
Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2

 , , , and
, , , and
Abstract
1. Introduction
2. Results
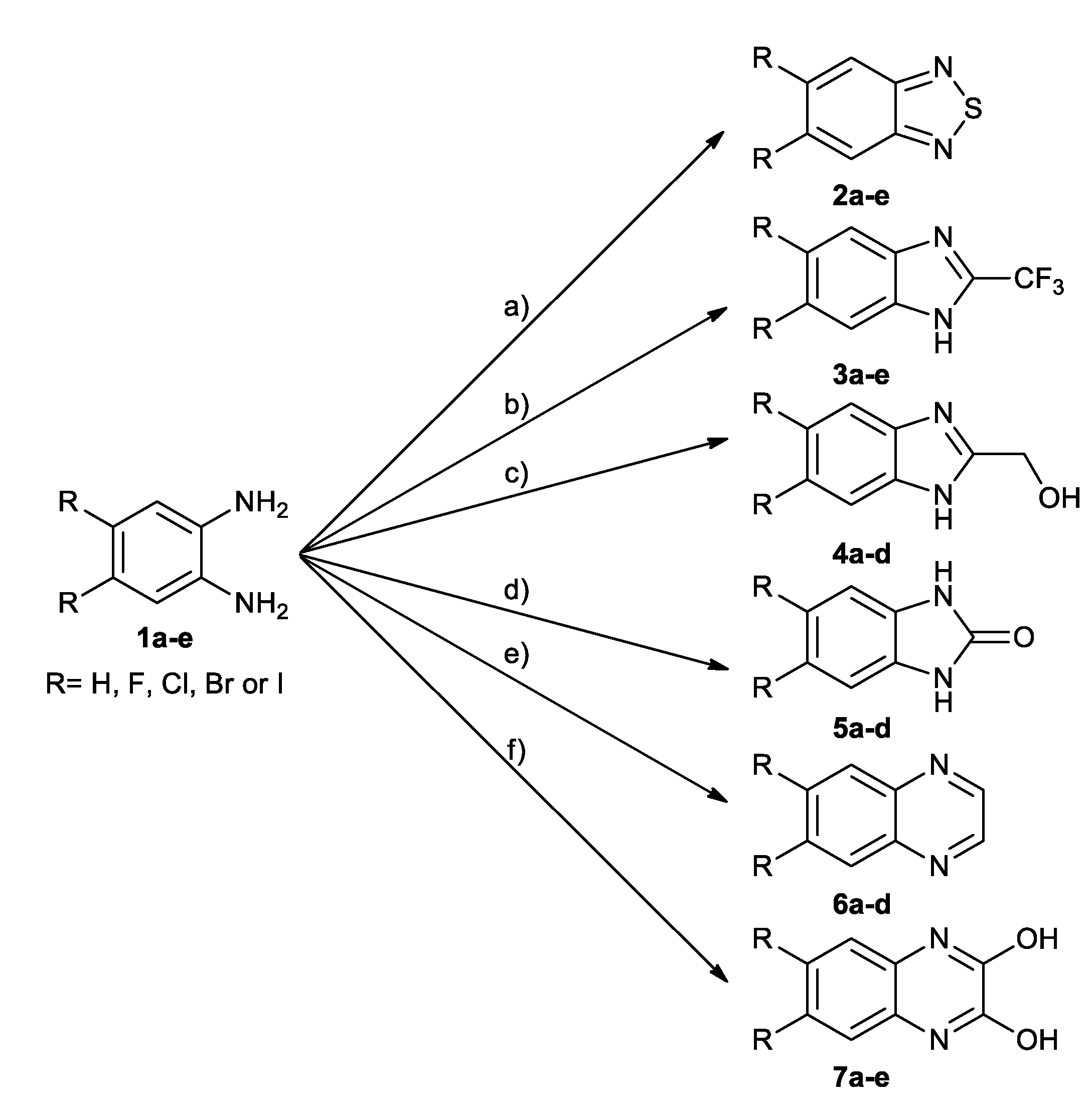
2.1. Chemistry
2.2. Hydrophobicity Data
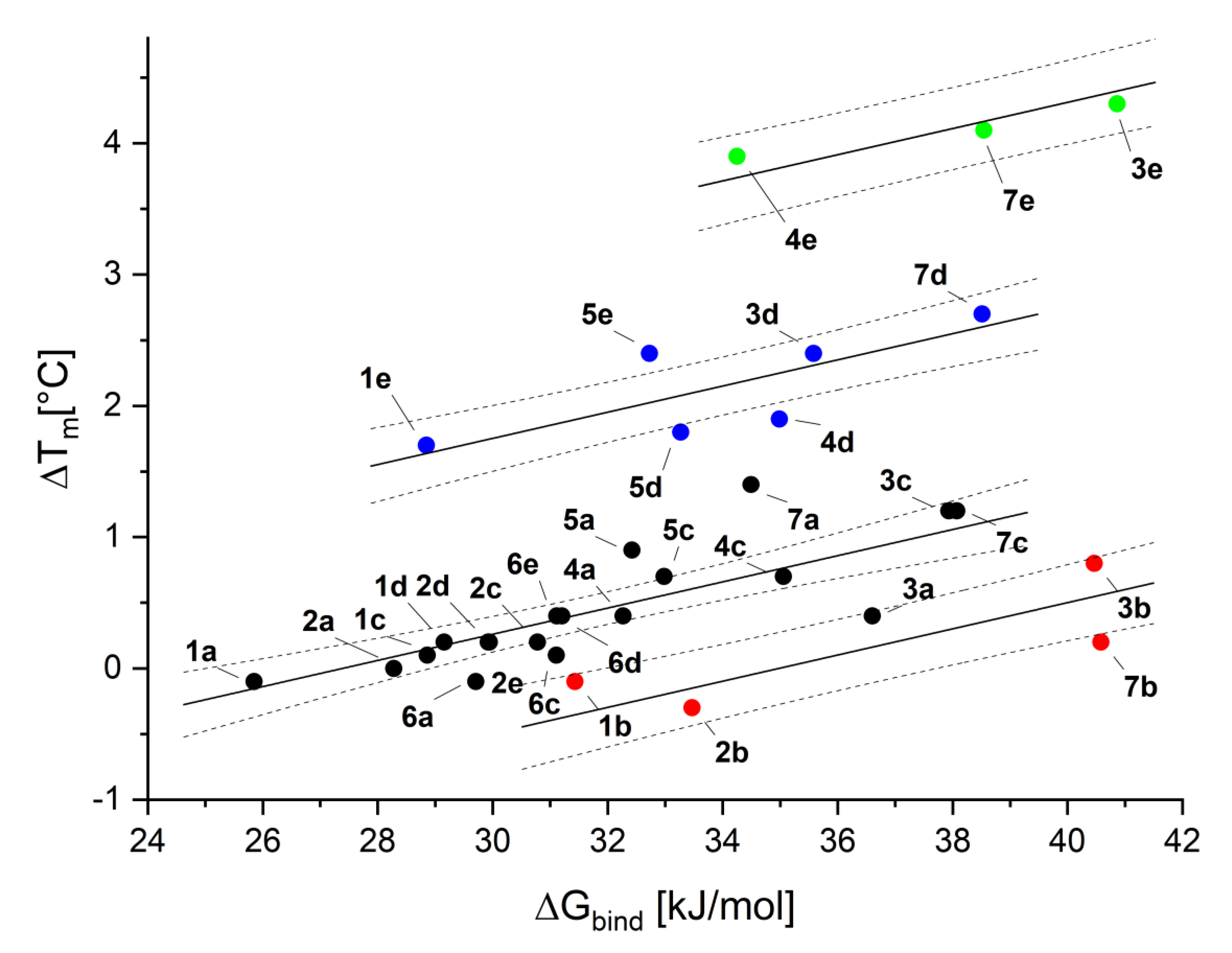
2.3. Binding Affinity—Thermal Shift Assay
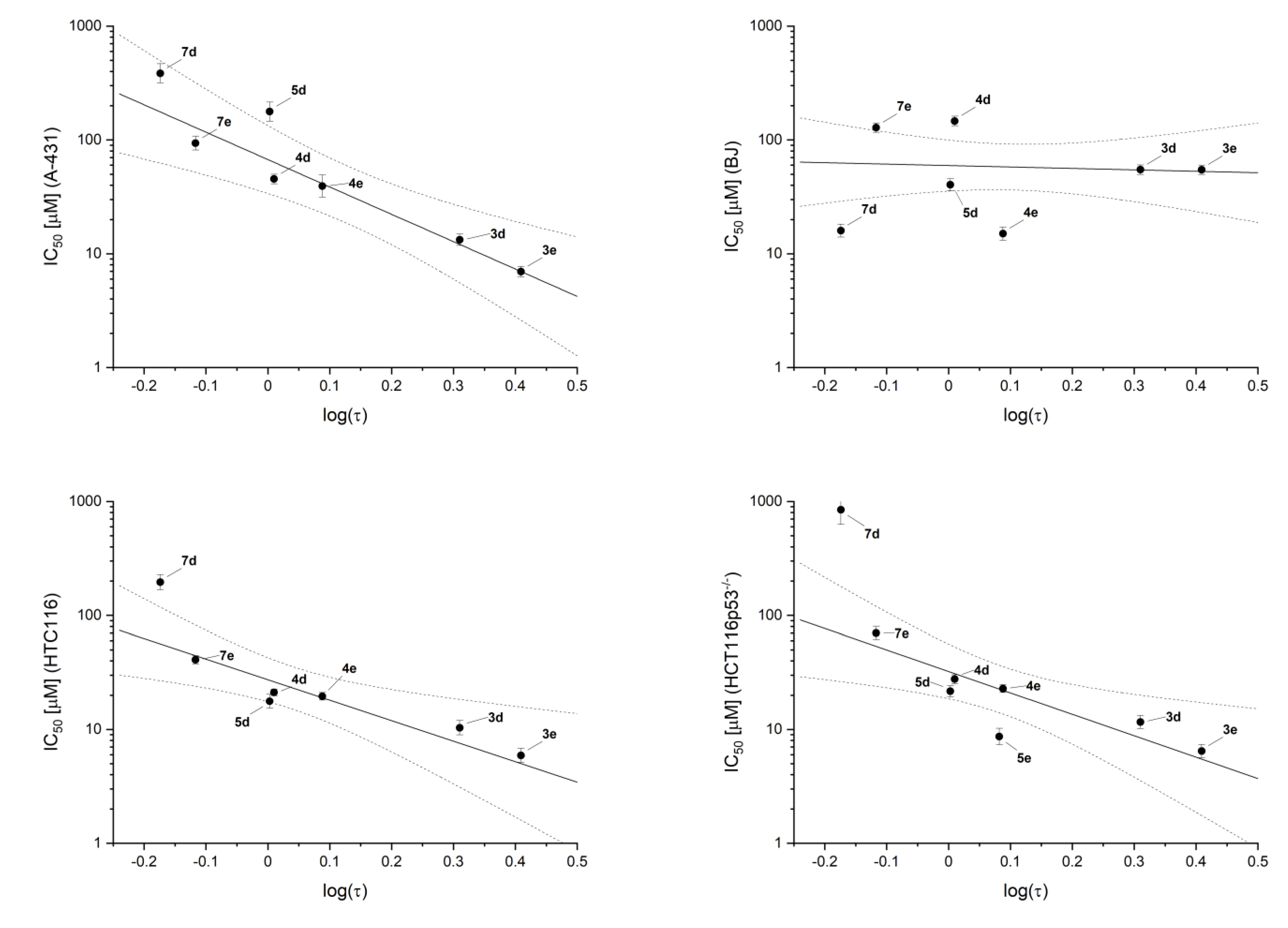
2.4. Inhibitory Activity
2.5. Cell Toxicity
2.6. Molecular Modeling
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. NanoDSF
4.3. Inhibitory Activity against Human CK2α
4.4. Cell Viability Assay
4.5. Chromatographic Hydrophobicity Index
4.6. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Gotz, C.; Montenarh, M. Protein kinase CK2 in development and differentiation. Biomed. Rep. 2017, 6, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Graham, K.C.; Litchfield, D.W. Interactions between the subunits of casein kinase II. J. Biol. Chem. 1995, 270, 13017–13021. [Google Scholar] [CrossRef]
- Krippner-Heidenreich, A.; Talanian, R.V.; Sekul, R.; Kraft, R.; Thole, H.; Ottleben, H.; Luscher, B. Targeting of the transcription factor Max during apoptosis: Phosphorylation-regulated cleavage by caspase-5 at an unusual glutamic acid residue in position P1. Biochem. J. 2001, 358, 705–715. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Montessuit, S.; Magnenat, E.; Vilbois, F.; Hochmann, A.; Journot, L.; Antonsson, B.; Martinou, J.C. Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell 2001, 8, 601–611. [Google Scholar] [CrossRef]
- Turowec, J.P.; Vilk, G.; Gabriel, M.; Litchfield, D.W. Characterizing the convergence of protein kinase CK2 and caspase-3 reveals isoform-specific phosphorylation of caspase-3 by CK2a’: Implications for pathological roles of CK2 in promoting cancer cell survival. Oncotarget 2013, 4, 560–571. [Google Scholar] [CrossRef]
