M. The importance of inter- and intramolecular van der Waals interactions in organic reactions: The dimerization of anthracene revisited. Angew. Chem. Int. Ed. 2006, 118, 641–645. [Google Scholar] [CrossRef]
- Hujo, W.; Grimme, S. Performance of the van der Waals density functional VV10 and (hybrid) GGA variants for thermochemistry and noncovalent interactions. J. Chem. Theory Comput. 2011, 7, 3866–3871. [Google Scholar] [CrossRef] [PubMed]
- Kupka, T.; Stachow, M.; Nieradka, M.; Kaminsky, J.; Pluta, T. Convergence of nuclear magnetic shieldings in the Kohn-Sham limit of several small molecules. J. Chem. Theory Comput. 2010, 6, 1580–1589. [Google Scholar] [CrossRef]
- Abraham, R.J.; Mobli, M. Modelling 1H NMR Spectra of Organic Compounds Theory, Applications and NMR Prediction Software; John Wiley & Sons Ltd.: Chichester, UK, 2008. [Google Scholar]
- Kaneko, F.; Yano, J.; Sato, K. Diversity in the fatty-acid conformation and chain packing of cis-unsaturated lipids. Curr. Opin. Struct. Biol. 1998, 8, 417–425. [Google Scholar] [CrossRef]
- Suzuki, M.; Ogaki, T.; Sato, K. Crystallization and transformation mechanisms of α, β and γ polymorphs of ultra-pure oleic acid. J. Am. Oil. Chem. Soc. 1985, 62, 1600–1604. [Google Scholar] [CrossRef]
- Kaneko, F.; Yamazaki, K.; Kitagawa, K.; Kikyo, T.; Kobayashi, M.; Sato, K.; Suzuki, M. Structure and crystallization behavior of the β phase of oleic acid. J. Phys. Chem. B 1997, 101, 1803–1809. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 0.9, Revision. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C | D | E | F | G | H | I | J | K | L | M | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Group | δexp (ppm) | δexp (ppm) | δexp (ppm) | δexp (ppm) | Group | δexp (ppm) | δexp (ppm) | Group | δexp (ppm) | δexp (ppm) | δexp (ppm) | δexp (ppm) | Group | δexp (ppm) | δexp (ppm) | δexp (ppm) |
| H1a | 5.15 | 5.1 | 5.09 | 5.05 | H1 | 1.8 | 1.84 | H9 | 2.09 | 2.12 | 2.18 | 2.17 | H11 | 6.10 | 6.19 | 6.48 |
| H1b | 5.24 | 5.23 | 5.17 | 5.18 | H2 | 5.7 | 5.69 | H10 | 5.70 | 5.74 | 5.48 | 5.40 | H12 | 6.10 | 6.40 | 6.48 |
| H2 | 6.8 | 6.36 | 6.74 | 6.3 | H3 | 5.83 | 6.12 | H11 | 6.10 | 6.50 | 6.43 | 5.98 | H10 | 6.04 | 6.01 | 6.08 |
| H3 | 6.0 | 6.22 | 5.93 | 6.16 | H4 | 6.5 | 6.10 | H12 | 6.18 | 5.98 | 6.13 | 6.37 | H13 | 6.04 | 6.12 | 6.08 |
| H4 | 6.0 | 6.22 | 5.93 | 6.16 | H5 | 6.5 | 6.10 | H13 | 6.41 | 6.16 | 5.96 | 6.16 | H9 | 5.66 | 5.4 | 5.46 |
| H5 | 6.8 | 6.36 | 6.74 | 6.3 | H6 | 5.83 | 6.12 | H14 | 6.02 | 6.46 | 6.52 | 6.07 | H14 | 5.66 | 5.74 | 5.46 |
| H6a | 5.15 | 5.1 | 5.09 | 5.05 | H7 | 5.7 | 5.69 | H15 | 5.47 | 5.56 | 5.75 | 5.70 | H2 | 2.37 | 2.40 | 2.37 |
| H6b | 5.24 | 5.23 | 5.17 | 5.18 | H8 | 1.8 | 1.84 | H16 | 1.76 | 1.77 | 1.80 | 1.77 | H8 | 2.10 | 2.20 | 2.22 |
