
The Neglected Nuclei


{kind=link}
{kind=link}
{kind=link}
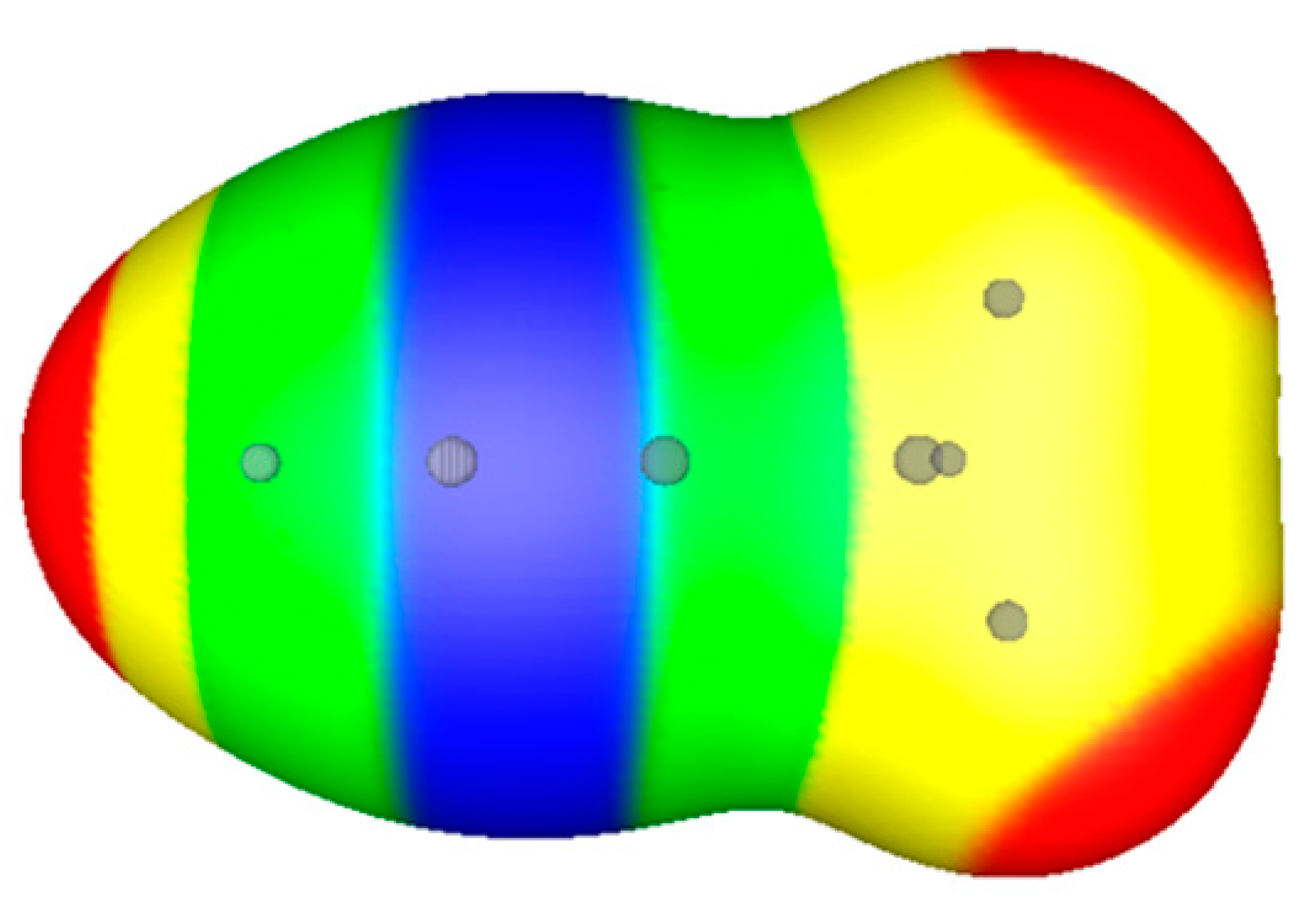{kind=link}
Abstract
1. The Born–Oppenheimer Approximation
2. Electrostatic Potentials and Electric Fields
3. Methods
4. Free Neutral Atoms
5. The Electrostatic Potential Does Not Necessarily Follow the Electronic Density
6. Through-Space Substituent Effects
7. Nitrogen Heterocycles

8. The Nuclear Potential
9. Energy Relationships
10. Discussion and Summary
- (1)
- Electrostatic potentials do not always follow the electronic density in a molecule. The potentials created by the nuclei must also be considered. Regions of high electronic density may have negative electrostatic potentials, but this is not necessarily the case;
- (2)
- Because of the nuclear contributions, the electrostatic potentials in certain regions may sometimes contradict what would be anticipated from electronegativities and electron donating or withdrawing tendencies of substituents and heteroatoms. This does not invalidate these tendencies; it just means that nuclear effects need to be explicitly considered;
- (3)
- The contours of the electronic density are frequently similar to the contours of the electrostatic potential due to the nuclei alone;
- (4)
- The electrostatic potential at a nucleus that is created by the electronic density and the other nuclei of a molecule is characteristic of that nucleus and varies very little with the molecular, atomic or ionic environment of that nucleus; and
- (5)
- Atomic and molecular energies can be expressed rigorously in terms of the electrostatic potentials at the nuclei of their constituent atoms. Electron–electron repulsion, including correlation, is taken into account implicitly rather than explicitly.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Born, M.; Oppenheimer, J.R. Zur Quantentheorie der Molekeln. Ann. Phys. 1927, 389, 457–484. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry; Wiley: Chichester, UK, 2002; pp. 100–101. [Google Scholar]
- Stewart, R.F. On the mapping of electrostatic properties from Bragg diffraction data. Chem. Phys. Lett. 1979, 65, 335–342. [Google Scholar] [CrossRef]
