
Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Enzymatic Synthesis of Xylose Oleate
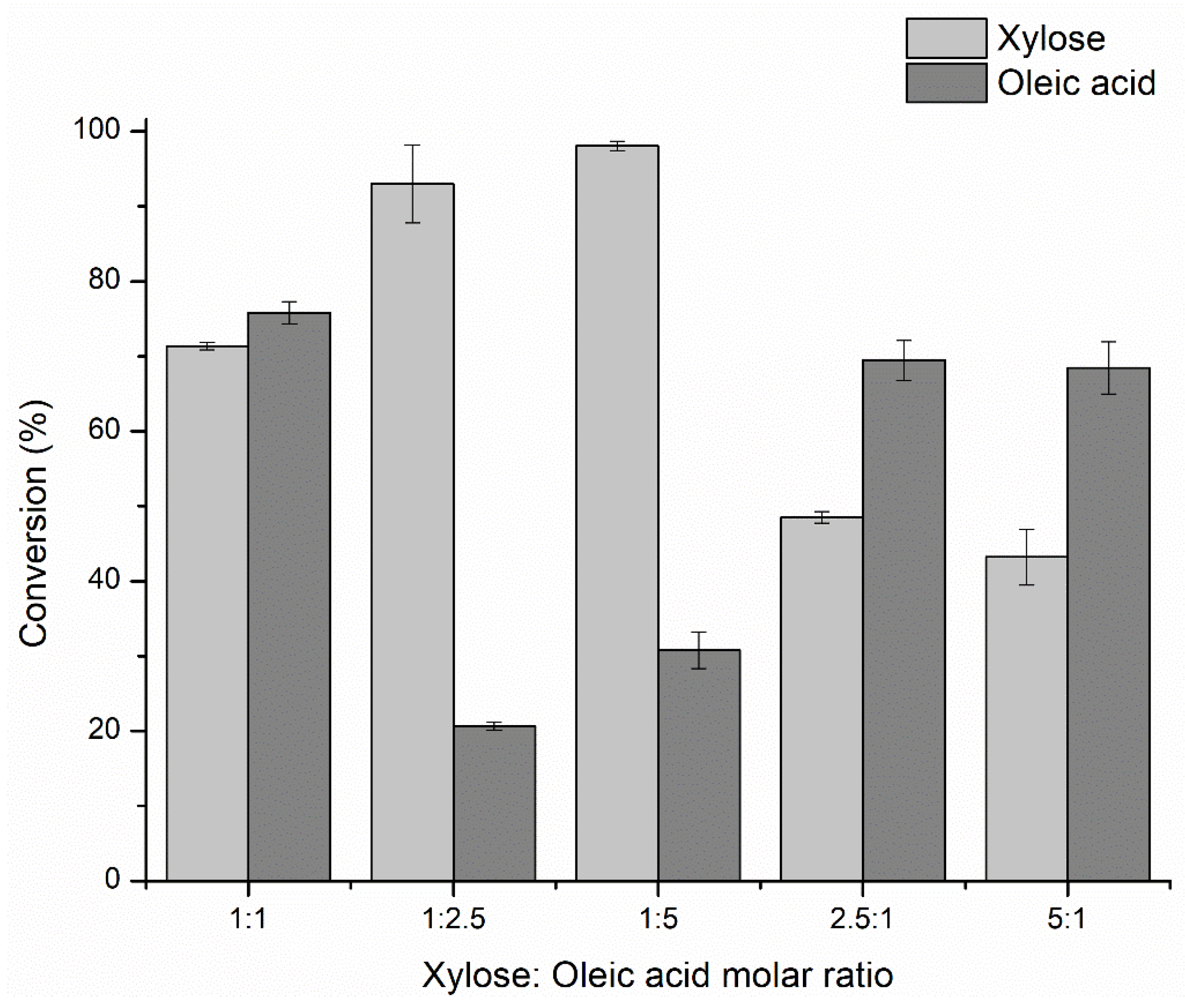
2.1.1. Effect of the Xylose: Oleic Acid Molar Ratio on the Process Performance
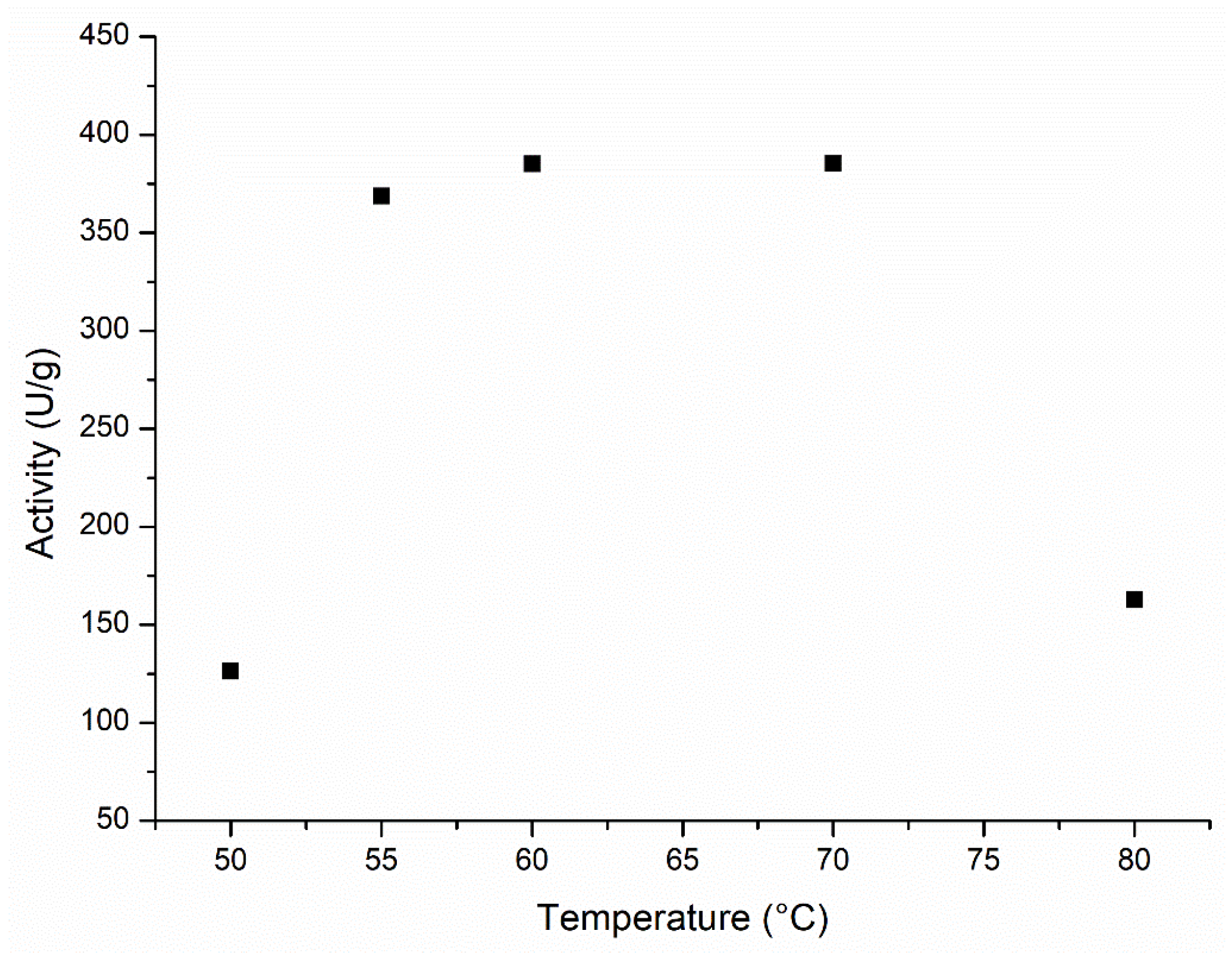
2.1.2. Effect of the Temperature on the Lipozyme 435 Activity Profile
2.1.3. Esterification Rates for Lipozyme 435
2.1.4. Conversion Profiles in the Synthesis of Xylose Oleate
2.1.5. Lipozyme 435 Operational Stability
2.2. Purification and Characterization of Xylose Oleate
2.3. Emulsion Capacity Assays
3. Materials and Methods
3.1. Materials
3.2. Enzymatic Synthesis of Xylose Oleate
3.2.1. Xylose Solubility in MEK
3.2.2. Determination of the Effect of Xylose: Oleic Acid Molar Ratio on the Reaction Performance
3.2.3. Effect of the Temperature on the Lipozyme 435 Activity Profile
3.2.4. Determination of Esterification Rates for Lipozyme 435
Determination of Initial Rates
Determination of Long-Term Rates
3.2.5. Determination of the Lipozyme 435 Operational Stability
3.3. Purification and Characterization of Xylose Oleate
3.4. Emulsion Capacity Assays
3.5. Standard Enzymatic Activities
3.5.1. Hydrolysis Activity of Lipozyme 435
3.5.2. Esterification Activity of Lipozyme 435
3.6. Chromatographic Analyses
3.6.1. Xylose Determination
3.6.2. Oleic Acid Determination
3.6.3. Furfural Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bidjou-Haiour, C.; Klai, N. Lipase catalyzed synthesis of fatty acid xylose esters and their surfactant properties. Asian J. Chem. 2013, 25, 4347–4350. [Google Scholar] [CrossRef]
- Chang, P.; Zhang, Z.; Tang, S. Lipase-catalyzed synthesis of sugar ester in mixed biphasic system of ionic liquids and supercritical carbon dioxide. J. Chin. Chem. Soc. 2018, 65, 452–458. [Google Scholar] [CrossRef]
- Ogawa, S.; Endo, A.; Kitahara, N.; Yamagishi, T.; Aoyagi, S.; Hara, S. Factors determining the reaction temperature of the solvent-free enzymatic synthesis of trehalose esters. Carbohydr. Res. 2019, 482, 107739. [Google Scholar] [CrossRef]
- Shin, D.W.; Mai, N.L.; Bae, S.W.; Koo, Y.M. Enhanced lipase-catalyzed synthesis of sugar fatty acid esters using supersaturated sugar solution in ionic liquids. Enzym. Microb. Technol. 2019, 126, 18–23. [Google Scholar] [CrossRef]
- Abdulmalek, E.; Hamidon, N.F.; Abdul Rahman, M.B. Optimization and characterization of lipase catalysed synthesis of xylose caproate ester in organic solvents. J. Mol. Catal. B Enzym. 2016, 132, 1–4. [Google Scholar] [CrossRef]
- Méline, T.; Muzard, M.; Deleu, M.; Rakotoarivonina, H.; Plantier-Royon, R.; Rémond, C. D-Xylose and L-arabinose laurate esters: Enzymatic synthesis, characterization and physico-chemical properties. Enzym. Microb. Technol. 2018, 112, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Shaw, J.F. Biocatalysis for the production of carbohydrate esters. New Biotechnol. 2009, 26, 109–116. [Google Scholar] [CrossRef]
- Neta, N.S.; Teixeira, J.A.; Rodrigues, L.R. Sugar ester surfactants: Enzymatic synthesis and applications in food industry. Crit. Rev. Food Sci. Nutr. 2015, 55, 595–610. [Google Scholar] [CrossRef]
