
Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions

 , , , and
, , , and 
Abstract
{kind=link}
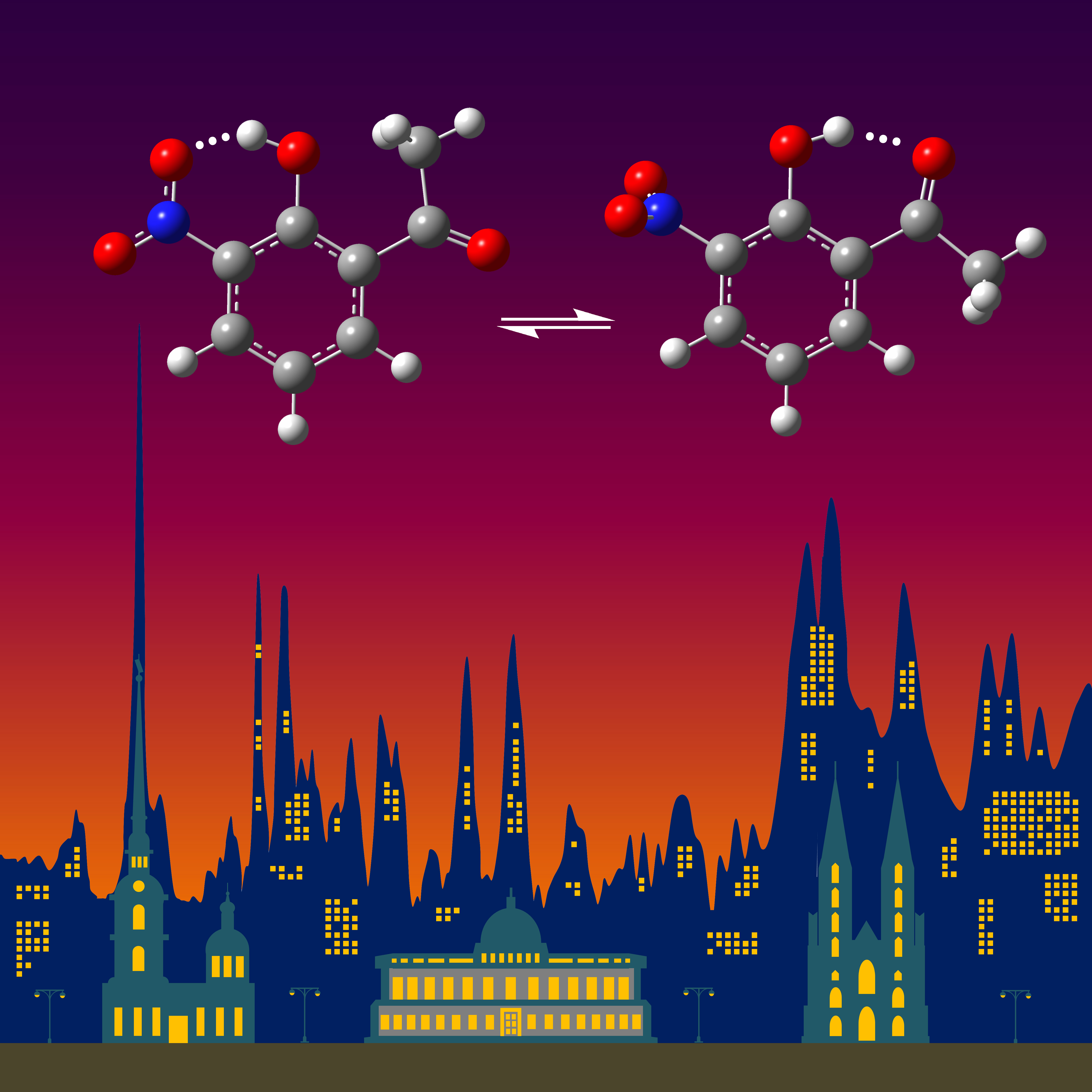
{kind=link}
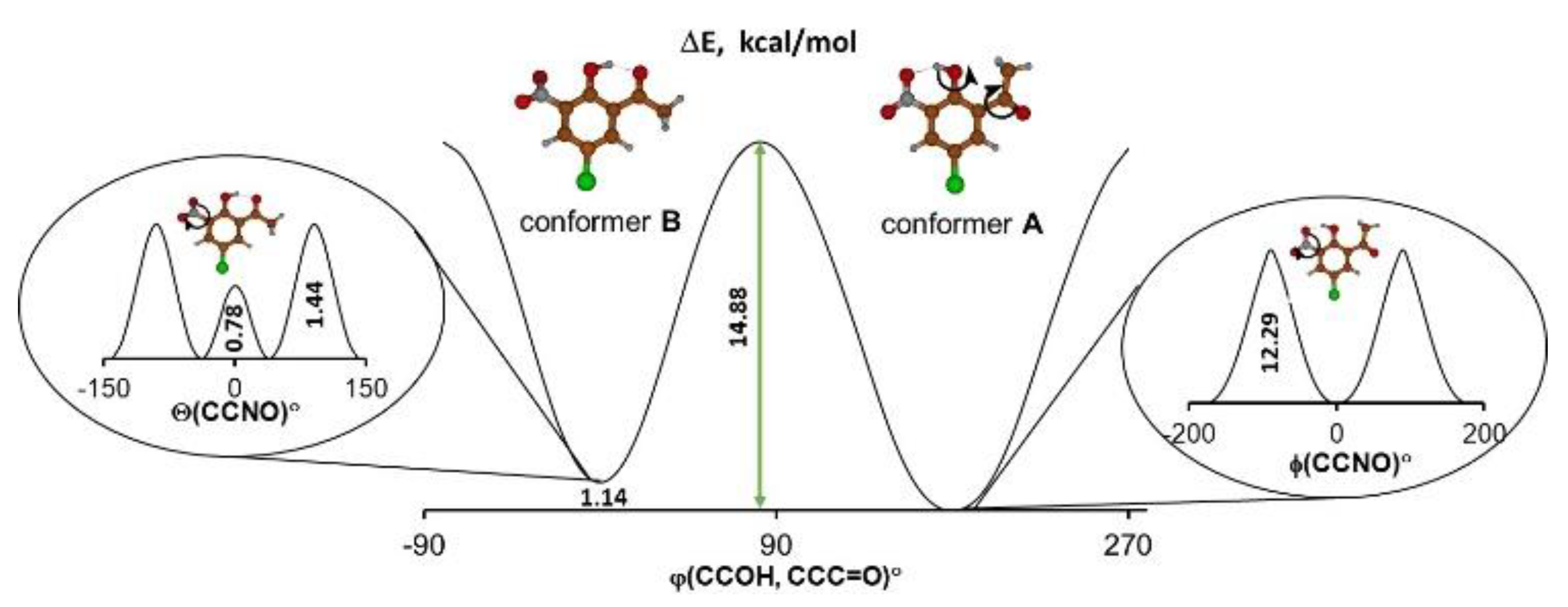
{kind=link}
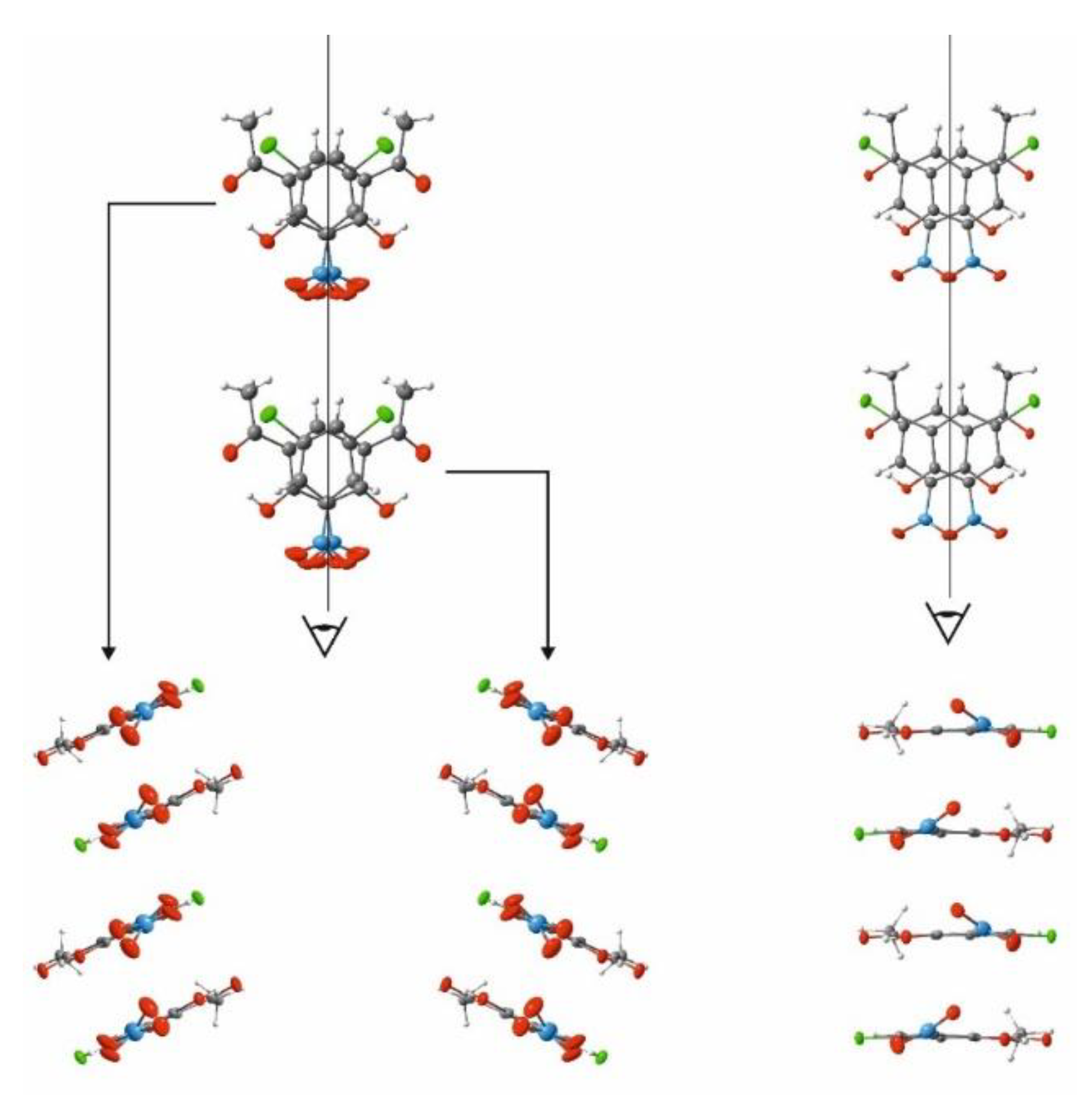
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
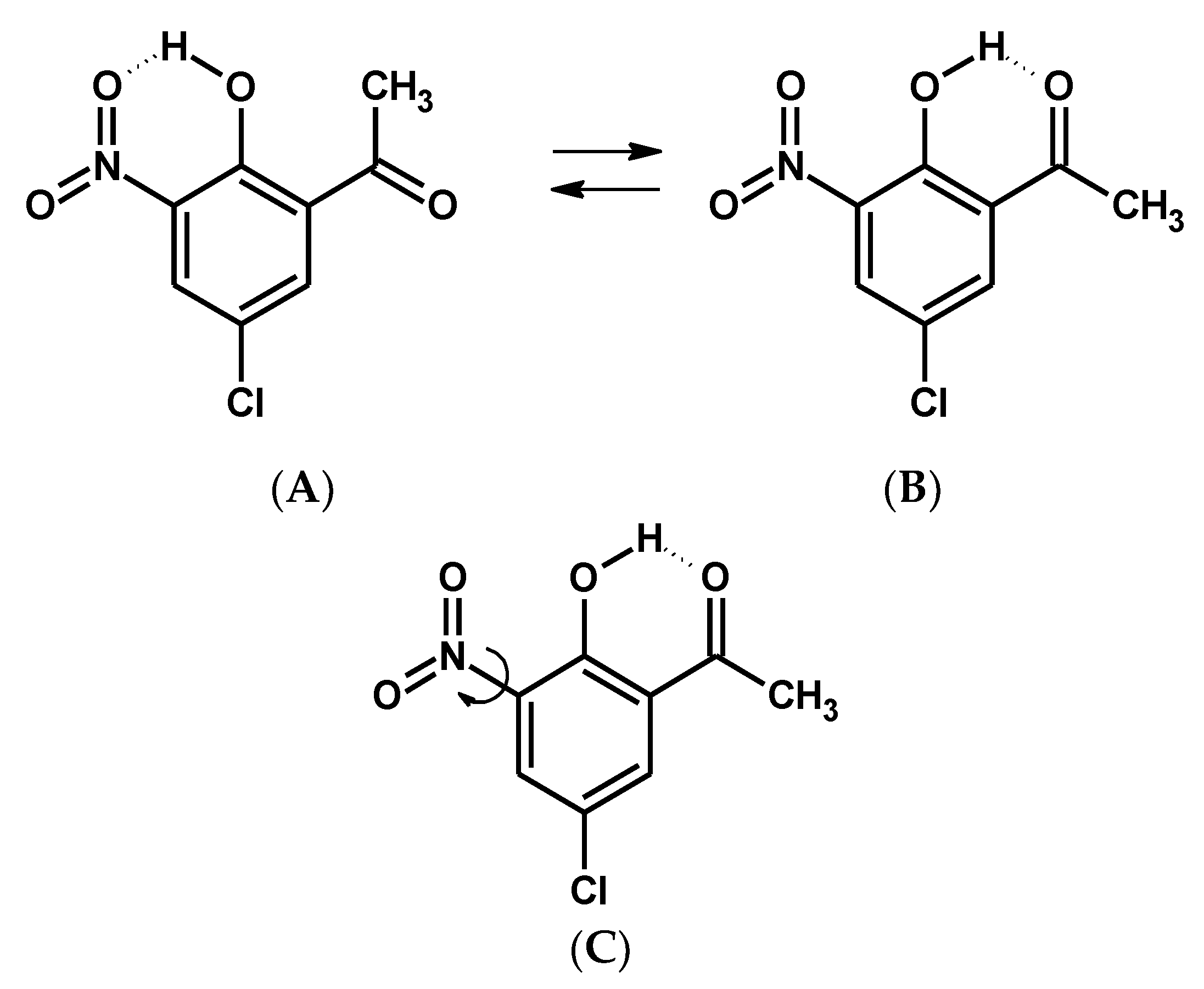
2.1. Detection of Polymorphs in the Solid State and Isomers under the Matrix Condition by Spectroscopic Methods
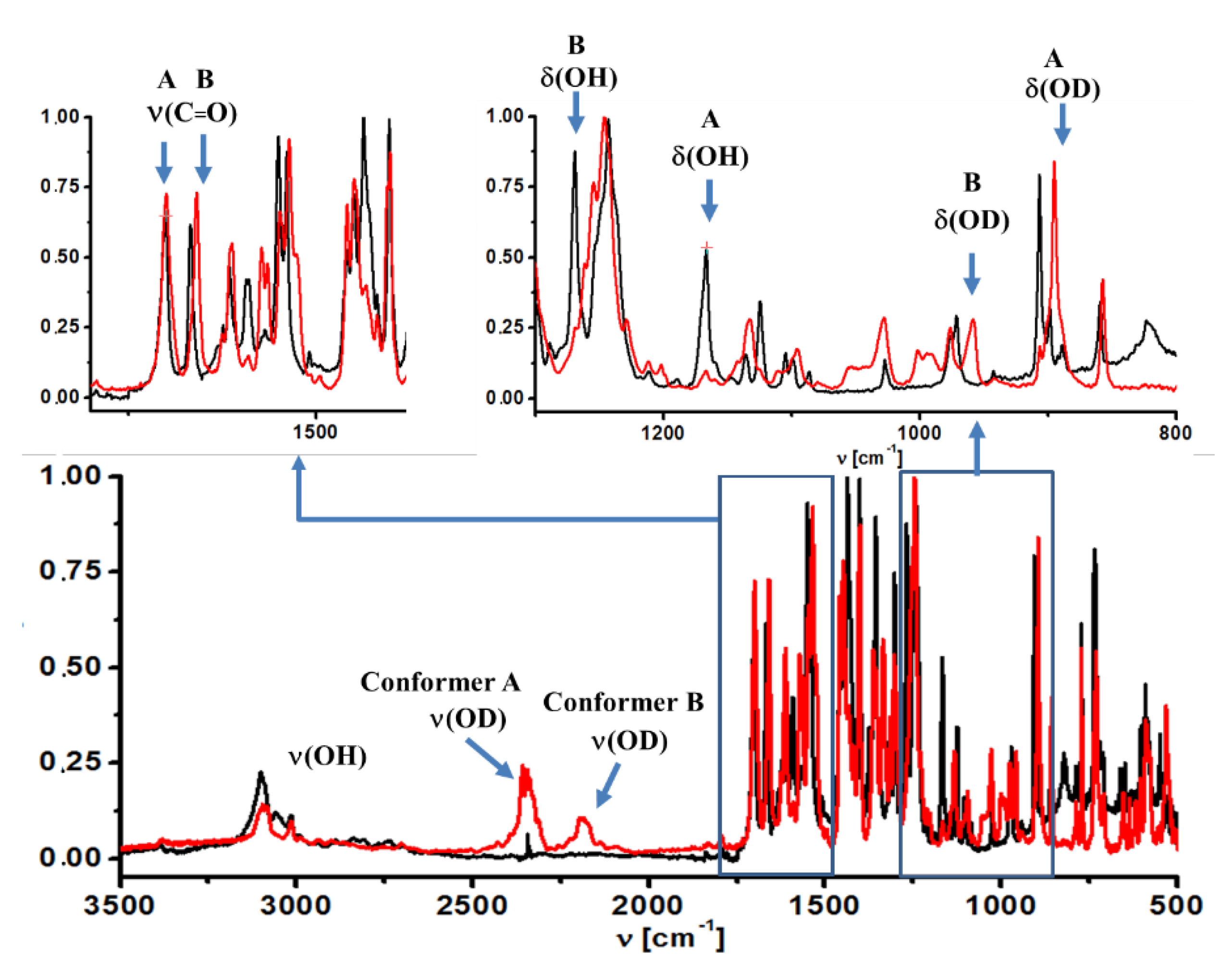
2.1.1. The ν(OH) Stretching Mode
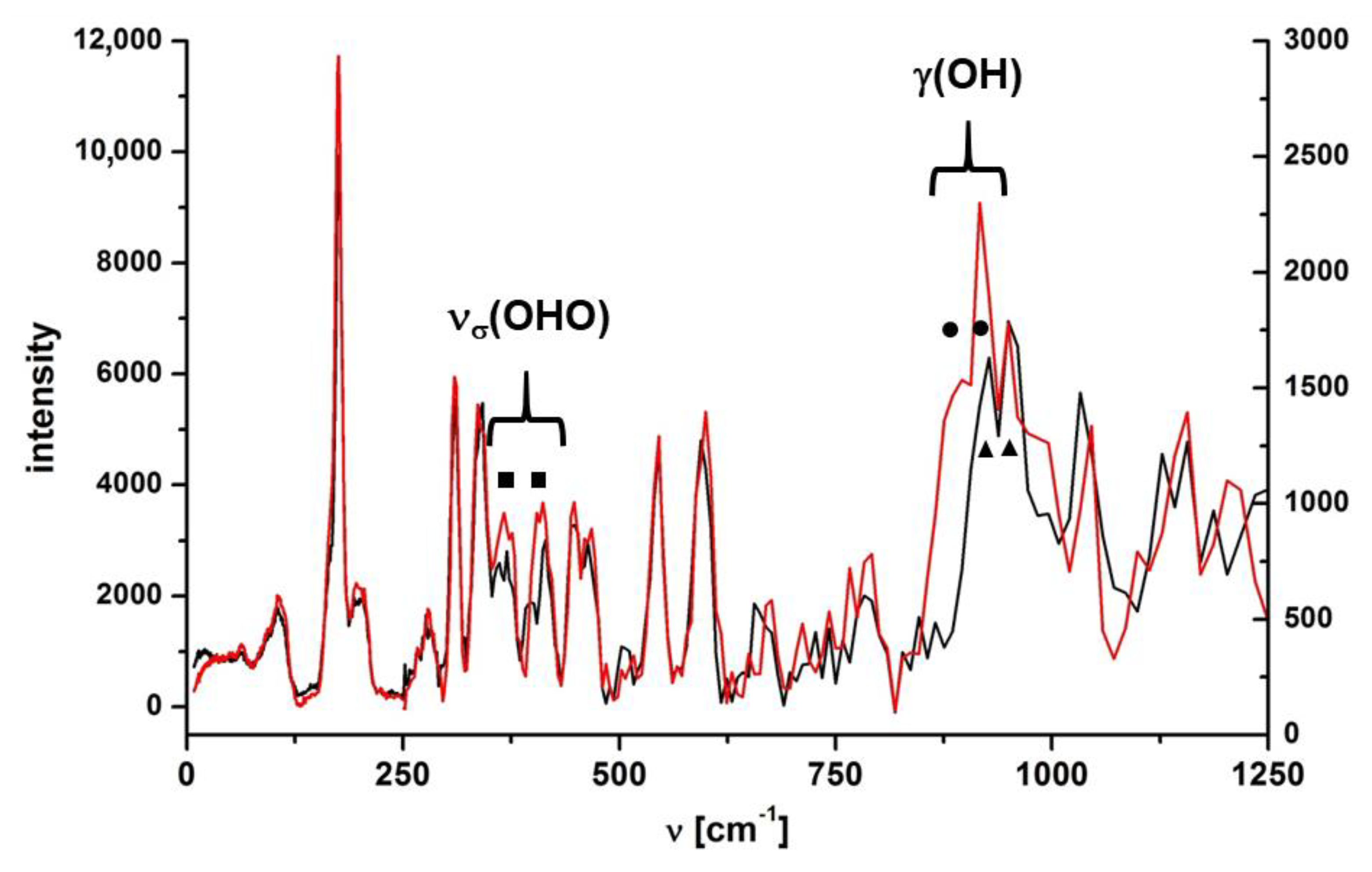
2.1.2. The δ(OH) and γ(OH) Bending Modes
2.1.3. The ν(C = O) Stretching Mode
2.1.4. Nitro Group Mode
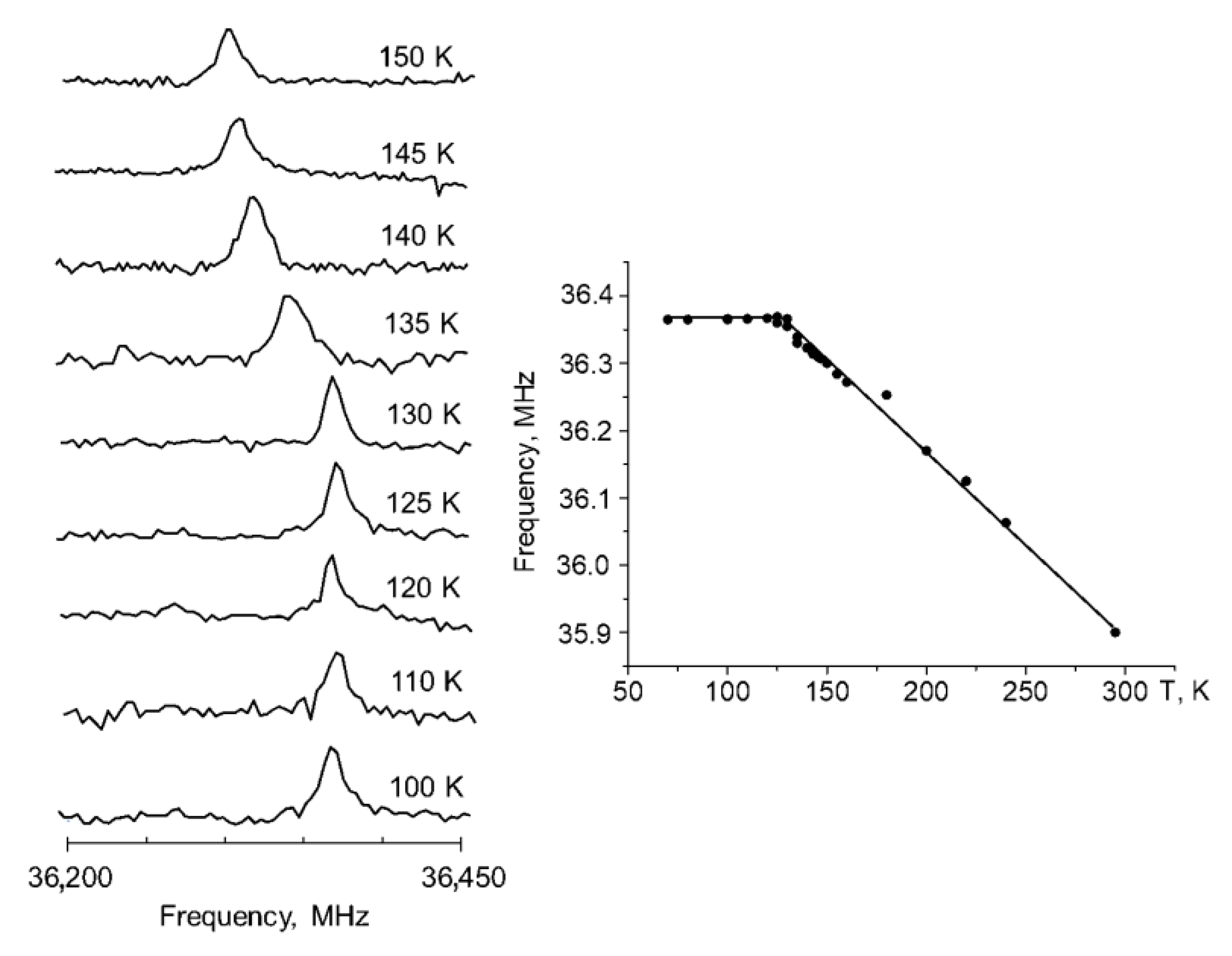
2.2. Spectral Changes under Phase Transition in the Solid State
3. Materials and Methods
3.1. Chemicals
3.2. Computational Details
3.3. Thermal Measurements
3.4. NQR Measurements
3.5. Crystallographic Details
3.6. IR and Raman Measurements
3.7. Incoherent Inelastic Neutron Scattering (IINS) Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bernstein, J. Polymorphism in Molecular Crystals, 2nd ed.; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.K.; Centore, R.; Causà, M.; Tuzi, A.; Borbone, F.; Naumov, P. Strong and Anomalous Thermal Expansion Precedes the Thermosalient Effect in Dynamic Molecular Crystals. Sci. Rep. 2016, 6, 29610. [Google Scholar] [CrossRef] [PubMed]
