
Cephalotaxine Inhibits the Survival of Leukemia Cells by Activating Mitochondrial Apoptosis Pathway and Inhibiting Autophagy Flow



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
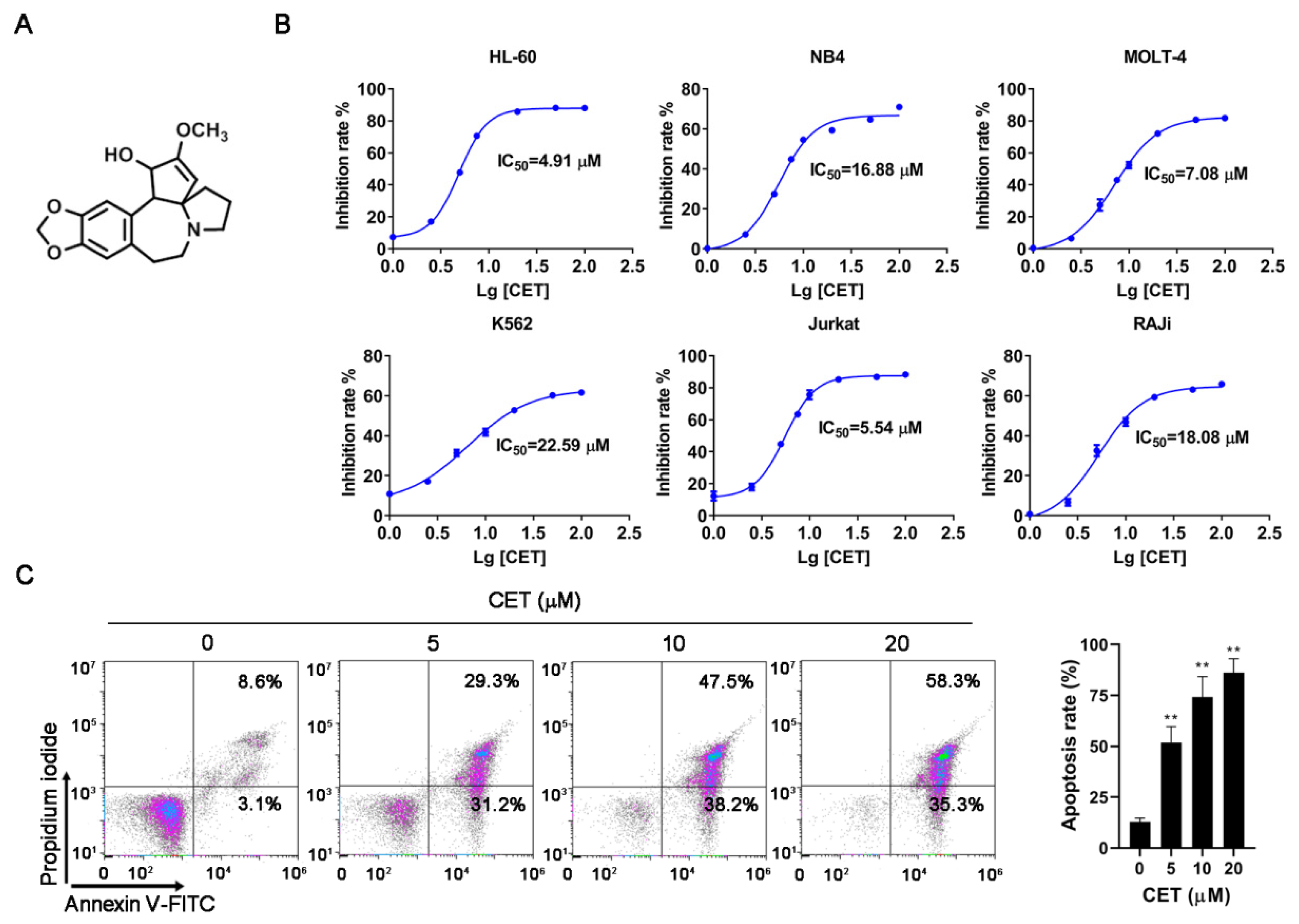
2.1. CET Induces Apoptosis of Leukemia Cells
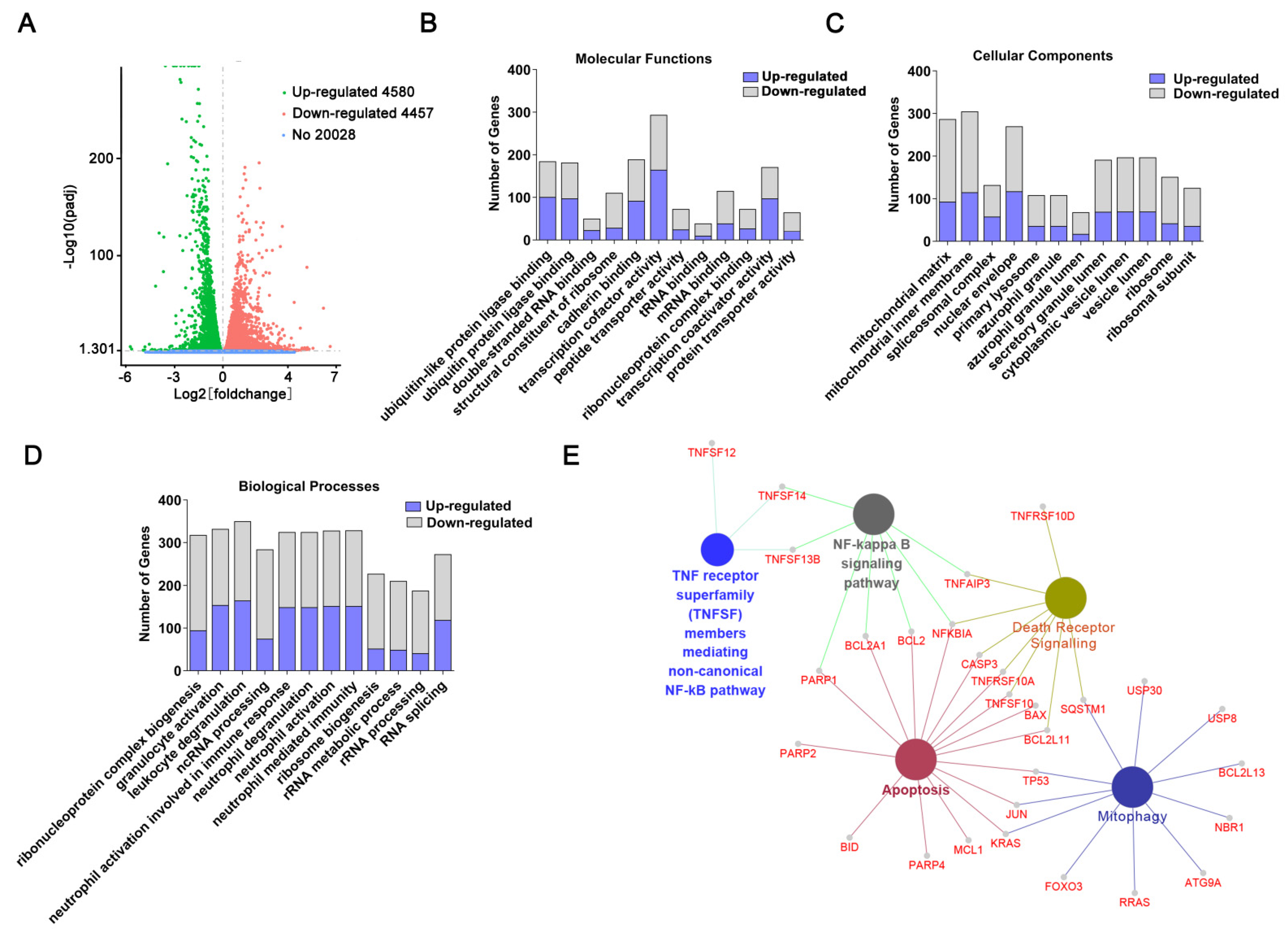
2.2. Transcriptome Sequencing-Coupled with Bioinformatics Analysis Reveals Signaling Networks Regulated by CET
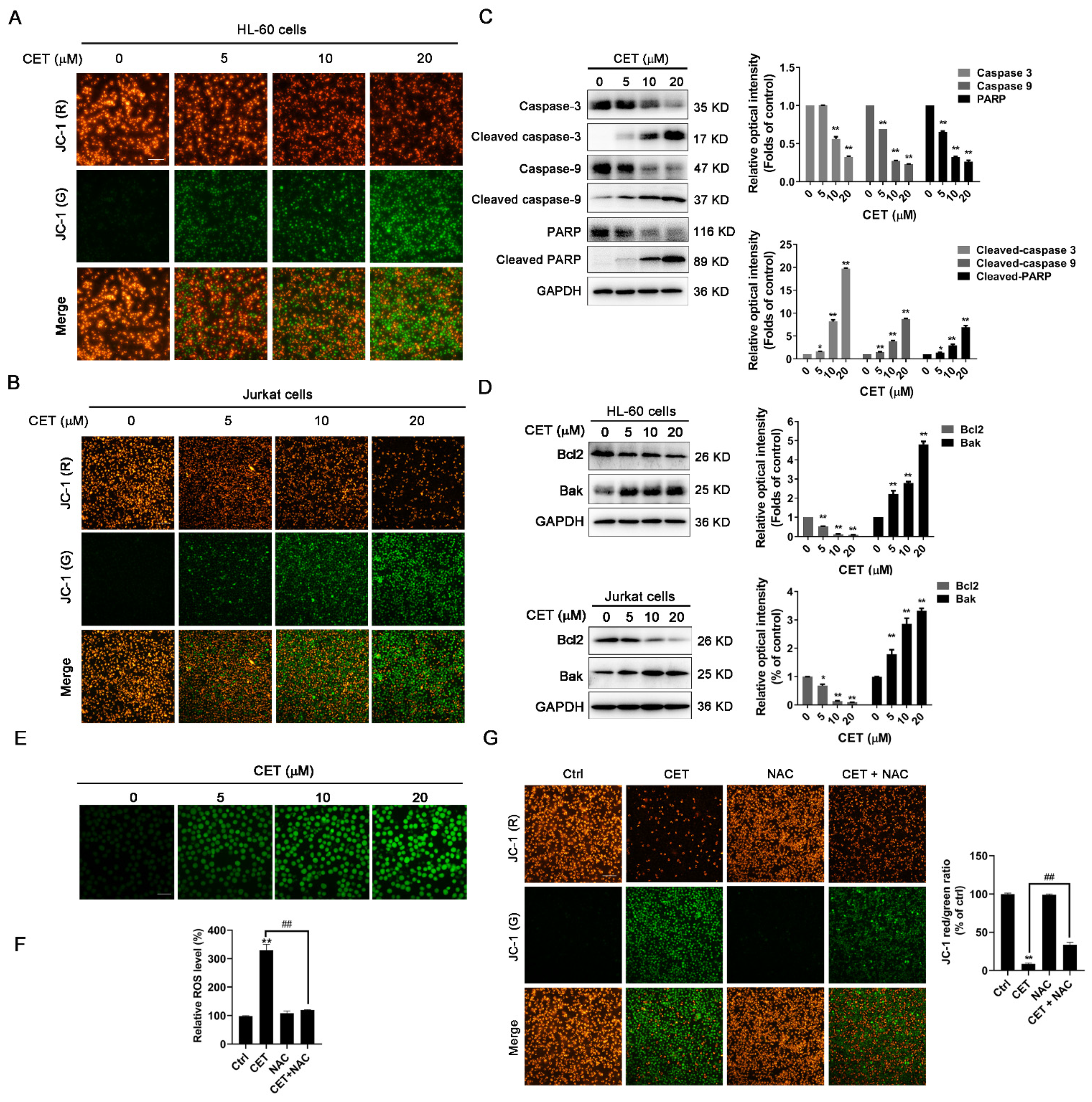
2.3. CET Activates the Mitochondria-Dependent Pathway in Leukemia Cells
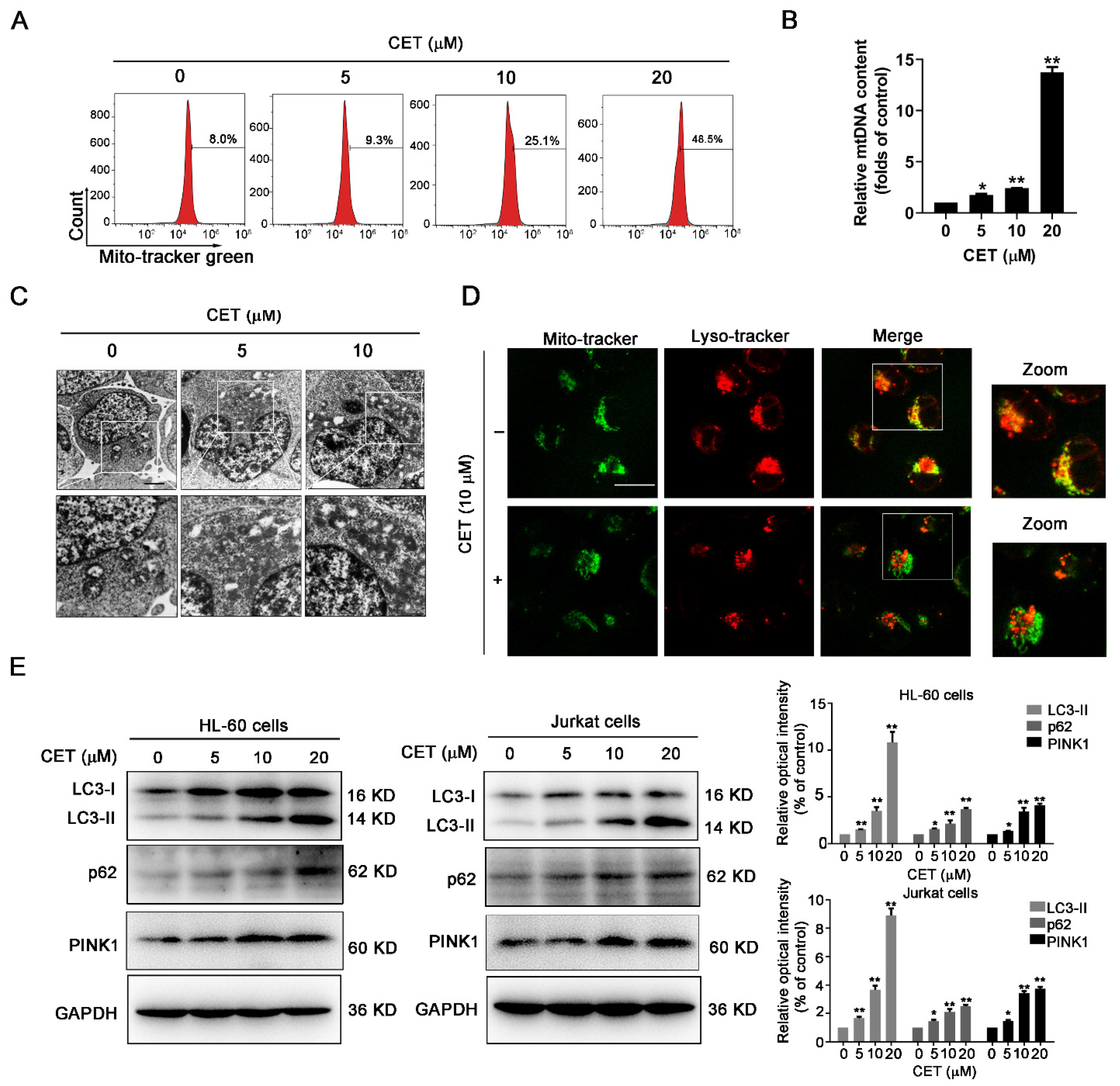
2.4. CET Interferes with Mitochondrial Homeostasis by Regulating Autophagy
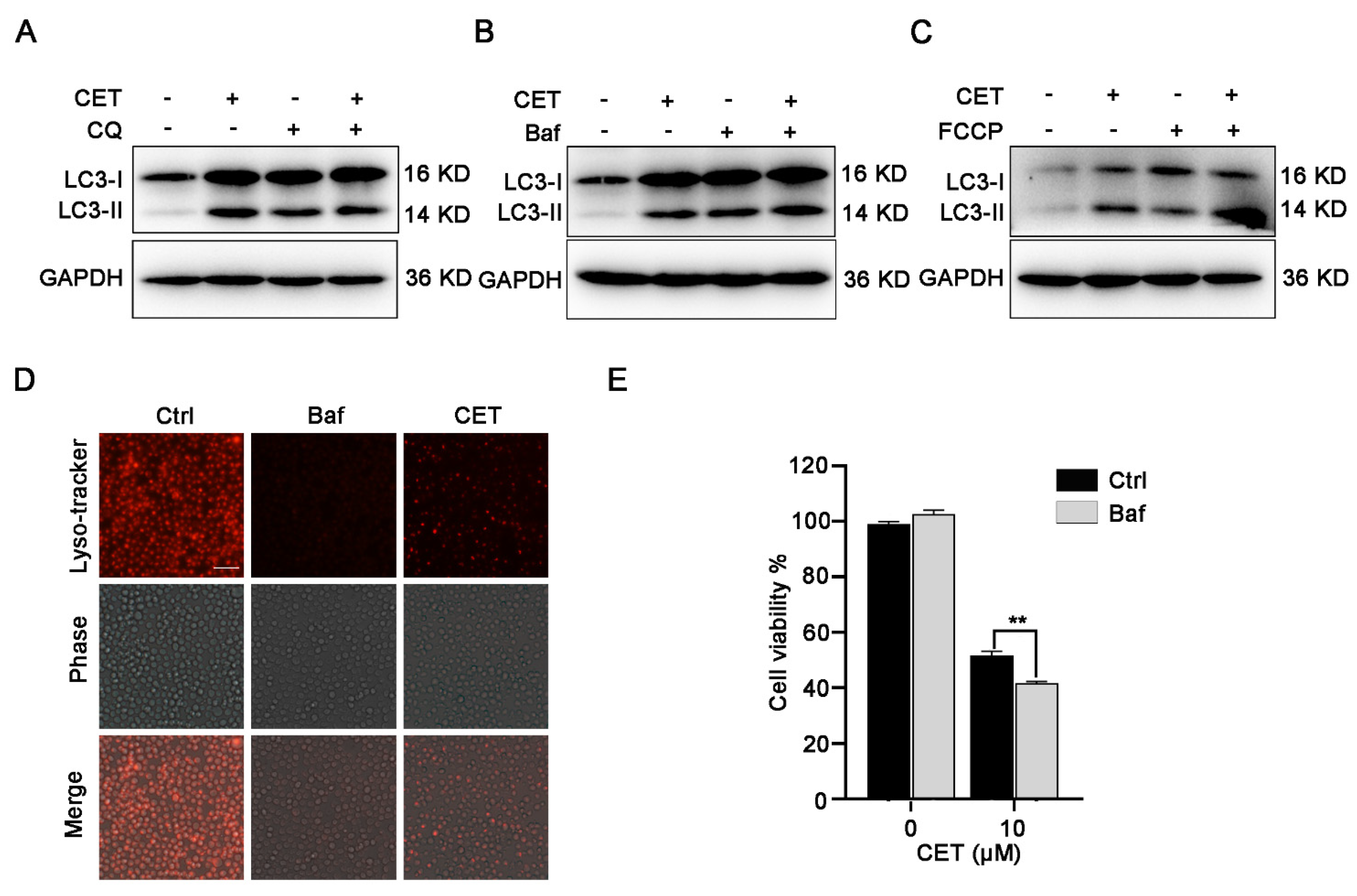
2.5. CET Inhibits Autophagy Flow
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Mitochondrial Membrane Potential
4.5. Measurement of ROS
4.6. Western Blot Analysis
4.7. RNA Sequencing Assay and Bioinformatics Analysis
4.8. Mitochondrial DNA (mtDNA) Analysis
4.9. Flow Cytometry Analysis