- Shi, Y.G.; Li, J.R.; Chu, Y.H. Enzyme-catalyzed regioselective synthesis of sucrose-based esters. J. Chem. Technol. Biotechnol. 2011, 86, 1457–1468. [Google Scholar] [CrossRef]
- Siebenhaller, S.; Hajek, T.; Muhle-Goll, C.; Himmelsbach, M.; Luy, B.; Kirschhöfer, F.; Brenner-Weiß, G.; Hahn, T.; Zibek, S.; Syldatk, C. Beechwood carbohydrates for enzymatic synthesis of sustainable glycolipids. Bioresour. Bioprocess. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Siebenhaller, S.; Kirchhoff, J.; Kirschhöfer, F.; Brenner-Weiß, G.; Muhle-Goll, C.; Luy, B.; Haitz, F.; Hahn, T.; Zibek, S.; Syldatk, C.; et al. Integrated process for the enzymatic production of fatty acid sugar esters completely based on lignocellulosic substrates. Front. Chem. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, V.; dos Santos, J.B.C.; Tardioli, P.W. Porcine pancreatic lipase hydrophobically adsorbed on octyl-silica: A robust biocatalyst for syntheses of xylose fatty acid esters. Biocatal. Biotransform. 2017, 35, 298–305. [Google Scholar] [CrossRef]
- De Lima, L.N.; Mendes, A.A.; Fernandez-Lafuente, R.; Tardioli, P.W.; Camargo Giordano, R.D.L. Performance of different immobilized lipases in the syntheses of short- and long-chain carboxylic acid esters by esterification reactions in organic media. Molecules 2018, 23, 766. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.Y.; Chen, Y.; Banwell, M.G.; Wang, Y.; Lan, P. Enzymatic preparation of a homologous series of long-chain 6- O -acylglucose esters and their evaluation as emulsifiers. J. Agric. Food Chem. 2018, 66, 3949–3956. [Google Scholar] [CrossRef]
- Bouzaouit, N.; Bidjou-Haiour, C. Optimization of lipase catalyzed synthesis of fatty acid xylose ester using statistical experimental designs. Pharma Chem. 2015, 7, 261–269. [Google Scholar]
- Gumel, A.M.; Annuar, M.S.M.; Heidelberg, T.; Chisti, Y. Lipase mediated synthesis of sugar fatty acid esters. Process Biochem. 2011, 46, 2079–2090. [Google Scholar] [CrossRef]
- Kapoor, M.; Gupta, M.N. Lipase promiscuity and its biochemical applications. Process Biochem. 2012, 47, 555–569. [Google Scholar] [CrossRef]
- Teng, Y.; Stewart, S.G.; Hai, Y.W.; Li, X.; Banwell, M.G.; Lan, P. Sucrose fatty acid esters: Synthesis, emulsifying capacities, biological activities and structure-property profiles. Crit. Rev. Food Sci. Nutr. 2020. [Google Scholar] [CrossRef]
- Kundys, A.; Białecka-Florjańczyk, E.; Fabiszewska, A.; Małajowicz, J. Candida antarctica lipase B as catalyst for cyclic esters synthesis, their polymerization and degradation of aliphatic polyesters. J. Polym. Environ. 2018, 26, 396–407. [Google Scholar] [CrossRef]
- Arcens, D.; Grau, E.; Grelier, S.; Cramail, H.; Peruch, F. 6-O-glucose palmitate synthesis with lipase: Investigation of some key parameters. Mol. Catal. 2018, 460, 63–68. [Google Scholar] [CrossRef]
- Ma, Y.R.; Banwell, M.G.; Yan, R.; Lan, P. Comparative study of the emulsifying properties of a homologous series of long-chain 6′-O-acylmaltose esters. J. Agric. Food Chem. 2018, 66, 8832–8840. [Google Scholar] [CrossRef]
- Gonçalves, M.C.P.; Kieckbusch, T.G.; Perna, R.F.; Fujimoto, J.T.; Morales, S.A.V.; Romanelli, J. Trends on enzyme immobilization researches based on bibliometric analysis. Process Biochem. 2019, 76, 95–110. [Google Scholar] [CrossRef]
