
Redox Potential and Crystal Chemistry of Hexanuclear Cluster Compounds



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods: Calculations of the Effective Ionic Charges (or BVS) of TMs in Cluster Compounds
3. Results and Discussion
3.1. Analysis of Available Electrochemical Data
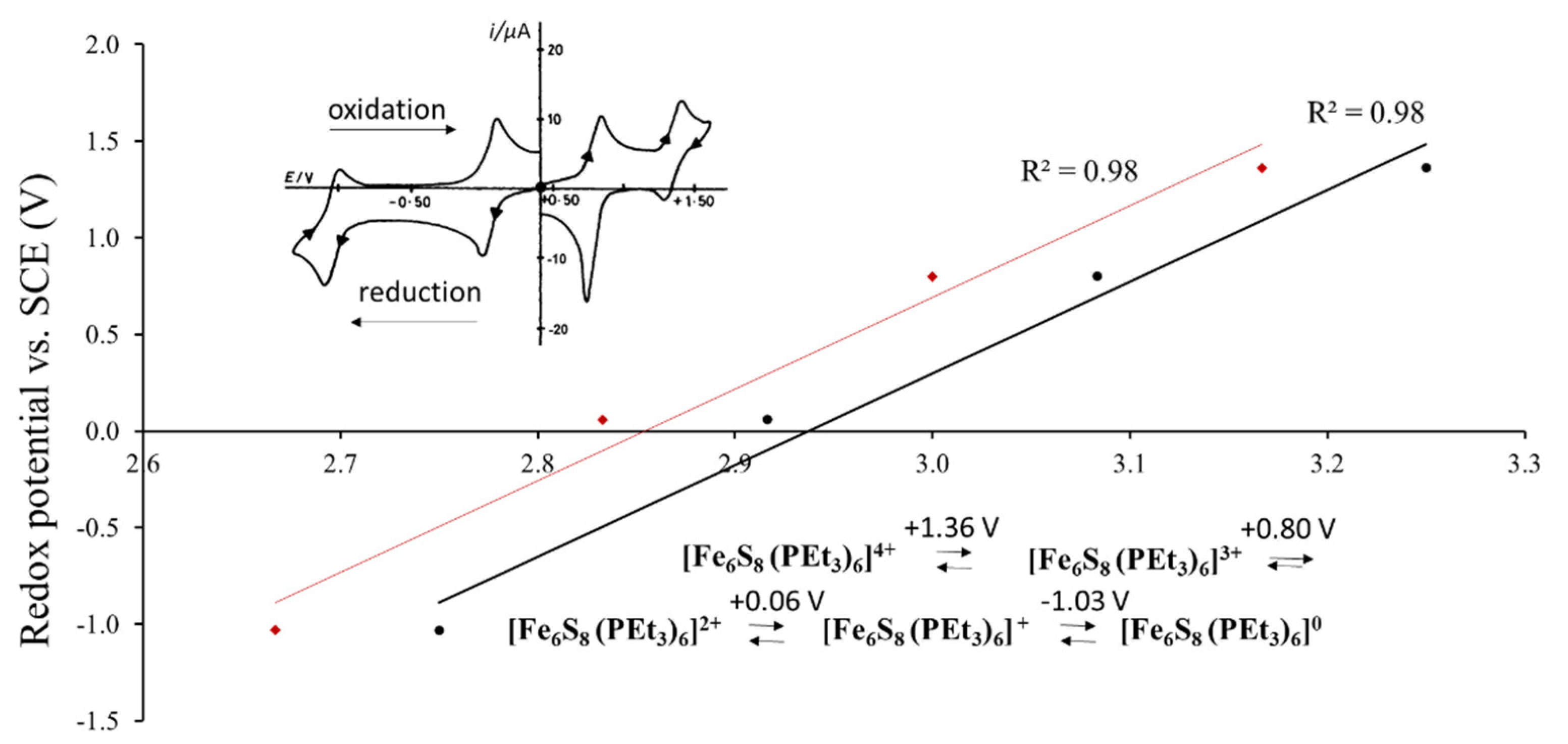
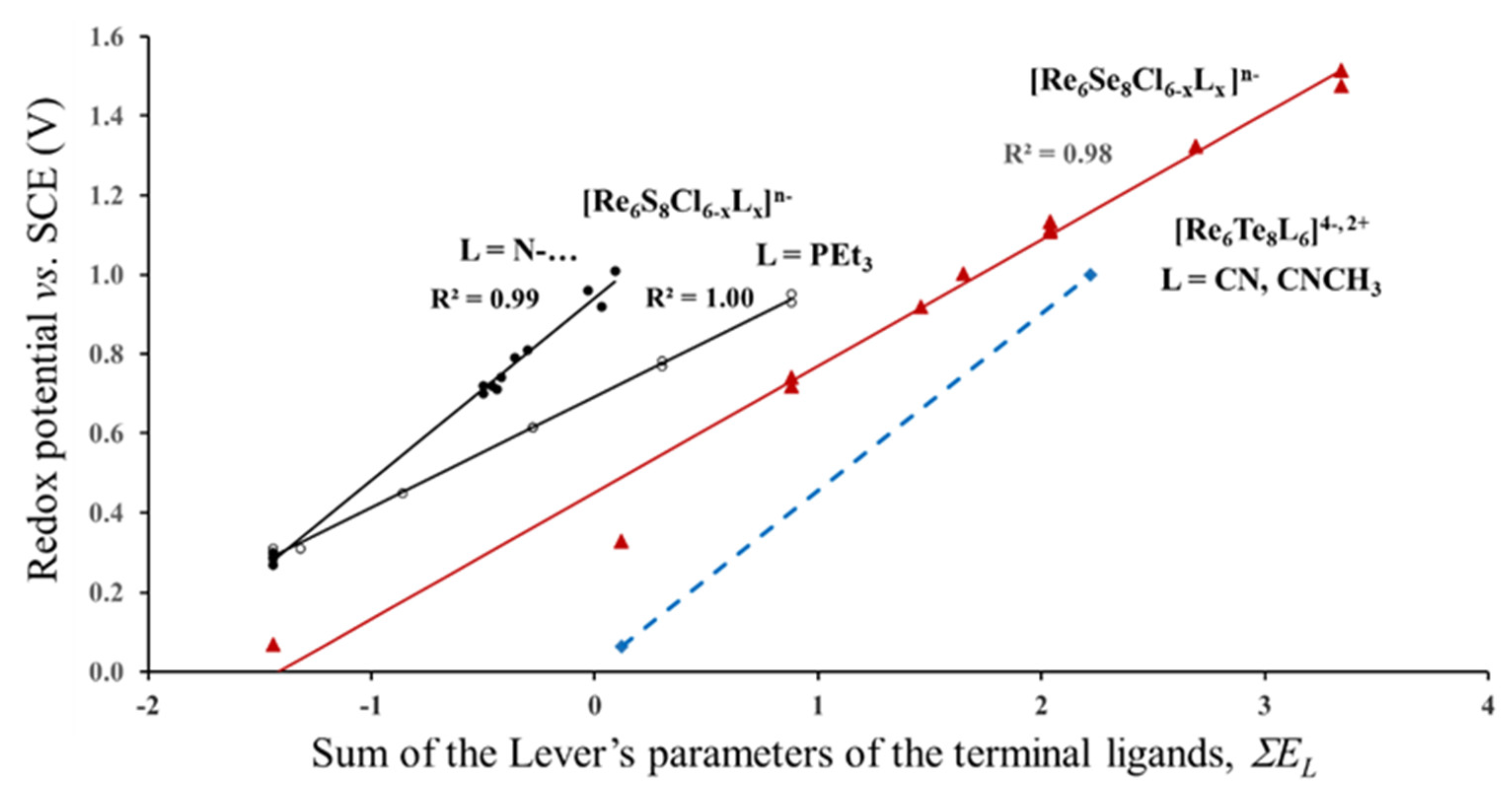
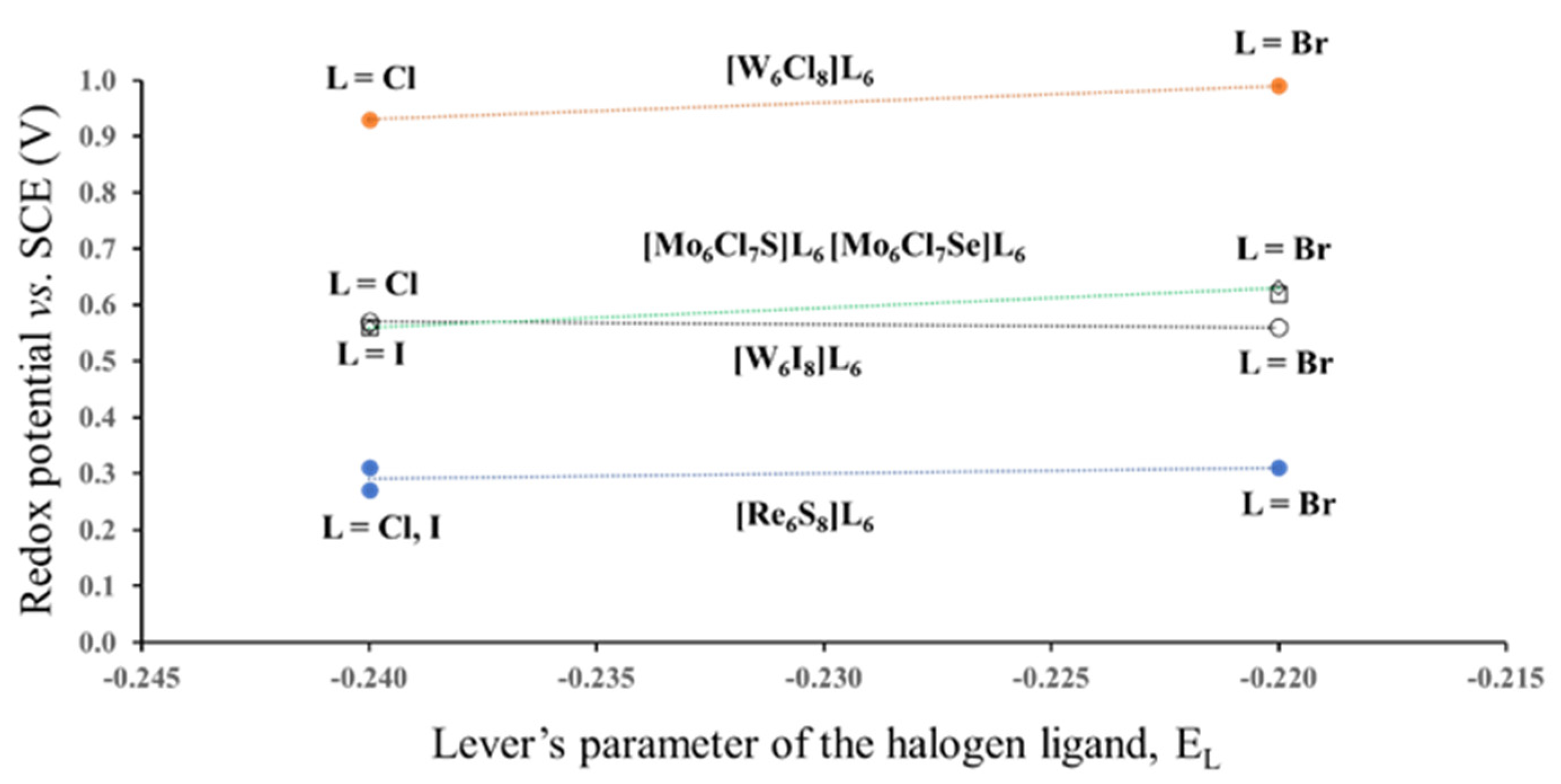
3.1.1. Case of the Constant Cluster Core and Different Outer Ligands
3.1.2. Case of the Fixed Lever’s Parameter for the Outer Ligands