- Nangia, A. Conformational polymorphism in organic crystals. Acc. Chem. Res. 2008, 41, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.M.; Steed, J.W. Packing Problems: High Z’ Crystal Structures and Their Relationship to Cocrystals, Inclusion Compounds, and Polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef]
- Hilfiker, R. Polymorphism: In the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Brittain, H.G. Polymorphism in Pharmaceutical Solids, 1st ed.; Marcel Dekker Inc.: New York, NY, USA, 1999. [Google Scholar]
- Yu, L. Polymorphism in Molecular Solids: An Extraordinary System of Red, Orange, and Yellow Crystals. Acc. Chem. Res. 2010, 43, 1257–1266. [Google Scholar] [CrossRef]
- Schmidtmann, M.; Gutmann, M.J.; Middlemiss, D.S.; Wilson, C.C. Towards proton transfer in hydrogen bonded molecular complexes: Joint experimental and theoretical modelling and an energy scale for polymorphism. CrystEngComm 2007, 9, 743–745. [Google Scholar] [CrossRef]
- Bilton, C.; Howard, J.A.K.; Laxmi Madhavi, N.N.; Nangia, A.; Desiraju, G.R.; Allen, F.H.; Wilson, C.C. When is a polymorph not a polymorph? Helical trimeric O–H⋯O synthons in trans-1,4-diethynylcyclohexane-1,4-diol. Chem. Commun. 1999, 1675–1676. [Google Scholar] [CrossRef]