- Politzer, P.; Truhlar, D.G. (Eds.) Chemical Applications of Atomic and Molecular Potentials; Plenum Press: New York, NY, USA, 1981. [Google Scholar]
- Klein, C.L.; Stevens, E.D. Charge density studies of drug molecules. In Structure and Reactivity; Liebman, J.F., Greenberg, A., Eds.; VCH Publishers: New York, NY, USA, 1988; pp. 26–64. [Google Scholar]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Mathematical modeling and physical reality in noncovalent interactions. J. Mol. Model. 2015, 21, 52. [Google Scholar] [CrossRef]
- Clark, T.; Politzer, P.; Murray, J.S. Correct electrostatic treatment of noncovalent interactions: The importance of polarization. WIREs Comput. Mol. Sci. 2015, 5, 169–177. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surface properties: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Delgado-Barrio, G.; Prat, R.F. Deformed Hartree-Fock solutions for atoms. III. Convergent iterative process and results for O−. Phys. Rev. A 1975, 12, 2288–2297. [Google Scholar] [CrossRef]
- Sen, K.D.; Politzer, P. Characteristic features of the electrostatic potentials of singly negative monoatomic ions. J. Chem. Phys. 1989, 90, 4370–4372. [Google Scholar] [CrossRef]
- Feynman, R.P. Forces in molecules. Phys. Rev. 1939, 56, 340–343. [Google Scholar] [CrossRef]
- Hirschfelder, J.O.; Eliason, M.A. Electrostatic Hellmann-Feynman theorem applied to the J. long-range interaction of two hydrogen atoms. J. Chem. Phys. 1967, 47, 1164–1169. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Chandra, A.K. A view of bond formation in terms of molecular charge densities. Can. J. Chem. 1968, 46, 953–966. [Google Scholar] [CrossRef]
- Hunt, K.L.C. Dispersion dipoles and dispersion forces: Proof of Feynman’s “conjecture” and generalization to interacting molecules of arbitrary symmetry. J. Chem. Phys. 1990, 92, 1180–1187. [Google Scholar] [CrossRef]
- Johnson, R.D., III. (Ed.) NIST Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database No. 101. Available online: http://cccbdb.nist.gov/ (accessed on 21 August 2020).