4.10. Transmission Electron Microscope (TEM) Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Heendeniya, S.N.; Keerthirathna, L.R.; Manawadu, C.K.; Dissanayake, I.H.; Ali, R.; Mashhour, A.; Alzahrani, H.; Godakumbura, P.; Boudjelal, M.; Peiris, D.C. Therapeutic efficacy of Nyctanthes arbor-tristis flowers to inhibit proliferation of acute and chronic primary human leukemia cells, with adipocyte differentiation and in silico analysis of interactions between survivin protein and selected secondary metabolites. Biomolecules 2020, 10, 165. [Google Scholar] [CrossRef]
- Djunic, I.; Virijevic, M.; Djurasinovic, V.; Novkovic, A.; Colovic, N.; Kraguljac-Kurtovic, N.; Vidovic, A.; Suvajdzic-Vukovic, N.; Tomin, D. Prognostic significance of CD56 antigen expression in patients with acute myeloid leukemia. Med. Oncol. 2012, 29, 2077–2082. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Keating, M.J.; Freireich, E.J. Toward the potential cure of leukemias in the next decade. Cancer 2018, 124, 4301–4313. [Google Scholar] [CrossRef]
- Scarfò, L.; Ferreri, A.J.; Ghia, P. Chronic lymphocytic leukaemia. Crit. Rev. Oncol. Hematol. 