- Gonçalves, M.C.P.; Romanelli, J.P.; Guimarães, J.R.; Vieira, A.C.; de Azevedo, B.P.; Tardioli, P.W. Reviewing research on the synthesis of CALB-catalyzed sugar esters incorporating systematic mapping principles. Crit. Rev. Biotechnol. 2021, 1–23. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Virgen-Ortíz, J.J.; dos Santos, J.C.S.; Berenguer-Murcia, Á.; Alcantara, A.R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Flores, L.F.; Beltran, H.I.; Arrieta-Baez, D.; Reyes-Duarte, D. Regioselective synthesis of lactulose esters by Candida antarctica and Thermomyces lanuginosus lipases. Catalysts 2017, 7, 263. [Google Scholar] [CrossRef]
- De Lima, L.N.; Vieira, G.N.A.; Kopp, W.; Tardioli, P.W.; Giordano, R.L.C. Mono- and heterofunctionalized silica magnetic microparticles (SMMPs) as new carriers for immobilization of lipases. J. Mol. Catal. B Enzym. 2016, 133, S491–S499. [Google Scholar] [CrossRef]
- Neta, N.D.A.S.; dos Santos, J.C.S.; de Oliveira Sancho, S.; Rodrigues, S.; Gonçalves, L.R.B.; Rodrigues, L.R.; Teixeira, J.A. Enzymatic synthesis of sugar esters and their potential as surface-active stabilizers of coconut milk emulsions. Food Hydrocoll. 2012, 27, 324–331. [Google Scholar] [CrossRef]
- Vescovi, V.; Kopp, W.; Guisán, J.M.; Giordano, R.L.C.; Mendes, A.A.; Tardioli, P.W. Improved catalytic properties of Candida antarctica lipase B multi-attached on tailor-made hydrophobic silica containing octyl and multifunctional amino- glutaraldehyde spacer arms. Process Biochem. 2016, 51, 2055–2066. [Google Scholar] [CrossRef]
- Rahman, M.; Arumugan, M.; Khairuddin, N.S.K.; Abdulmalek, E.; Basri, M.; Salleh, A. Microwave assisted enzymatic S-synthesis of fatty acid sugar ester in ionic liquid-tert-butanol biphasic solvent system. Asian J. Chem. 2012, 24, 5058–5062. [Google Scholar]
- Sutili, F.K.; Nogueira, D.D.O.; Leite, S.G.F.; Miranda, L.S.M.; De Souza, R.O.M.A. Lipase immobilized in microemulsion based organogels (MBGs) as an efficient catalyst for continuous-flow esterification of protected fructose. RSC Adv. 2015, 5, 37287–37291. [Google Scholar] [CrossRef]
- Siódmiak, T.; Mangelings, D.; Vander Heyden, Y.; Ziegler-Borowska, M.; Marszałł, M.P. High enantioselective Novozym 435-catalyzed esterification of (R,S)-flurbiprofen monitored with a chiral stationary phase. Appl. Biochem. Biotechnol. 2015, 175, 2769–2785. [Google Scholar] [CrossRef]
- Findrik, Z.; Megyeri, G.; Gubicza, L.; Bélafi-Bakó, K.; Nemestóthy, N.; Sudar, M. Lipase catalyzed synthesis of glucose palmitate in ionic liquid. J. Clean. Prod. 2016, 112, 1106–1111. [Google Scholar] [CrossRef]
- Mai, N.L.; Ahn, K.; Bae, S.W.; Shin, D.W.; Morya, V.K.; Koo, Y.M. Ionic liquids as novel solvents for the synthesis of sugar fatty acid ester. Biotechnol. J. 2014, 9, 1565–1572. [Google Scholar] [CrossRef]
- Neta, N.S.; Peres, A.M.; Teixeira, J.A.; Rodrigues, L.R. Maximization of fructose esters synthesis by response surface methodology. New Biotechnol. 2011, 28, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, V.M.; Boeriu, C.G.; Zaccheria, F.; Ravasio, N. Synthesis and characterization of arabinose-palmitic acid esters by enzymatic esterification. Mol. Catal. 2017, 433, 383–390. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, A.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef]