- Barnes, A.J. Matrix isolation studies of hydrogen bonding—An historical perspective. J. Mol. Struct. 2018, 1163, 77–85. [Google Scholar] [CrossRef]
- Barnes, A.J.; Orville-Thomas, W.J.; Müller, A.; Gaufrès, R. (Eds.) Matrix Isolation Spectroscopy; Springer Publishing Company: Dordrecht, The Netherlands, 1981. [Google Scholar]
- Nogueira, B.A.; Ildiz, G.O.; Canotilho, J.; Eusebio, M.E.S.; Henriques, M.S.C.; Paixao, J.A.; Fausto, R. 5-Methylhydantoin: From Isolated Molecules in a Low-Temperature Argon Matrix to Solid State Polymorphs Characterization. J. Phys. Chem. A 2017, 121, 5267–5279. [Google Scholar] [CrossRef]
- Avadanei, M.; Kus’, N.; Cozan, V.; Fausto, R. Structure and Photochemistry of N-Salicylidene-p-carboxyaniline Isolated in Solid Argon. J. Phys. Chem. A 2015, 119, 9121–9132. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons, with Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005. [Google Scholar]
- Marques, M.P.M.; Valero, R.; Parker, S.F.; Tomkinson, J.; Batista de Carvalho, L.A.E. Polymorphism in Cisplatin Anticancer Drug. J. Phys. Chem. B 2013, 117, 6421–6429. [Google Scholar] [CrossRef] [PubMed]