| H15 | 2.10 | 2.20 | 2.22 | |||||||||||||
| H3 | 1.65 | 1.64 | 1.65 | |||||||||||||
| H4 | 1.39 | 1.41 | 1.39 | |||||||||||||
| H5 | 1.39 | 1.41 | 1.39 | |||||||||||||
| H6 | 1.39 | 1.41 | 1.39 | |||||||||||||
| H7 | 1.39 | 1.41 | 1.39 | |||||||||||||
| H16 | 1.33 | 1.33 | 1.32 | |||||||||||||
| H17 | 1.33 | 1.33 | 1.32 | |||||||||||||
| H18 | 0.91 | 0.97 | 0.94 | |||||||||||||
| Compound | Conformer | B3LYP/6–31+G(d) | APFD/6–31+G(d) | ||||
|---|---|---|---|---|---|---|---|
| φc7c8c9c10 (°) | φc13c14c15c16 (°) | ΔG (kcal/mol) (% Population) | φc7c8c9c10 (°) | φc13c14c15c16 (°) | ΔG (kcal/mol) (% Population) | ||
| 9E,11E,13E Isomer (β-Eleostearic acid) | A | 120.26 (S) | 118.59 (S) | +0.36 (33.41) | 119.21 (S) | 117.99 (S) | +0.16 (38.68) |
| B | 120.11 (S) | −119.97 (S’) | 0.00 (61.35) | 118.73 (S) | −118.59 (S’) | 0.00 (50.67) | |
| C | −0.78 (Syn) | −0.03 (Syn) | +3.00 (0.39) | −0.59 (Syn) | −0.11 (Syn) | +2.13 (1.39) | |
| D | −117.53 (S’) | −0.83 (Syn) | +1.89 (2.53) | −116.94 (S’) | −0.62 (Syn) | +1.38 (4.94) | |
| E | −0.75 (Syn) | −118.55 (S’) | +1.94 (2.32) | −0.58 (Syn) | −117.27 (S’) | +1.46 (4.31) | |
| 9Z,11E,13E Isomer (β-Eleostearic acid) | A | 116.67 (S) | 119.18 (S) | +0.03 (25.07) | 111.25 (S) | 117.87 (S) | 0.00 (36.53) |
| B | −118.92 (S’) | −119.37 (S’) | +0.15 (20.48) | −114.31 (S’) | −117.53 (S’) | +0.48 (16.25) | |
| C | 116.88 (S) | −119.16 (S’) | +0.02 (25.50) | 112.96 (S) | −117.54 (S’) | +0.18 (26.96) | |
| D | −118.88 (S’) | 119.28 (S) | 0.00 (26.38) | −113.83 (S’) | 118.32 (S) | +0.47 (16.52) | |
| E | −119.43 (S’) | −0.76 (Syn) | +1.38 (2.57) | −113.80 (S’) | −0.22 (Syn) | +1.35 (3.74) | |
| 9Z,11E,13Z Isomer (Punicic acid) | A | 117.46 (S) | 119.33 (S) | 0.00 (38.54) | 113.45 (S) | 114.70 (S) | 0.00 (44.84) |
| B | 117.76 (S) | −118.63 (S’) | +0.08 (33.67) | 112.18 (S) | −112.33 (S’) | +0.31 (26.57) | |
| C | −119.22 (S’) | 119.55 (S) | +0.27 (24.43) | −113.43 (S’) | 113.54 (S) | +0.27 (28.43) | |
| D | 1.52 | −121.09 (S’) | +3.83 (1.51) | 5.34 | −110.17 (S’) | +3.77 (0.08) | |
| E | 117.81 (S) | 2.02 (Syn) | +3.63 (1.85) | 111.01 (S) | 4.55 (Syn) | +3.78 (0.08) | |
| 10E,12E,14Z-Hexadecatrienyl acetate | A | 120.04 (S) | +0.28 (25.59) | 118.73 (S) | +0.08 (28.88) | ||
| B | −119.39 (S’) | 0.00 (41.05) | −118.73 (S’) | +0.03 (31.42) | |||
| C | 120.03 (S) | +0.20 (29.29) | 118.73 (S) | 0.00 (33.05) | |||
| D | 1.56 (Syn) | +1.37 (4.07) | 1.16 (Syn) | +0.95 (6.65) | |||
| 10E,12Z,14Z-Hexadecatrienyl acetate | A | 119.67 (S) | +0.44 (22.44) | 118.45 (S) | +0.20 (26.06) | ||
| B | −119.64 (S’) | 0.00 (47.15) | −118.86 (S’) | 0.00 (36.52) | |||
| C | 119.56 (S) | +0.35 (26.12) | 118.28 (S) | +0.12 (29.82) | |||