- De Castro, H.F.; Mendes, A.A.; dos Santos, J.C.; de Aguiar, C.L. Modificação de óleos e gorduras por biotransformação. Quim. Nova 2004, 27, 146–156. [Google Scholar] [CrossRef]
- Kumari, A.; Mahapatra, P.; Garlapati, V.K.; Banerjee, R. Enzymatic transesterification of Jatropha oil. Biotechnol. Biofuels 2009, 2, 1. [Google Scholar] [CrossRef]
- Fallavena, L.P.; Antunes, F.H.F.; Alves, J.S.; Paludo, N.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Ultrasound technology and molecular sieves improve the thermodynamically controlled esterification of butyric acid mediated by immobilized lipase from Rhizomucor miehei. RSC Adv. 2014, 4, 8675–8681. [Google Scholar] [CrossRef]
- Li, L.; Ji, F.; Wang, J.; Li, Y.; Bao, Y. Esterification degree of fructose laurate exerted by Candida antarctica lipase B in organic solvents. Enzym. Microb. Technol. 2015, 69, 46–53. [Google Scholar] [CrossRef]
- Kasche, V. Mechanism and yields in enzyme catalysed equilibrium and kinetically controlled synthesis of β-lactam antibiotics, peptides and other condensation products. Enzym. Microb. Technol. 1986, 8, 4–16. [Google Scholar] [CrossRef]
- Halling, P.J. Thermodynamic predictions for biocatalysis in nonconventional media: Theory, tests, and recommendations for experimental design and analysis. Enzym. Microb. Technol. 1994, 16, 178–206. [Google Scholar] [CrossRef]
- Castillo, E.; Dossat, V.; Marty, A.; Stéphane Condoret, J.; Combes, D. The role of silica gel in lipase-catalyzed esterification reactions of high-polar substrates. J. Am. Oil Chem. Soc. 1997, 74, 77–85. [Google Scholar] [CrossRef]
- Colombié, S.; Tweddell, R.J.; Condoret, J.S.; Marty, A. Water activity control: A way to improve the efficiency of continuous lipase esterification. Biotechnol. Bioeng. 1998, 60, 362–368. [Google Scholar] [CrossRef]
- Dossat, V.; Combes, D.; Marty, A. Continuous enzymatic transesterification of high oleic sunflower oil in a packed bed reactor: Influence of the glycerol production. Enzym. Microb. Technol. 1999, 25, 194–200. [Google Scholar] [CrossRef]
- Marty, A.; Dossat, V.; Condoret, J.S. Continuous operation of lipase-catalyzed reactions in nonaqueous solvents: Influence of the production of hydrophilic compounds. Biotechnol. Bioeng. 1997, 56, 232–237. [Google Scholar] [CrossRef]
- Séverac, E.; Galy, O.; Turon, F.; Pantel, C.A.; Condoret, J.S.; Monsan, P.; Marty, A. Selection of CalB immobilization method to be used in continuous oil transesterification: Analysis of the economical impact. Enzym. Microb. Technol. 2011, 48, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.B.; Schein, M.F.; Friedrich, J.L.R.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Ultrasound-assisted butyl acetate synthesis catalyzed by Novozym 435: Enhanced activity and operational stability. Ultrason. Sonochem. 2013, 20, 1155–1160. [Google Scholar] [CrossRef]