- Rivera, S.A.; Allis, D.G.; Hudson, B.S. Importance of Vibrational Zero-Point Energy Contribution to the Relative Polymorph Energies of Hydrogen-Bonded Species. Cryst. Growth Des. 2008, 8, 3905–3907. [Google Scholar] [CrossRef]
- Johnson, M.R.; Trommsdorff, H.P. Vibrational spectra of crystalline formic and acetic acid isotopologues by inelastic neutron scattering and numerical simulations. Chem. Phys. 2009, 355, 118–122. [Google Scholar] [CrossRef]
- Seliger, J.; Zagar, V. Hydrogen bonding and proton transfer in cocrystals of 4,4’-bipyridyl and organic acids studied using nuclear quadrupole resonance. Phys. Chem. Chem. Phys. 2014, 16, 18141–18147. [Google Scholar] [CrossRef] [PubMed]
- Seliger, J.; Zagar, V.; Asaji, T.; Gotoh, K.; Ishida, H. A N-14 nuclear quadrupole resonance study of phase transitions and molecular dynamics in hydrogen bonded organic antiferroelectrics 55DMBP-H(2)ca and 1,5-NPD-H(2)ca. Phys. Chem. Chem. Phys. 2011, 13, 9165–9172. [Google Scholar] [CrossRef] [PubMed]
- Orville-Thomas, W.J. Internal Rotation in Molecules; John Wiley & Sons, Inc.: New York, NY, USA, 1974. [Google Scholar]
- Filarowski, A.; Kochel, A.; Koll, A.; Bator, G.; Mukjerhee, S. Phase transition and intramolecular hydrogen bonding in nitro derivatives of ortho-hydroxy acetophenones. J. Mol. Struct. 2006, 785, 7–13. [Google Scholar] [CrossRef]