- Riman, S.; Rizkallah, R.; Kassardjian, A.; Alexander, K.E.; Luscher, B.; Hurt, M.M. Phosphorylation of the Transcription Factor YY1 by CK2 alpha Prevents Cleavage by Caspase 7 during Apoptosis. Mol. Cell. Biol. 2012, 32, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wu, J.; Unger, G.M.; Kren, B.T.; Ahmed, K. CK2 Suppression of Apoptosis and Its Implication in Cancer Biology and Therapy. In Protein Kinase CK2; Pinna, L.A., Ed.; Wiley: Hoboken, NJ, USA, 2013; pp. 319–343. [Google Scholar]
- Ruzzene, M.; Pinna, L.A. Addiction to protein kinase CK2: A common denominator of diverse cancer cells? BBA Proteins Proteom. 2010, 1804, 499–504. [Google Scholar] [CrossRef]
- Duncan, J.S.; Litchfield, D.W. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. BBA Proteins Proteom. 2008, 1784, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Zandomeni, R.; Zandomeni, M.C.; Shugar, D.; Weinmann, R. Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J. Biol. Chem. 1986, 261, 3414–3419. [Google Scholar] [CrossRef]
- Zien, P.; Duncan, J.S.; Skierski, J.; Bretner, M.; Litchfield, D.W.; Shugar, D. Tetrabromobenzotriazole (TBBt) and tetrabromobenzimidazole (TBBz) as selective inhibitors of protein kinase CK2: Evaluation of their effects on cells and different molecular forms of human CK2. BBA Proteins Proteom. 2005, 1754, 271–280. [Google Scholar] [CrossRef]
- Pagano, M.A.; Andrzejewska, M.; Ruzzene, M.; Sarno, S.; Cesaro, L.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J. Med. Chem. 2004, 47, 6239–6247. [Google Scholar] [CrossRef] [PubMed]
- Janeczko, M.; Orzeszko, A.; Kazimierczuk, Z.; Szyszka, R.; Baier, A. CK2 alpha and CK2 alpha’ subunits differ in their sensitivity to 4,5,6,7-tetrabromo- and 4,5,6,7-tetraiodo-1H-benzimidazole derivatives. Eur. J. Med. Chem. 2012, 47, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Sarno, S.; Papinutto, E.; Franchin, C.; Bain, J.; Elliott, M.; Meggio, F.; Kazimierczuk, Z.; Orzeszko, A.; Zanotti, G.; Battistutta, R.; et al. ATP site-directed inhibitors of protein kinase CK2: An update. Curr. Top. Med. Chem. 2011, 11, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Mazzorana, M.; Pinna, L.A.; Battistutta, R. A structural insight into CK2 inhibition. Mol. Cell. Biochem. 2008, 316, 57–62. [Google Scholar] [CrossRef]