2016, 104, 169–182. [Google Scholar] [CrossRef]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Győrffy, B.; Gabrielson, E.; Saxena, N.K.; et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic modulation of autophagy in leukaemia and lymphoma. Cells 2019, 8, 103. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016, 7, e2361. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, V.; Mora, R.; Park, Y.J.; Plass, C.; Chiramel, A.I.; Bartenschlager, R.; Döhner, H.; Stilgenbauer, S.; Pscherer, A.; Lichter, P.; et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Lett. 2012, 72, 1763–1772. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Wang, Z.H.; Zhu, S.C.; Zhang, G.S.; Liu, S.F. Inhibition of autophagy enhances the anticancer activity of bortezomib in B-cell acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2015, 5, 639–650. [Google Scholar] [PubMed]
- Yuan, N.; Lin, S.; Zhang, S.P.; Lin, W.W.; Cao, Y.; Xu, F.; Fang, Y.X.; Wang, Z.; Zhang, H.; Li, X.; et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblas-tic leukemia. Haematologica 2015, 100, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J. Homoharringtonine and omacetaxine for myeloid hematological malignancies. J. Hematol. Oncol. 2014, 7, 2. [Google Scholar] [CrossRef]
- Lai, Z.Z.; Ho, Y.J.; Lu, J.W. Cephalotaxine inhibits Zika infection by impeding viral replication and stability. Biochem. Biophys. Res. Commun. 2020, 522, 1052–1058. [Google Scholar] [CrossRef]
- Romero, M.R.; Serrano, M.A.; Efferth, T.; Alvarez, M.; Marin, J.J. Effect of cantharidin, cephalotaxine and homoharringtonine on “in vitro” models of hepatitis B virus (HBV) and bovine viral diarrhoea virus (BVDV) replication. Planta Med. 2007, 73, 552–558. [Google Scholar] [CrossRef]
- Efferth, T.; Sauerbrey, A.; Halatsch, M.E.; Ross, D.D.; Gebhart, E. Molecular modes of action of cephalotaxine and homoharringtonine from the coniferous tree Cephalotaxus hainanensis in human tumor cell lines. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 367, 56–67. [Google Scholar] [CrossRef]
- He, L.; Gu, K. Tanshinone IIA regulates colorectal cancer apoptosis via attenuation of Parkin-mediated mitophagy by suppressing AMPK/Skp2 pathways. Mol. Med. Rep. 2018, 18, 1692–1703. [Google Scholar] [CrossRef]