- Kalenik, J.; Majerz, I.; Sobczyk, L.; Grech, E.; Habeeb, M.M.M. 35Cl nuclear quadrupole resonance and infrared studies of hydrogen-bonded adducts of 2-chloro-4-nitrobenzoic acid. J. Chem. Soc. Faraday Trans. I 1989, 85, 3187–3193. [Google Scholar] [CrossRef]
- Apih, T.; Gregorovic, A.; Zagar, V.; Seliger, J. Nuclear Quadrupole Resonance Study of Proton and Deuteron Migration in Short Strong Hydrogen Bonds Formed in Molecular Complex 3,5-Dinitrobenzoic Acid-Nicotinic Acid and in Deuterated 3,5-Pyridinedicarboxylic Acid. J. Phys. Chem. C 2016, 120, 9992–10000. [Google Scholar] [CrossRef]
- Asaji, T.; Hoshino, M.; Ishida, H.; Konnai, A.; Shinoda, Y.; Seliger, J.; Žagar, V. Phase transition and proton exchange in 1,3-diazinium hydrogen chloranilate monohydrate. Hyperfine Interact. 2010, 198, 85–91. [Google Scholar] [CrossRef]
- Tobu, Y.; Ikeda, R.; Nihei, T.A.; Gotoh, K.; Ishida, H.; Asaji, T. Temperature dependence of one-dimensional hydrogen bonding in morpholinium hydrogen chloranilate studied by Cl-35 nuclear quadrupole resonance and multi-temperature X-ray diffraction. Phys. Chem. Chem. Phys. 2012, 14, 12347–12354. [Google Scholar] [CrossRef]