- De Moliner, E.; Brown, N.R.; Johnson, L.N. Alternative binding modes of an inhibitor to two different kinases. Eur. J. Biochem. 2003, 270, 3174–3181. [Google Scholar] [CrossRef]
- Battistutta, R.; Mazzorana, M.; Cendron, L.; Bortolato, A.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Moro, S.; Pinna, L.A. The ATP-binding site of protein kinase CK2 holds a positive electrostatic area and conserved water molecules. ChemBioChem 2007, 8, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, J.; Winiewska, M.; Czapinska, H.; Poznanska, A.; Shugar, D. Halogen bonds involved in binding of halogenated ligands by protein kinases. Acta Biochim. Pol. 2016, 63, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, N.; Olsen, B.; Raaf, J.; Bretner, M.; Issinger, O.G.; Niefind, K. Structure of the human protein kinase CK2 catalytic subunit CK2alpha’ and interaction thermodynamics with the regulatory subunit CK2beta. J. Mol. Biol. 2011, 407, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Czapinska, H.; Winiewska-Szajewska, M.; Szymaniec-Rutkowska, A.; Piasecka, A.; Bochtler, M.; Poznanski, J. Halogen Atoms in the Protein-Ligand System. Structural and Thermodynamic Studies of the Binding of Bromobenzotriazoles by the Catalytic Subunit of Human Protein Kinase CK2. J. Phys. Chem. B 2021, 125, 2491–2503. [Google Scholar] [CrossRef]
- Szymaniec-Rutkowska, A.; Bugajska, E.; Kasperowicz, S.; Mieczkowska, K.; Maciejewska, A.M.; Poznanski, J. Does the partial molar volume of a solute reflect the free energy of hydrophobic solvation? J. Mol. Liq. 2019, 293, 111527. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen bonding in halocarbon-protein complexes: A structural survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Shinada, N.K.; de Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356. [Google Scholar] [CrossRef]