- Paludo, N.; Alves, J.S.; Altmann, C.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. The combined use of ultrasound and molecular sieves improves the synthesis of ethyl butyrate catalyzed by immobilized Thermomyces lanuginosus lipase. Ultrason. Sonochem. 2015, 22, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.B.; Friedrich, J.L.R.; Cavalheiro, J.C.; Garcia-Galan, C.; Barbosa, O.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Improved production of butyl butyrate with lipase from Thermomyces lanuginosus immobilized on styrene-divinylbenzene beads. Bioresour. Technol. 2013, 134, 417–422. [Google Scholar] [CrossRef]
- Graebin, G.; Martins, B.; Garcia-galan, C.; Fernandez-lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Immobilization of lipase B from Candida antarctica on porous styrene—divinylbenzene beads improves butyl acetate synthesis. Biotechnol. Prog. 2012, 28, 406–412. [Google Scholar] [CrossRef]
- Poppe, J.K.; Garcia-Galan, C.; Matte, C.R.; Fernandez-Lafuente, R.; Rodrigues, R.C.; Ayub, M.A.Z. Optimization of synthesis of fatty acid methyl esters catalyzed by lipase B from Candida antarctica immobilized on hydrophobic supports. J. Mol. Catal. B Enzym. 2013, 94, 51–56. [Google Scholar] [CrossRef]
- Alves, J.S.; Garcia-Galan, C.; Schein, M.F.; Silva, A.M.; Barbosa, O.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Combined effects of ultrasound and immobilization protocol on butyl acetate synthesis catalyzed by CALB. Molecules 2014, 19, 9562–9576. [Google Scholar] [CrossRef]
- Tarahomjoo, S.; Alemzadeh, I. Surfactant production by an enzymatic method. Enzym. Microb. Technol. 2003, 33, 33–37. [Google Scholar] [CrossRef]
- Tsukamoto, J.; Haebel, S.; Valen, G.P.; Peter, M.G.; Franco, T.T. Enzymatic direct synthesis of acrylic acid esters of mono- and disaccharides. J. Chem. Technol. Biotechnol. 2008, 83, 1486–1492. [Google Scholar] [CrossRef]
- Hernandez, K.; Garcia-Verdugo, E.; Porcar, R.; Fernandez-Lafuente, R. Hydrolysis of triacetin catalyzed by immobilized lipases: Effect of the immobilization protocol and experimental conditions on diacetin yield. Enzym. Microb. Technol. 2011, 48, 510–517. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Palomo, J.M.; Cocca, J.; Mateo, C.; Moro, P.; Terreni, M.; Fernandez-Lafuente, R.; Guisan, J.M. Regio-selective deprotection of peracetylated sugars via lipase hydrolysis. Tetrahedron 2003, 59, 5705–5711. [Google Scholar] [CrossRef]
- Filice, M.; Bavaro, T.; Fernandez-Lafuente, R.; Pregnolato, M.; Guisan, J.M.; Palomo, J.M.; Terreni, M. Chemo-biocatalytic regioselective one-pot synthesis of different deprotected monosaccharides. Catal. Today 2009, 140, 11–18. [Google Scholar] [CrossRef]
- Jönsson, L.J.; Martín, C. Bioresource technology pretreatment of lignocellulose: Formation of inhibitory by-products and strategies for minimizing their effects. Bioresour. Technol. 2016, 199, 103–112. [Google Scholar] [CrossRef]
- Vulfson, E.N.; Halling, P.J.; Holland, H.L.; Rhee, J.S.; Kwon, S.J.; Han, J.J. Water activity control for lipase-catalyzed reactions in nonaqueous media. Enzym. Nonaqueous Solvents 2003, 15, 135–150. [Google Scholar] [CrossRef]