- Dolinenkov, P.; Korneva, I.; Sinyavsky, N. The Distribution Change of Relaxation Times in Cl-35 NQR for Phase Transitions in p-Dichlorobenzene. Appl. Magn. Reson. 2015, 46, 17–24. [Google Scholar] [CrossRef]
- Kovacs, A.; Izvekov, V.; Keresztury, G.; Pongor, G. Vibrational analysis of 2-nitrophenol. A joint FT-IR, FT-Raman and scaled quantum mechanical study. Chem. Phys. 1998, 238, 231–243. [Google Scholar] [CrossRef]
- Pająk, J.; Maes, G.; De Borggraeve, W.M.; Boens, N.; Filarowski, A. Matrix-isolation FT-IR and theoretical investigation of the competitive intramolecular hydrogen bonding in 5-methyl-3-nitro-2-hydroxyacetophenone. J. Mol. Struct. 2008, 880, 86–96. [Google Scholar] [CrossRef]
- Konopacka, A.; Filarowski, A.; Pawełka, Z. Solvent influence on the transformation of intramolecular hydrogen bonds in 2-hydroxy-5-methyl-3-nitroacetophenone. J. Solution Chem. 2005, 34, 929–945. [Google Scholar] [CrossRef]
- Sobczyk, L.; Chudoba, D.M.; Tolstoy, T.M.; Filarowski, A. Some Brief Notes on Theoretical and Experimental Investigations of Intramolecular Hydrogen Bonding. Molecules 2016, 21, 1657. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, J.; Parker, S.F.; Braden, D.A.; Hudson, B.S. Inelastic neutron scattering spectra of the transverse acoustic modes of the normal alkanes. Phys. Chem. Chem. Phys. 2002, 4, 716–721. [Google Scholar] [CrossRef]