- Voth, A.R.; Khuu, P.; Oishi, K.; Ho, P.S. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. [Google Scholar] [CrossRef]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of Halogen Bonding in Solution: Substituent, Structural, and Solvent Effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef]
- Voth, A.R.; Hays, F.A.; Ho, P.S. Directing macromolecular conformation through halogen bonds. Proc. Natl. Acad. Sci. USA 2007, 104, 6188–6193. [Google Scholar] [CrossRef]
- Winiewska, M.; Bugajska, E.; Poznanski, J. ITC-derived binding affinity may be biased due to titrant (nano)-aggregation. Binding of halogenated benzotriazoles to the catalytic domain of human protein kinase CK2. PLoS ONE 2017, 12, e173260. [Google Scholar] [CrossRef]
- Wasik, R.; Winska, P.; Poznanski, J.; Shugar, D. Synthesis and Physico-Chemical Properties in Aqueous Medium of All Possible Isomeric Brom Analogues of Benzo-1H-Triazole, Potential Inhibitors of Protein Kinases. J. Phys. Chem. B 2012, 116, 7259–7268. [Google Scholar] [CrossRef] [PubMed]
- Wasik, R.; Lebska, M.; Felczak, K.; Poznanski, J.; Shugar, D. Relative Role of Halogen Bonds and Hydrophobic Interactions in Inhibition of Human Protein Kinase CK2 alpha by Tetrabromobenzotriazole and Some C(5)-Substituted Analogues. J. Phys. Chem. B 2010, 114, 10601–10611. [Google Scholar] [CrossRef] [PubMed]
- Wasik, R.; Winska, P.; Poznanski, J.; Shugar, D. Isomeric Mono-, Di-, and Tri-Bromobenzo-1H-Triazoles as Inhibitors of Human Protein Kinase CK2alpha. PLoS ONE 2012, 7, e48898. [Google Scholar] [CrossRef] [PubMed]