- Gandhi, N.N.; Patil, N.S.; Sawant, S.B.; Joshi, J.B.; Wangikar, P.P.; Mukesh, E.D. Lipase-catalyzed esterification. Catal. Rev. Sci. Eng. 2000, 42, 439–480. [Google Scholar] [CrossRef]
- Mensah, P.; Carta, G. Adsorptive control of water in esterification with immobilized enzymes. Continuous operation in a periodic counter-current reactor. Biotechnol. Bioeng. 1999, 66, 137–146. [Google Scholar] [CrossRef]
- Schmidell, W.; Lima, U.A.; Aquarone, E.; Borzani, W. Biotecnologia Industrial, 1st ed.; Edgard Blucher Ltda.: São Paulo, Brazil, 2001. [Google Scholar]
- Ljunger, G.; Adlercreutz, P.; Mattiasson, B. Enzymatic synthesis of octyl-β-glucoside in octanol at controlled water activity. Enzym. Microb. Technol. 1994, 16, 751–755. [Google Scholar] [CrossRef]
- Lortie, R. Enzyme catalyzed esterification. Biotechnol. Adv. 1997, 15, 1–15. [Google Scholar] [CrossRef]
- Oguntimein, G.B.; Erdmann, H.; Schmid, R.D. Lipase catalysed synthesis of sugar ester media. Biotechnol. Lett. 1993, 15, 175–180. [Google Scholar] [CrossRef]
- Schlotterbeck, A.; Lang, S.; Wray, V.; Wagner, F. Lipase-catalyzed monoacylation of fructose. Biotechnol. Lett. 1993, 15, 61–64. [Google Scholar] [CrossRef]
- Zaidan, U.H.; Abdul Rahman, M.B.; Othman, S.S.; Basri, M.; Abdulmalek, E.; Abdul Rahman, R.N.Z.R.; Salleh, A.B. Biocatalytic production of lactose ester catalysed by mica-based immobilised lipase. Food Chem. 2012, 131, 199–205. [Google Scholar] [CrossRef]
- Li, L.; Ji, F.; Wang, J.; Jiang, B.; Li, Y.; Bao, Y. Efficient mono-acylation of fructose by lipase-catalyzed esterification in ionic liquid co-solvents. Carbohydr. Res. 2015, 416, 51–58. [Google Scholar] [CrossRef]
- Dang, H.T.; Obiri, O.; Hayes, D.G. Feed batch addition of saccharide during saccharide-fatty acid esterification catalyzed by immobilized lipase: Time course, water activity, and kinetic model. J. Am. Oil Chem. Soc. 2005, 82, 487–493. [Google Scholar] [CrossRef]
- Ward, O.P.; Fang, J.; Li, Z. Lipase-catalyzed synthesis of a sugar ester containing arachidonic acid. Enzym. Microb. Technol. 1997, 20, 52–56. [Google Scholar] [CrossRef]
- An, D.; Zhang, X.; Liang, F.; Xian, M.; Feng, D.; Ye, Z. Synthesis, surface properties of glucosyl esters from renewable materials for use as biosurfactants. Colloids Surf. A 2019, 577, 257–264. [Google Scholar] [CrossRef]
- Rueda, N.; Dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved performance of lipases immobilized on heterofunctional octyl-glyoxyl agarose beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Manoel, E.A.; dos Santos, J.C.S.; Freire, D.M.G.; Rueda, N.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports involves the open form of the enzyme. Enzym. Microb. Technol. 2015, 71, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Hirata, D.B.; Torrestiana-Sanchez, B.; Rosales-Quintero, A.; Fernandez-Lafuente, R. Relevance of substrates and products on the desorption of lipases physically adsorbed on hydrophobic supports. Enzym. Microb. Technol. 2017, 96, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.B.; Graebin, N.G.; Lorenzoni, A.S.G.; Fernandez-Lafuente, R.; Ayub, M.A.Z.; Rodrigues, R.C. Rapid and high yields of synthesis of butyl acetate catalyzed by Novozym 435: Reaction optimization by response surface methodology. Process Biochem. 2011, 46, 2311–2316. [Google Scholar] [CrossRef]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar] [CrossRef]