- Wei, R.; Cao, J.; Yao, S. Matrine promotes liver cancer cell apoptosis by inhibiting mitophagy and PINK1/Parkin pathways. Cell Stress Chaperones. 2018, 23, 1295–1309. [Google Scholar] [CrossRef]
- León-Aparicio, D.; Salvador, C.; Aparicio-Trejo, O.E.; Briones-Herrera, A.; Pedraza-Chaverri, J.; Vaca, L.; Sampieri, A.; Padilla-Flores, T.; López-González, Z.; León-Contreras, J.C.; et al. Novel potassium channels in kidney mitochondria: The hyperpolarization-activated and cyclic nucleotide-gated HCN channels. Int. J. Mol. Sci. 2019, 20, 4995. [Google Scholar] [CrossRef]
- Li, H.; Pan, Y.; Wu, H.; Yu, S.; Wang, J.; Zheng, J.; Wang, C.; Li, J.; Jiang, J. Inhibition of excessive mitophagy by N-acetyl-L-tryptophan confers hepatoprotection against Ischemia-Reperfusion injury in rats. PeerJ 2020, 8, e8665. [Google Scholar] [CrossRef] [PubMed]
- Messling, S.; Matthias, J.; Xiong, Q.; Fischer, S.; Eichinger, L. The two Dictyostelium discoideum autophagy 8 proteins have distinct autophagic functions. Eur. J. Cell Biol. 2017, 96, 312–324. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Desagher, S.; Martinou, J.C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N. BCL-2 family proteins: Critical checkpoints of apoptotic cell death. Clin. Cancer Res. 2007, 13, 7254–7263. [Google Scholar] [CrossRef]