- Winiewska, M.; Kucinska, K.; Makowska, M.; Poznanski, J.; Shugar, D. Thermodynamics parameters for binding of halogenated benzotriazole inhibitors of human protein kinase CK2 alpha. BBA Proteins Proteom. 2015, 1854, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Winiewska, M.; Makowska, M.; Maj, P.; Wielechowska, M.; Bretner, M.; Poznanski, J.; Shugar, D. Thermodynamic parameters for binding of some halogenated inhibitors of human protein kinase CK2. Biochem. Biophys. Res. Commun. 2015, 456, 282–287. [Google Scholar] [CrossRef]
- Marzec, E.; Poznański, J.; Paprocki, D. Thermodynamic contribution of iodine atom to the binding of heterogeneously polyhalogenated benzotriazoles by the catalytic subunit of human protein kinase CK2. IUBMB Life 2020, 72, 1203–1210. [Google Scholar] [CrossRef]
- Neto, B.A.D.; Lopes, A.S.A.; Ebeling, G.; Goncalves, R.S.; Costa, V.E.U.; Quina, F.H.; Dupont, J. Photophysical and electrochemical properties of pi-extended molecular 2,1,3-benzothiadiazoles. Tetrahedron 2005, 61, 10975–10982. [Google Scholar] [CrossRef]
- Rene, O.; Souverneva, A.; Magnuson, S.R.; Fauber, B.P. Efficient syntheses of 2-fluoroalkylbenzimidazoles and -benzothiazoles. Tetrahedron Lett. 2013, 54, 201–204. [Google Scholar] [CrossRef]
- Alasmary, F.A.S.; Snelling, A.M.; Zain, M.E.; Alafeefy, A.M.; Awaad, A.S.; Karodia, N. Synthesis and Evaluation of Selected Benzimidazole Derivatives as Potential Antimicrobial Agents. Molecules 2015, 20, 15206–15223. [Google Scholar] [CrossRef]
- Kilchmann, F.; Marcaida, M.J.; Kotak, S.; Schick, T.; Boss, S.D.; Awale, M.; Gonczy, P.; Reymond, J.L. Discovery of a Selective Aurora A Kinase Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 7188–7211. [Google Scholar] [CrossRef]
- Valko, K.; Bevan, C.; Reynolds, D. Chromatographic hydrophobicity index by fast-gradient RP HPLC: A high-throughput alternative to log P/log D. Anal. Chem. 1997, 69, 2022–2029. [Google Scholar] [CrossRef]
- Kasperowicz, S.; Marzec, E.; Maciejewska, A.M.; Trzybiński, D.; Bretner, M.; Woźniak, K.; Poznański, J.; Mieczkowska, K. A competition between hydrophobic and electrostatic interactions in protein–ligand systems. Binding of heterogeneously halogenated benzotriazoles by the catalytic subunit of human protein kinase CK2. IUBMB Life 2020, 72, 1211–1219. [Google Scholar] [CrossRef]