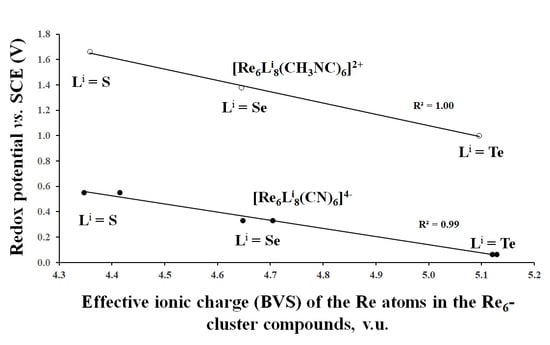
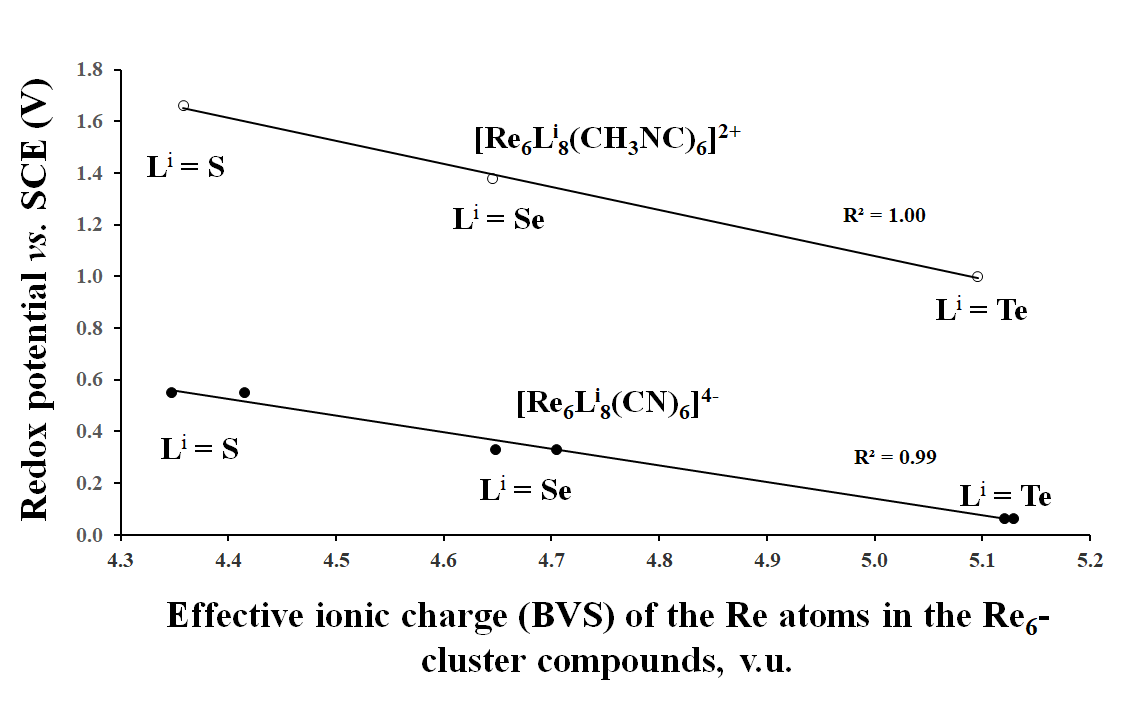
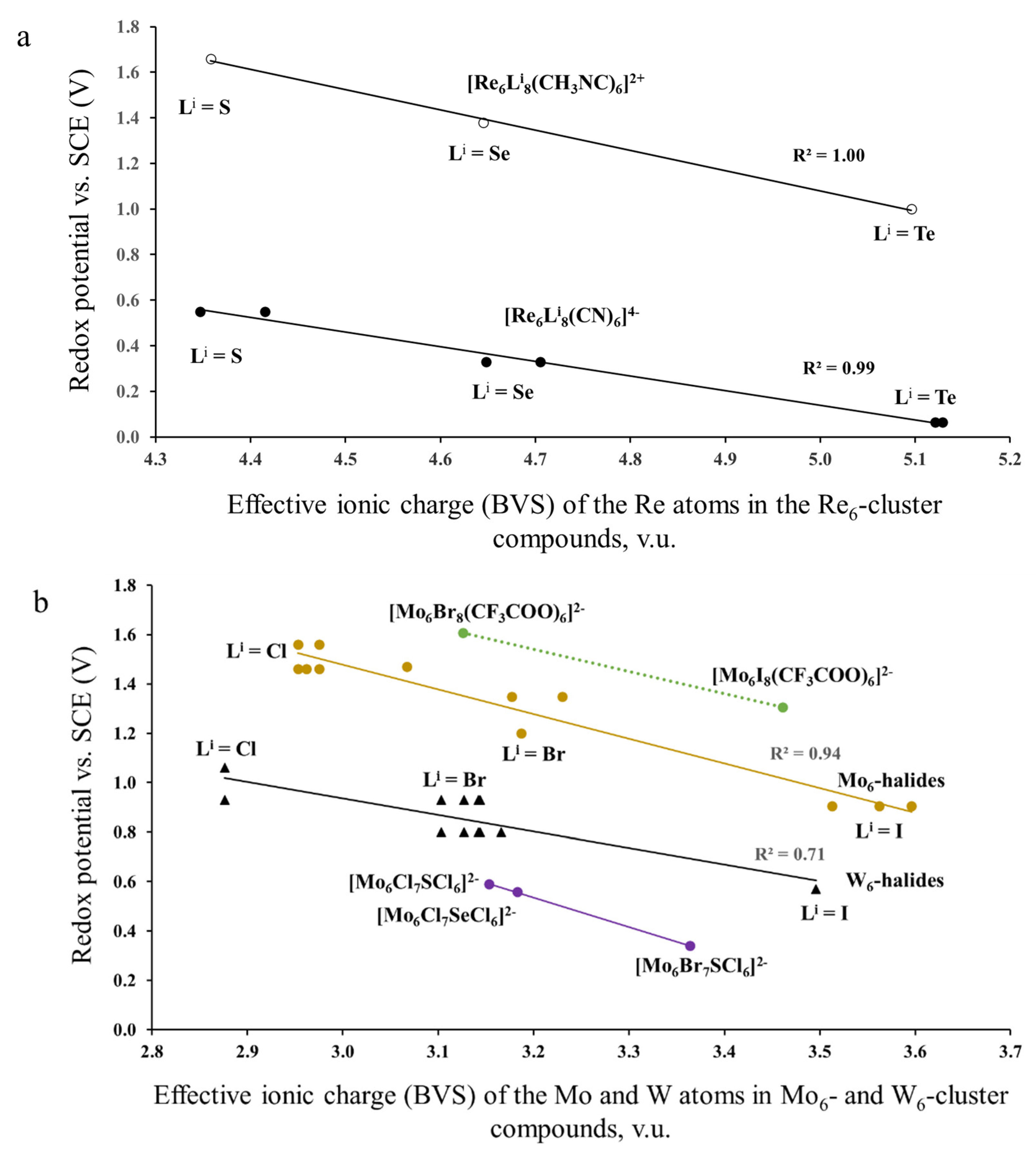
3.2. Correlation Between the Effective Ionic Charge (BVS) of TMs and the Redox Potential
3.3. Steric and Electrostatic Effects in Cluster