- Marques, M.P.M.; Batista de Carvalho, L.A.E.; Valero, R.; Machado, N.F.L.; Parker, S.F. An inelastic neutron scattering study of dietary phenolic acids. Phys. Chem. Chem. Phys. 2014, 16, 7491–7500. [Google Scholar] [CrossRef]
- Davydov, A.S. Teorya Molekularnyzh Eksitonov; Nauka: Moscow, Russia, 1968. [Google Scholar]
- Howard, J.; Tomkinson, J.; Eckert, J.; Goldstone, J.A.; Taylor, A.D. Inelastic neutron scattering studies of some intramolecular hydrogen bonded complexes: A new correlation of γ(OHO) vs R (OO). J. Chem. Phys. 1983, 78, 3150–3155. [Google Scholar] [CrossRef]
- Novak, A. Hydrogen bonding in solids correlation of spectroscopic and crystallographic data. Struct. Bonding 1974, 18, 177–216. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Martin, J.M.L.; Van Alsenoy, C. Gar2ped; University of Antwerp: Antwerpen, Belgium, 1995. [Google Scholar]
- Oxford Diffraction Poland Sp. CryslAlis CCD; CryslAlis RED Version 1.171.13 beta (Release 14.11.2003) Copyright 1995−2003; Oxford Diffraction Ltd.: Abingdon, UK, 2003. [Google Scholar]