- Poznanski, J.; Poznanska, A.; Shugar, D. A Protein Data Bank Survey Reveals Shortening of Intermolecular Hydrogen Bonds in Ligand-Protein Complexes When a Halogenated Ligand Is an H-Bond Donor. PLoS ONE 2014, 9, e99984. [Google Scholar] [CrossRef] [PubMed]
- Marzec, E.; Switalska, M.; Winiewska-Szajewska, M.; Wojcik, J.; Wietrzyk, J.; Maciejewska, A.M.; Poznanski, J.; Mieczkowski, A. The halogenation of natural flavonoids, baicalein and chrysin, enhances their affinity to human protein kinase CK2. IUBMB Life 2020, 72, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, M.; Yepez-Mulia, L.; Cedillo-Rivera, R.; Tapia, A.; Vilpo, L.; Vilpo, J.; Kazimierczuk, Z. Synthesis, antiprotozoal and anticancer activity of substituted 2-trifluoromethyl- and 2-pentafluoroethylbenzimidazoles. Eur. J. Med. Chem. 2002, 37, 973–978. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; Mazzorana, M.; Sarno, S.; Kazimierczuk, Z.; Zanotti, G.; Pinna, L.A. Inspecting the structure-activity relationship of protein kinase CK2 inhibitors derived from tetrabromo-benzimidazole. Chem. Biol. 2005, 12, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R. Structural bases of protein kinase CK2 inhibition. Cell. Mol. Life Sci. 2009, 66, 1868–1889. [Google Scholar] [CrossRef] [PubMed]
- Hernandes, M.Z.; Cavalcanti, S.M.; Moreira, D.R.; de Azevedo Junior, W.F.; Leite, A.C. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug. Targets 2010, 11, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Battistutta, R.; De Moliner, E.; Sarno, S.; Zanotti, G.; Pinna, L.A. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001, 10, 2200–2206. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, September 5th–9th, 2005. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, M.; Pagano, M.A.; Meggio, F.; Brunati, A.M.; Kazimierczuk, Z. Polyhalogenobenzimidazoles: Synthesis and their inhibitory activity against casein kinases. Biorg. Med. Chem. 2003, 11, 3997–4002. [Google Scholar] [CrossRef]