Compounds and Their Influence on the Redox Potential
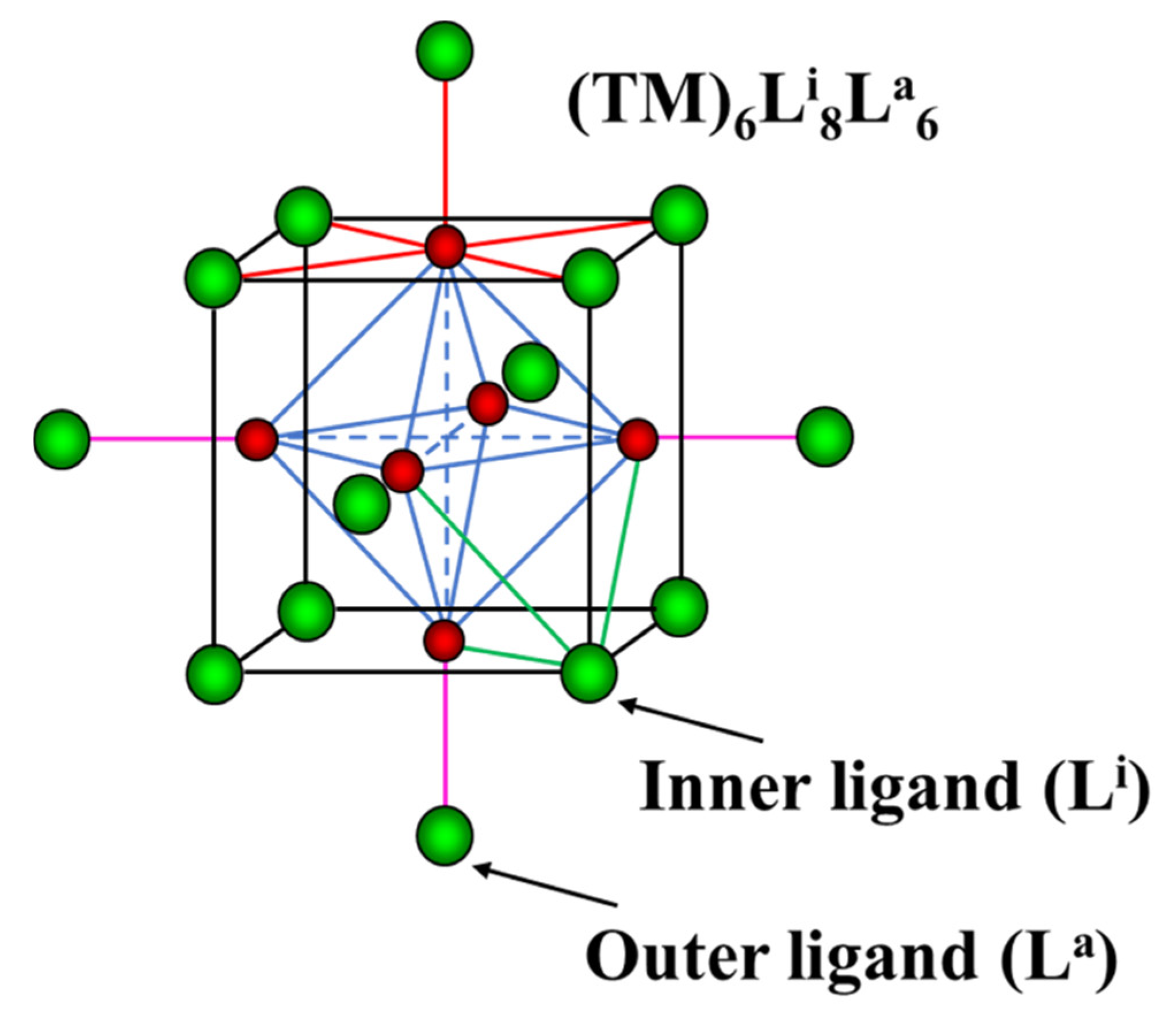
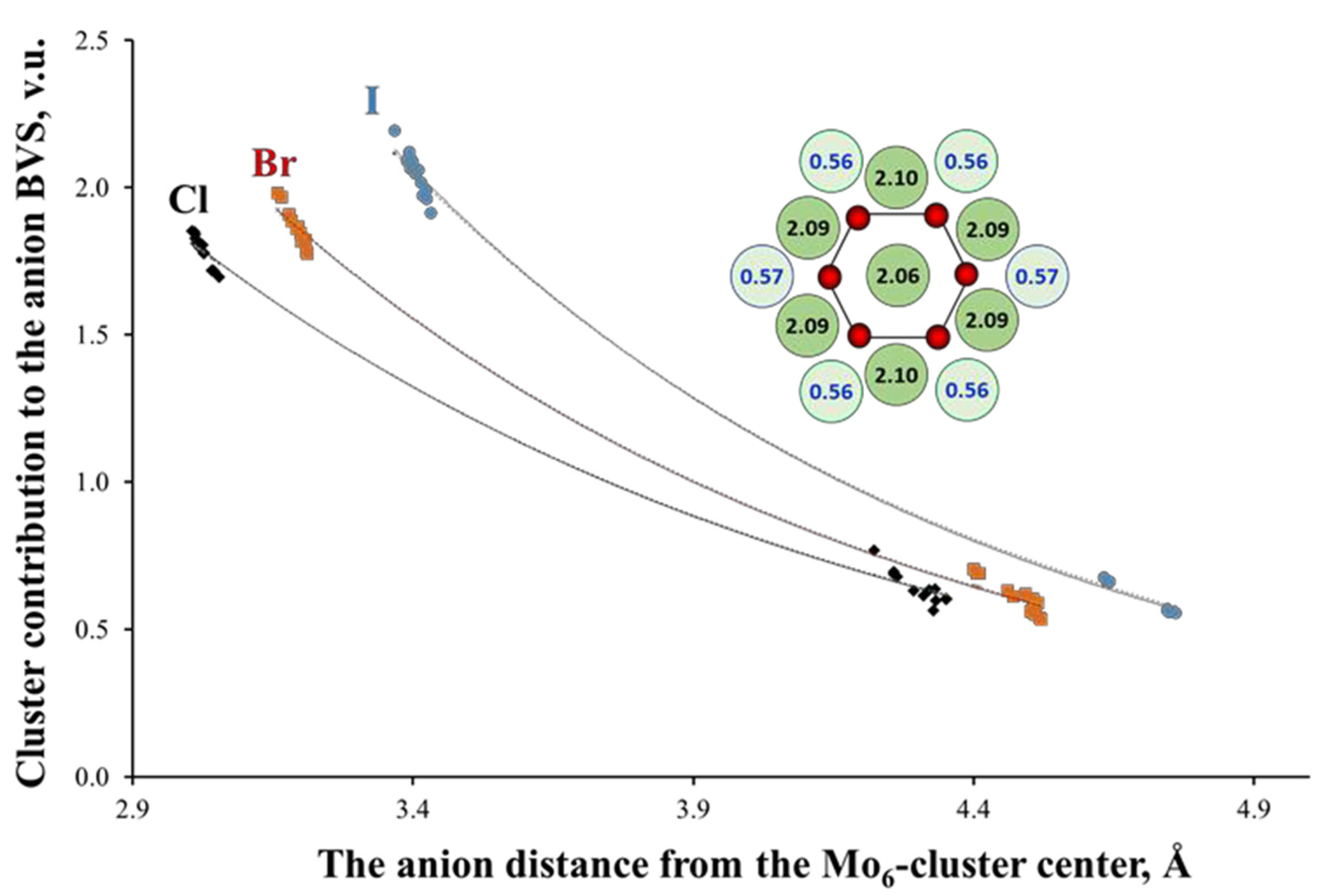
3.3.1. Basic Crystal Chemistry of Cluster Compounds
3.3.2. Rationalization of the Factors Affecting the Redox Potential of Cluster Compounds
- According to Equation (1) proposed by Lever, in organic solvents, the constants, SM and IM, are insensitive to the net charge of the species [9]. Why is this not the case for the compounds under study? In other words, why Equation (1) in our case is not universal for the redox couple of a given metal? In which cases will the redox potential depend solely on the Lever’s parameters?
- Why does the position of a given ligand in the cluster surrounding (inner or outer site) change effectively the redox potential of cluster compounds?