- Estaquier, J.; Vallette, F.; Vayssiere, J.L.; Mignotte, B. The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 2012, 942, 157–183. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Hadwen, J.; Schock, S.; Mears, A.; Yang, R.; Charron, P.; Zhang, L.; Xi, H.S.; MacKenzie, A. Transcriptomic RNAseq drug screen in cerebrocortical cultures: Toward novel neurogenetic disease therapies. Hum. Mol. Genet. 2018, 27, 3206–3217. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Q.; Dahan, A.; Xue, J.; Wei, L.; Tan, W.; Zhang, G. Transcriptomic analyses reveal the molecular mechanisms of schisandrin B alleviates CCl4-induced liver fibrosis in rats by RNA-sequencing. Chem. Biol. Interact. 2019, 309, 108675. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Flavell, R.A. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8670–8675. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Guo, Q.; Zheng, S.; Liu, Y.; Yang, H.; Zhao, M.; Yao, L.; Zeng, K.; Tu, P. Cephalotaxine Inhibits the Survival of Leukemia Cells by Activating Mitochondrial Apoptosis Pathway and Inhibiting Autophagy Flow. Molecules 2021, 26, 2996. https://doi.org/10.3390/molecules26102996
Liu T, Guo Q, Zheng S, Liu Y, Yang H, Zhao M, Yao L, Zeng K, Tu P. Cephalotaxine Inhibits the Survival of Leukemia Cells by Activating Mitochondrial Apoptosis Pathway and Inhibiting Autophagy Flow. Molecules. 2021; 26(10):2996. https://doi.org/10.3390/molecules26102996
Chicago/Turabian StyleLiu, Tingting, Qiang Guo, Shuze Zheng, Yang Liu, Heng Yang, Meimei Zhao, Lu Yao, Kewu Zeng, and Pengfei Tu. 2021. "Cephalotaxine Inhibits the Survival of Leukemia Cells by Activating Mitochondrial Apoptosis Pathway and Inhibiting Autophagy Flow" Molecules 26, no. 10: 2996. https://doi.org/10.3390/molecules26102996
APA StyleLiu, T., Guo, Q., Zheng, S., Liu, Y., Yang, H., Zhao, M., Yao, L., Zeng, K., & Tu, P. (2021). Cephalotaxine Inhibits the Survival of Leukemia Cells by Activating Mitochondrial Apoptosis Pathway and Inhibiting Autophagy Flow. Molecules, 26(10), 2996. https://doi.org/10.3390/molecules26102996