- Gianoncelli, A.; Cozza, G.; Orzeszko, A.; Meggio, F.; Kazimierczuk, Z.; Pinna, L.A. Tetraiodobenzimidazoles are potent inhibitors of protein kinase CK2. Biorg. Med. Chem. 2009, 17, 7281–7289. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Structure | R | Compound | ΔTm [°C] | Tret [min] | log(τ) | ΔGbind [kJ/mol] |
|---|---|---|---|---|---|---|---|
| 1 |  | H | 1a | −0.1 ± 0.1 | 6.12 | −0.651 | 25.9 |
| 2 | F | 1b | −0.1 ± 0.1 | 6.28 | −0.591 | 31.4 | |
| 3 | Cl | 1c | 0.1 ± 0.1 | 7.52 | −0.298 | 28.5 | |
| 4 | Br | 1d | 0.2 ± 0.1 | 8.10 | −0.208 | 29.2 | |
| 5 | I | 1e | 1.7 ± 0.1 | 9.03 | −0.093 | 28.9 | |
| 6 |  | H | 2a | 0.0 ± 0.1 | 10.10 | 0.009 | 28.3 |
| 7 | F | 2b | −0.3 ± 0.1 | 12.38 | 0.169 | 33.5 | |
| 8 | Cl | 2c | 0.2 ± 0.1 | 12.55 | 0.179 | 30.8 | |
| 9 | Br | 2d | 0.2 ± 0.1 | 19.45 | 0.461 | 29.9 | |
| 10 | I | 2e | 0.2 ± 0.1 | 19.70 | 0.468 | 29.9 | |
| 11 |  | H | 3a | 0.4 ± 0.1 | 7.68 | −0.270 | 36.6 |
| 12 | F | 3b | 0.8 ± 0.1 | 9.02 | −0.095 | 40.5 | |
| 13 | Cl | 3c | 1.2 ± 0.1 | 13.57 | 0.234 | 37.9 | |
| 14 | Br | 3d | 2.4 ± 0.1 | 15.20 | 0.310 | 35.6 | |
| 15 | I | 3e | 4.3 ± 0.1 | 17.83 | 0.409 | 40.9 | |
| 16 |  | H | 4a | 0.4 ± 0.1 | 6.32 | −0.579 | 31.4 |
| 17 | Cl | 4c | 0.7 ± 0.1 | 9.17 | −0.079 | 33.8 | |
| 18 | Br | 4d | 1.9 ± 0.1 | 10.12 | 0.010 | 32.7 | |
| 19 | I | 4e | 3.7 ± 0.1 | 11.12 | 0.088 | 32.2 | |
| 20 |  | H | 5a | 0.8 ± 0.1 | 6.28 | −0.591 | 32.4 |
| 21 | Cl | 5c | 0.8 ± 0.1 | 9.12 | −0.084 | 33.0 | |
| 22 | Br | 5d | 1.8 ± 0.1 | 10.03 | 0.003 | 33.3 | |
| 23 | I | 5e | 2.4 ± 0.1 | 11.03 | 0.082 | 32.7 | |
| 24 |  | H | 6a | −0.1 ± 0.1 | 7.53 | −0.295 | 29.7 |
| 25 | Cl | 6c | 0.1 ± 0.1 | 20.62 | 0.495 | 31.1 | |