- Which ligand property is responsible of the changes in the effective ionic charge of TM?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Roos, G.; De Proft, F.; Geerlings, P. Electron Capture by the Thiyl Radical and Disulfide Bond: Ligand Effects on the Reduction Potential. Chem. A Eur. J. 2013, 19, 5050–5060. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, T.; Umakoshi, K.; Sasaki, Y.; Ishizaka, S.; Kim, H.B.; Kitamura, N. Emission and Metal- and Ligand-Centered-Redox Characteristics of the Hexarhenium(III) Clusters trans- and cis-[Re6(μ3-S)8Cl4(L)2]2−, Where L Is a Pyridine Derivative or Pyrazine. Inorg. Chem. 2000, 39, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, M.A.; Brylev, K.A.; Abramov, P.A.; Sakuda, E.; Akagi, S.; Ito, A.; Kitamura, N.; Sokolov, M.N. Synthetic Tuning of Redox, Spectroscopic, and Photophysical Properties of {Mo6I8}4+ Core Cluster Complexes by Terminal Carboxylate Ligands. Inorg. Chem. 2016, 55, 8437–8445. [Google Scholar] [CrossRef]
- Goddard, C.A.; Long, J.R.; Holm, R.H. Synthesis and Characterization of Four Consecutive Members of the Five-Member [Fe6S8(PEt3)6]n+ (n = 0−4) Cluster Electron Transfer Series. Inorg. Chem. 1996, 35, 4347–4354. [Google Scholar] [CrossRef]
- Gabriel, J.-P.; Boubekeur, K.; Uriel, S.; Batail, P. Chemistry of Hexanuclear Rhenium Chalcohalide Clusters. Chem. Rev. 2001, 101, 2037–2066. [Google Scholar] [CrossRef]
- Cecconi, F.; Ghilardi, C.A.; Midollini, S.; Orlandini, A.; Zanello, P. Redox behaviour of the iron–sulphur cluster [Fe6(µ3-S)8(PEt3)6]2+. Synthesis and crystal structure of the new paramagnetic monopositive species [Fe6(µ3-S)8(PEt3)6]+ as its [PF6]– salt. J. Chem. Hencec. Dalt. Trans. 1987, 831–835. [Google Scholar] [CrossRef]
- Zietlow, T.C.; Schaefer, W.P.; Sadeghi, B.; Hua, N.; Gray, H.B. Hexanuclear tungsten cluster structures: Tetradecachlorohexatungstate(2-), tetradecabromohexatungstate(2-), and tetradecaiodohexatungstate(2-) relevance to unusual emissive behavior. Inorg. Chem. 1986, 25, 2195–2198. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L. Characterization of Coordination Compounds by Electrochemical Parameters. Eur. J. Inorg. Chem. 2007, 2007, 1473–1482. [Google Scholar] [CrossRef]
- Lever, A.B.P. Electrochemical parametrization of metal complex redox potentials, using the ruthenium(III)/ruthenium(II) couple to generate a ligand electrochemical series. Inorg. Chem. 1990, 29, 1271–1285. [Google Scholar] [CrossRef]
- Szczepura, L.F.; Oh, M.K.; Knott, S.A. Synthesis and electrochemical study of the first tetrazolate hexanuclear rhenium cluster complex. Chem. Commun. 2007, 4617–4619. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y. Recent Progress in the Chemistry of Rhenium Cluster Complexes and Its Relevance to the Prospect of Corresponding Technetium Chemistry. ChemInform 2007, 38. [Google Scholar] [CrossRef]
- Rojas-Poblete, M.; Carreño, A.; Gacitúa, M.; Páez-Hernández, D.; Rabanal-León, W.A.; Arratia-Pérez, R. Electrochemical behaviors and relativistic DFT calculations to understand the terminal ligand influence on the [Re6(μ3-Q)8X6]4− clusters. New J. Chem. 2018, 42, 5471–5478. [Google Scholar] [CrossRef]
- Rabanal-León, W.A.; Murillo-López, J.A.; Páez-Hernández, D.; Arratia-Pérez, R. Understanding the Influence of Terminal Ligands on the Electronic Structure and Bonding Nature in [Re6(μ3-Q8)]2+ Clusters. J. Phys. Chem. A 2014, 118, 11083–11089. [Google Scholar] [CrossRef]
- Roos, B.O.; Borin, A.C.; Gagliardi, L. Reaching the Maximum Multiplicity of the Covalent Chemical Bond. Angew. Chem. Int. Ed. 2007, 46, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Isnard, O. Bond-valence model for metal cluster compounds. II. Matrix effect. Acta Crystallogr. Sect. B 2013, 69, 426–438. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Gatti, C. Metal–Metal Bond in the Light of Pauling’s Rules. Molecules 2021, 26, 304. [Google Scholar] [CrossRef]
- Brown, I.D. The Chemical Bond in Inorganic Chemistry: The Bond Valence Model; International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK, 2016; ISBN 9780191060663. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry; George Fisher Baker Non-Resident Lecture Series; Cornell University Press: Ithaca, NY, USA, 1960; ISBN 9780801403330. [Google Scholar]