- Sheldrick, G.M. SHELXS97 Program for Solution of Crystal Structure; University of Goettingen: Goettingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXl97 Program for Refinement of Crystal Structure; University of Goettingen: Goettingen, Germany, 1997. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hetmańczyk, Ł.; Szklarz, P.; Kwocz, A.; Wierzejewska, M.; Pagacz-Kostrzewa, M.; Melnikov, M.Y.; Tolstoy, P.M.; Filarowski, A. Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions. Molecules 2021, 26, 3109. https://doi.org/10.3390/molecules26113109
Hetmańczyk Ł, Szklarz P, Kwocz A, Wierzejewska M, Pagacz-Kostrzewa M, Melnikov MY, Tolstoy PM, Filarowski A. Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions. Molecules. 2021; 26(11):3109. https://doi.org/10.3390/molecules26113109
Chicago/Turabian StyleHetmańczyk, Łukasz, Przemysław Szklarz, Agnieszka Kwocz, Maria Wierzejewska, Magdalena Pagacz-Kostrzewa, Mikhail Ya. Melnikov, Peter M. Tolstoy, and Aleksander Filarowski. 2021. "Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions" Molecules 26, no. 11: 3109. https://doi.org/10.3390/molecules26113109
APA StyleHetmańczyk, Ł., Szklarz, P., Kwocz, A., Wierzejewska, M., Pagacz-Kostrzewa, M., Melnikov, M. Y., Tolstoy, P. M., & Filarowski, A. (2021). Polymorphism and Conformational Equilibrium of Nitro-Acetophenone in Solid State and under Matrix Conditions. Molecules, 26(11), 3109. https://doi.org/10.3390/molecules26113109