- Liu, W.L.; Lo, S.H.; Singco, B.; Yang, C.C.; Huang, H.Y.; Lin, C.H. Novel trypsin–FITC@MOF bioreactor efficiently catalyzes protein digestion. J. Mater. Chem. B 2013, 1, 928–932. [Google Scholar] [CrossRef]
- Idris, A.; Bukhari, A. Immobilized Candida antarctica lipase B: Hydration, stripping off and application in ring opening polyester synthesis. Biotechnol. Adv. 2012, 30, 550–563. [Google Scholar] [CrossRef]
- Bilal, M.; Zhao, Y.; Noreen, S.; Shah, S.Z.H.; Bharagava, R.N.; Iqbal, H.M.N. Modifying bio-catalytic properties of enzymes for efficient biocatalysis: A review from immobilization strategies viewpoint. Biocatal. Biotransform. 2019, 37, 159–182. [Google Scholar] [CrossRef]
- Barbosa, O.; Ruiz, M.; Ortiz, C.; Fernández, M.; Torres, R.; Fernandez-Lafuente, R. Modulation of the properties of immobilized CALB by chemical modification with 2,3,4-trinitrobenzenesulfonate or ethylendiamine. Advantages of using adsorbed lipases on hydrophobic supports. Process Biochem. 2012, 47, 867–876. [Google Scholar] [CrossRef]
- Rueda, N.; dos Santos, J.C.S.; Ortiz, C.; Torres, R.; Barbosa, O.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R. Chemical modification in the design of immobilized enzyme biocatalysts: Drawbacks and opportunities. Chem. Rec. 2016, 16, 1436–1455. [Google Scholar] [CrossRef]
- Wagner, F.W.; Walton, M.A.D.; Lincoln, R.S.M.; Lincoln, V.H.S. Separation and Purification of Sugar Esters. U.S. Patent No. 4,983,731, 8 January 1991. [Google Scholar]
- Ai, M.; Xiao, N.; Jiang, A. Molecular structural modification of duck egg white protein conjugates with monosaccharides for improving emulsifying capacity. Food Hydrocoll. 2021, 111, 106271. [Google Scholar] [CrossRef]
- El-Laithy, H.M.; Shoukry, O.; Mahran, L.G. Novel sugar esters proniosomes for transdermal delivery of vinpocetine: Preclinical and clinical studies. Eur. J. Pharm. Biopharm. 2011, 77, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J. Enzymatic synthesis of isomaltotriose palmitate and evaluation of its emulsifying property. Enzym. Microb. Technol. 2017, 101, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, R.M.F.; Silva, D.P.D.; Paz, M.C.D.F.; de Queiroz, J.C.F. Screening, production and characterization of biosurfactants from caatinga´s filamentous Fungi. Int. J. Pharm. Sci. Invent. 2017, 6, 23–28. [Google Scholar]
- Beisson, F.; Tiss, A.; Rivière, C.; Verger, R. Methods for lipase detection and assay: A critical review. Eur. J. Lipid Sci. Technol. 2000, 102, 133–153. [Google Scholar] [CrossRef]
- Kopp, W.; Silva, F.A.; Lima, L.N.; Masunaga, S.H.; Tardioli, P.W.; Giordano, R.C.; Araújo-Moreira, F.M.; Giordano, R.L.C. Synthesis and characterization of robust magnetic carriers for bioprocess applications. Mater. Sci. Eng. B Solid-State Mater. Adv. Technol. 2015, 193, 217–228. [Google Scholar] [CrossRef]
- Agilent. The Essential Chromatography and Spectroscopy Catalog, 2011/2012. Available online: https://www.agilent.com/en/promotions/catalog (accessed on 1 May 2021).