- Brown, I.D. Recent Developments in the Methods and Applications of the Bond Valence Model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Dixit, M.; Kosa, M.; Major, D.T.; Levi, E.; Aurbach, D. Is it True That the Normal Valence-Length Correlation Is Irrelevant for Metal–Metal Bonds? Chem. A Eur. J. 2016, 22, 5269–5276. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Gatti, C. Steric and Electrostatic Effects in Compounds with Centered Clusters Quantified by Bond Order Analysis. Cryst. Growth Des. 2020, 20, 2115–2122. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. Sect. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Gatti, C. Bond Order Conservation Principle and Peculiarities of the Metal–Metal Bonding. Inorg. Chem. 2018, 57, 15550–15557. [Google Scholar] [CrossRef]
- Long, J.R.; McCarty, L.S.; Holm, R.H. A Solid-State Route to Molecular Clusters: Access to the Solution Chemistry of [Re6Q8]2+ (Q = S, Se) Core-Containing Clusters via Dimensional Reduction. J. Am. Chem. Hencec. 1996, 118, 4603–4616. [Google Scholar] [CrossRef]
- Orto, P.J.; Nichol, G.S.; Okumura, N.; Evans, D.H.; Arratia-Pérez, R.; Ramirez-Tagle, R.; Wang, R.; Zheng, Z. Cluster carbonyls of the [Re6(μ3-Se)8]2+ core: Synthesis, structural characterization, and computational analysis. Dalton Trans. 2008, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Yamamoto, N.; Nagase, T.; Tsuboi, T.; Kobayashi, K.; Yamagata, T.; Imoto, H.; Unoura, K. Molecular models of the superconducting chevrel phases: Syntheses and structures of [Mo6X8(PEt3)6] and [PPN][Mo6X8(PEt3)6] (X = S, Se; PPN =(Ph3P)2N). Inorg. Chem. 1990, 29, 764–770. [Google Scholar] [CrossRef]
- Yoshimura, T.; Umakoshi, K.; Sasaki, Y.; Sykes, A.G. Synthesis, Structures, and Redox Properties of Octa(μ3-sulfido)hexarhenium(III) Complexes Having Terminal Pyridine Ligands. Inorg. Chem. 1999, 38, 5557–5564. [Google Scholar] [CrossRef]
- Mikhaylov, M.A.; Mironova, A.D.; Brylev, K.A.; Sukhikh, T.S.; Eltsov, I.V.; Stass, D.V.; Gushchin, A.L.; Kitamura, N.; Sokolov, M.N. Functionalization of [Re6Q8(CN)6]4− clusters by methylation of cyanide ligands. New J. Chem. 2019, 43, 16338–16348. [Google Scholar] [CrossRef]
- Kirakci, K.; Kubát, P.; Langmaier, J.; Polívka, T.; Fuciman, M.; Fejfarová, K.; Lang, K. A comparative study of the redox and excited state properties of (nBu4N)2[Mo6X14] and (nBu4N)2[Mo6X8(CF3COO)6] (X = Cl, Br, or I). Dalton Trans. 2013, 42, 7224–7232. [Google Scholar] [CrossRef]
- Ebihara, M.; Toriumi, K.; Saito, K. Syntheses and properties of monochalcogenide-substituted hexamolybdenum halide clusters. Inorg. Chem. 1988, 27, 13–18. [Google Scholar] [CrossRef]
- Jackson, J.A.; Turro, C.; Newsham, M.D.; Nocera, D.G. Oxygen quenching of electronically excited hexanuclear molybdenum and tungsten halide clusters. J. Phys. Chem. 1990, 94, 4500–4507. [Google Scholar] [CrossRef]
- Brown, I.D. Chemical and steric constraints in inorganic solids. Acta Crystallogr. Sect. B 1992, 48, 553–572. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Gatti, C. A revisit of the bond valence model makes it universal. Phys. Chem. Chem. Phys. 2020, 22, 13839–13849. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Gatti, C. Do the basic crystal chemistry principles agree with a plethora of recent quantum chemistry data? IUCrJ 2018, 5, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Isnard, O. Electronic Effect Related to the Nonuniform Distribution of Ionic Charges in Metal–Cluster Chalcogenide Halides. Eur. J. Inorg. Chem. 2014, 2014, 3736–3746. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, E.; Aurbach, D.; Gatti, C. Redox Potential and Crystal Chemistry of Hexanuclear Cluster Compounds. Molecules 2021, 26, 3069. https://doi.org/10.3390/molecules26113069
Levi E, Aurbach D, Gatti C. Redox Potential and Crystal Chemistry of Hexanuclear Cluster Compounds. Molecules. 2021; 26(11):3069. https://doi.org/10.3390/molecules26113069
Chicago/Turabian StyleLevi, Elena, Doron Aurbach, and Carlo Gatti. 2021. "Redox Potential and Crystal Chemistry of Hexanuclear Cluster Compounds" Molecules 26, no. 11: 3069. https://doi.org/10.3390/molecules26113069
APA StyleLevi, E., Aurbach, D., & Gatti, C. (2021). Redox Potential and Crystal Chemistry of Hexanuclear Cluster Compounds. Molecules, 26(11), 3069. https://doi.org/10.3390/molecules26113069