| 26 | Br | 6d | 0.4 ± 0.1 | 24.37 | 0.588 | 31.2 | |
| 27 | I | 6e | 0.4 ± 0.1 | 25.73 | 0.618 | 31.1 | |
| 28 |  | H | 7a | 0.1 ± 0.1 | 6.00 | −0.699 | 34.5 |
| 29 | F | 7b | 0.2 ± 0.1 | 6.42 | −0.548 | 40.6 | |
| 30 | Cl | 7c | 1.2 ± 0.1 | 7.97 | −0.227 | 38.1 | |
| 31 | Br | 7d | 2.7 ± 0.1 | 8.35 | −0.174 | 38.5 | |
| 32 | I | 7e | 4.1 ± 0.1 | 8.82 | −0.117 | 38.5 |
| Compound | IC50 [µM] | ||||
|---|---|---|---|---|---|
| hCK2α Assay | A-431 | BJ | HTC116 | HCT116p53−/− | |
| 3d | 15 ± 3 | 13 ± 2 | 55 ± 6 | 10 ± 2 | 12 ± 2 |
| 3e | 7 ± 4 | 7 ± 1 | 55 ± 5 | 6 ± 1 | 6 ± 1 |
| 4d | >1000 | 45 ± 5 | 145 ± 15 | 21 ± 1 | 28 ± 3 |
| 4e | 25 ± 5 | 39 ± 10 | 15 ± 2 | 20 ± 2 | 23 ± 2 |
| 5d | >1000 | 180 ± 40 | 40 ± 5 | 18 ± 3 | 22 ± 3 |
| 5e | 13 ± 2 | - | - | - | - |
| 7d | 13 ± 4 | 385 ± 82 | 16 ± 2 | 195 ± 35 | 850 ± 300 |
| 7e | 2 ± 1 | 94 ± 14 | 128 ± 13 | 41 ± 4 | 70 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winiewska-Szajewska, M.; Maciejewska, A.M.; Speina, E.; Poznański, J.; Paprocki, D. Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2. Molecules 2021, 26, 3163. https://doi.org/10.3390/molecules26113163
Winiewska-Szajewska M, Maciejewska AM, Speina E, Poznański J, Paprocki D. Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2. Molecules. 2021; 26(11):3163. https://doi.org/10.3390/molecules26113163
Chicago/Turabian StyleWiniewska-Szajewska, Maria, Agnieszka Monika Maciejewska, Elżbieta Speina, Jarosław Poznański, and Daniel Paprocki. 2021. "Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2" Molecules 26, no. 11: 3163. https://doi.org/10.3390/molecules26113163
APA StyleWiniewska-Szajewska, M., Maciejewska, A. M., Speina, E., Poznański, J., & Paprocki, D. (2021). Synthesis of Novel Halogenated Heterocycles Based on o-Phenylenediamine and Their Interactions with the Catalytic Subunit of Protein Kinase CK2. Molecules, 26(11), 3163. https://doi.org/10.3390/molecules26113163