| D | 1.22 (Syn) | +1.42 (4.29) | 0.87 (Syn) | +0.93 (7.60) | |||
| 10Z,12Z,14E-Hexadecatrienyl acetate | A | 119.01 (S) | 0.00 (30.52) | 112.30 (S) | +0.04 (20.48) | ||
| B | −119.51 (S’) | +0.51 (12.91) | −113.47 (S’) | +0.10 (18.51) | |||
| C | 118.80 (S) | 0.00 (30.52) | 112.24 (S) | 0.00 (21.92) | |||
| D | −119.50 (S’) | +0.48 (13.57) | −113.16 (S’) | +0.02 (21.19) | |||
| 10Z,12E,14E-Hexadecatrienyl acetate | A | 119.05 (S) | 0.00 (31.98) | 113.03 (S) | 0.00 (33.79) | ||
| B | −121.15 (S’) | +0.54 (12.85) | −116.62 (S’) | +0.63 (11.67) | |||
| C | 118.73 (S) | +0.04 (29.89) | 113.00 (S) | +0.04 (31.59) | |||
| D | −121.06 (S’) | +0.56 (12.43) | −116.57 (S’) | +0.65 (11.28) | |||
| Minimization Method | Correlation Coefficient (R2) | Mean Square Error | Intercept | Slope |
|---|---|---|---|---|
| B3LYP/6–31+G(d) a | 0.873 | 0.037 | −0.002 | 1.062 |
| APFD/6–31+G(d) a | 0.868 (0.825) | 0.041(0.044) | −0.102 (0.518) | 1.085 (0.956) |
| B3LYP/6–31+G(d) b | 0.984 | 0.005 | −0.387 | 1.127 |
| APFD/6–31+G(d) b | 0.982 (0.942) | 0.005 (0.015) | −0.506 (0.138) | 1.153 (1.020) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Venianakis, T.; Oikonomaki, C.; Siskos, M.G.; Primikyri, A.; Gerothanassis, I.P. DFT Calculations of 1H NMR Chemical Shifts of Geometric Isomers of Conjugated Linolenic Acids, Hexadecatrienyl Pheromones, and Model Triene-Containing Compounds: Structures in Solution and Revision of NMR Assignments. Molecules 2021, 26, 3477. https://doi.org/10.3390/molecules26113477
Venianakis T, Oikonomaki C, Siskos MG, Primikyri A, Gerothanassis IP. DFT Calculations of 1H NMR Chemical Shifts of Geometric Isomers of Conjugated Linolenic Acids, Hexadecatrienyl Pheromones, and Model Triene-Containing Compounds: Structures in Solution and Revision of NMR Assignments. Molecules. 2021; 26(11):3477. https://doi.org/10.3390/molecules26113477
Chicago/Turabian StyleVenianakis, Themistoklis, Christina Oikonomaki, Michael G. Siskos, Alexandra Primikyri, and Ioannis P. Gerothanassis. 2021. "DFT Calculations of 1H NMR Chemical Shifts of Geometric Isomers of Conjugated Linolenic Acids, Hexadecatrienyl Pheromones, and Model Triene-Containing Compounds: Structures in Solution and Revision of NMR Assignments" Molecules 26, no. 11: 3477. https://doi.org/10.3390/molecules26113477
APA StyleVenianakis, T., Oikonomaki, C., Siskos, M. G., Primikyri, A., & Gerothanassis, I. P. (2021). DFT Calculations of 1H NMR Chemical Shifts of Geometric Isomers of Conjugated Linolenic Acids, Hexadecatrienyl Pheromones, and Model Triene-Containing Compounds: Structures in Solution and Revision of NMR Assignments. Molecules, 26(11), 3477. https://doi.org/10.3390/molecules26113477