- Milessi, T.S.; Perez, C.L.; Zangirolami, T.C.; Corradini, F.A.S.; Sandri, J.P.; Moreno, M.R.F.; Giordano, R.C.; Thevelein, J.M.; Giordano, R.L.C. Biotechnology for biofuels repeated batches as a strategy for high 2G ethanol production from undetoxified hemicellulose hydrolysate using immobilized cells of recombinant Saccharomyces cerevisiae in a fixed—bed reactor. Biotechnol. Biofuels 2020, 13, 1–12. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
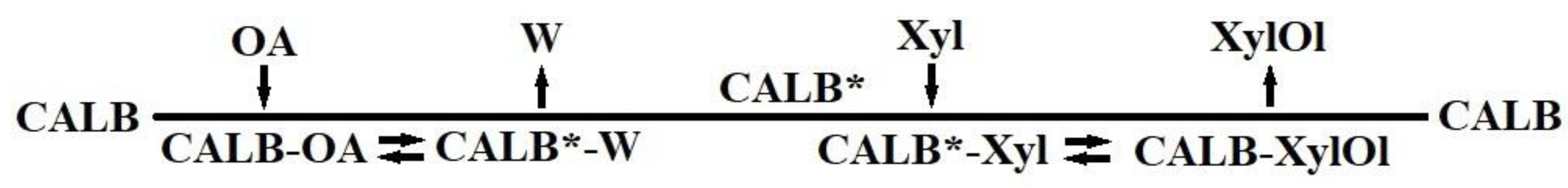{kind=link}
| Acyl Donor | MR * | Solvent | Conditions | Biocatalyst | Conversions * | References |
|---|---|---|---|---|---|---|
| Capric acid | 1:4 | DMSO; ACT | 60 °C, 24 h | N435 | 64 (FAC) | [5] |
| Oleic/Lauric acids | 1:5 | T-but | 46–55 °C, 48 h | CALB IM T2-350 CALB-SMMP-O CALB-SMMP-OG | XC: 52/54 (46 °C); 65 (55 °C) 50 (46 °C); 61/66 (55 °C) 57/50 (46 °C); 69/66 (55 °C) | [27] |
| Vinyl laurate | 1:3 | 2M2B; HEX; THF | 50 °C, 4 h | N435 | 74.8 (XC) | [6] |
| Butyric/caprylic/oleic acids | 1:5 | T-but, T-pent | 60 °C, 24 h | PPL-OS | 70 (XC) | [12] |
| Acrylic acid | 1:4.2 | T-but | 55 °C, 48 h | CALB IM N | 4.2 (XC) | [56] |
| Oleic acid | 1:5 | MEK | 60 °C, 24 h | L435 | 98 (XC), 31 (FAC) | This study |
| Parameters | |
|---|---|
| Vmax (mmol/L/min) | 1.57 |
| Km(OA) (mM) | 1120.40 |
| Km(Xyl) (mM) | 754.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, M.C.P.; Amaral, J.C.; Fernandez-Lafuente, R.; Sousa Junior, R.d.; Tardioli, P.W. Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone. Molecules 2021, 26, 3317. https://doi.org/10.3390/molecules26113317
Gonçalves MCP, Amaral JC, Fernandez-Lafuente R, Sousa Junior Rd, Tardioli PW. Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone. Molecules. 2021; 26(11):3317. https://doi.org/10.3390/molecules26113317
Chicago/Turabian StyleGonçalves, Maria Carolina Pereira, Jéssica Cristina Amaral, Roberto Fernandez-Lafuente, Ruy de Sousa Junior, and Paulo Waldir Tardioli. 2021. "Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone" Molecules 26, no. 11: 3317. https://doi.org/10.3390/molecules26113317
APA StyleGonçalves, M. C. P., Amaral, J. C., Fernandez-Lafuente, R., Sousa Junior, R. d., & Tardioli, P. W. (2021). Lipozyme 435-Mediated Synthesis of Xylose Oleate in Methyl Ethyl Ketone. Molecules, 26(11), 3317. https://doi.org/10.3390/molecules26113317