- Politzer, P.; Murray, J.S. Molecular electrostatic potentials and chemical reactivity. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; VCH Publishers: New York, NY, USA, 1991; Volume 2, pp. 273–312. [Google Scholar]
- Shusterman, G.P.; Shusterman, A.J. Teaching chemistry with electron density models. J. Chem. Educ. 1997, 74, 771–776. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. Unexpected substituent effects in face-to-face π-stacking interactions. J. Phys. Chem. A 2003, 107, 8377–8379. [Google Scholar] [CrossRef]
- Gorteau, V.; Bollot, G.; Mareda, J.; Perez-Velasco, A.; Matile, S. Rigid oligonaphthalenediimide rods as transmembrane anion-π slides. J. Am. Chem. Soc. 2006, 128, 14788–14789. [Google Scholar] [CrossRef]
- Laughrey, Z.R.; Kiehna, S.E.; Riemen, A.J.; Waters, M.L. Carbohydrate-π interactions: What are they worth? J. Am. Chem. Soc. 2008, 130, 14625–14633. [Google Scholar] [CrossRef]
- Ringer, A.L.; Sherrilll, C.D. Substituent effects in sandwich configurations of multiply substituted benzene dimers are not solely governed by electrostatic control. J. Am. Chem. Soc. 2009, 131, 4574–4575. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Through-space effects of substituents dominate molecular electrostatic potentials of substituted arenes. J. Chem. Theory Comput. 2009, 5, 2301–2312. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Are anion/π interactions actually a case of simple charge-dipole interactions? J. Phys. Chem. A 2010, 114, 8658–8664. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole interactions: Perspectives and misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Politzer, P.; Kasten, S.D. Analysis of the charge distributions in molecules of the types XCCH and XCN. J. Phys. Chem. 1976, 80, 283–287. [Google Scholar] [CrossRef]
- Exner, O. Correlation Analysis of Chemcal Data; Plenum Press: New York, NY, USA, 1988. [Google Scholar]
- Wheeler, S.E.; Houk, K.N. Substituent effects in cation/π interactions and electrostatic potentials above the centers of substituted benzenes are due primarily to through-space effects of the substituents. J. Am. Chem. Soc. 2009, 131, 3126–3127. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Shields, Z.P.-I.; Seybold, P.G.; Politzer, P. Intuitive and counterintuitive noncovalent interactions of aromatic π regions with the hydrogen and the nitrogen of HCN. J. Comput. Sci. 2015, 10, 209–216. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Bloom, J.W.G. Anion-π interactions and positive electrostatic potentials of N-heterocycles arise from the positions of the nuclei, not changes in the π-electron distribution. Chem. Commun. 2014, 50, 11118–11121. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Murray, J.S. Computational analysis of relative stabilities of polyazine N-oxides. Struct. Chem. 2013, 24, 1965–1974. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989; p. 51. [Google Scholar]
- Parr, R.G.; Berk, A. The bare-nuclear potential as harbinger for the electron density in a molecule. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Politzer, P., Truhlar, D.G., Eds.; Plenum Press: New York, NY, USA, 1981; pp. 51–62. [Google Scholar]
- Politzer, P.; Zilles, B.A. Some observations concerning electronic densities, electrostatic potentials and chemical potentials. Croat. Chem. Acta 1984, 57, 1055–1064. [Google Scholar]
- Tal, Y.; Bader, R.F.W.; Erkku, J. Structural homeomorphism between the electronic charge density and the nuclear potential of a molecular system. Phys. Rev. A 1980, 21, 1–11. [Google Scholar] [CrossRef]
- Politzer, P. Observations on the Significance o the Electrostatic Potentials at the Nuclei o Atoms and Molecules. Israel J. Chem. 1980, 19, 224–232. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Murray, J.S. The fundamental significance of electrostatic potentials at nuclei. In Reviews of Modern Quantum Chemistry: A Celebration of the Contributions of Robert G. Parr; Sen, K.D., Ed.; World Scientific: Singapore, 2002; Volume 1, pp. 63–84. [Google Scholar]
- Politzer, P.; Murray, J.S. Electrostatic potentials at the nuclei of atoms and molecules. Theor. Chem. Acc. 2021, 140, 7. [Google Scholar] [CrossRef]
- Wilson, E.B., Jr. Four-dimensional electron density function. J. Chem. Phys. 1962, 36, 2232–2233. [Google Scholar] [CrossRef]
- Hellmann, H. Einführung in Die Quantenchemie; Deuticke: Leipzig, Germany, 1937; p. 285. [Google Scholar]
- Politzer, P.; Parr, R.G. Some new energy formulas for atoms and molecules. J. Chem. Phys. 1974, 61, 4258–4262. [Google Scholar] [CrossRef]
- Politzer, P. Atomic and molecular energies as functionals of the electrostatic potential. Theor. Chem. Acc. 2004, 111, 395–399. [Google Scholar] [CrossRef]
- Foldy, L.L. A note on atomic binding energies. Phys. Rev. 1951, 83, 397–399. [Google Scholar] [CrossRef]
- Politzer, P. Atomic and molecular energy and energy difference formulae based upon electrostatic potentials at nuclei. In Single-Particle Density in Physics and Chemistry; March, N.H., Deb, B.M., Eds.; Academic Press: New York, NY, USA, 1987; Chapter 3; pp. 59–72. [Google Scholar]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Levy, M.; Clement, S.C.; Tal, Y. Correlation energies from Hartree-Fock electrostatic potentials at nuclei and generation of electrostatic potentials from asymptotic and zero-order information. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Politzer, P., Truhlar, D.G., Eds.; Plenum Press: New York, NY, USA, 1981; Chapter 3; pp. 29–50. [Google Scholar]
- Levy, M.; Tal, Y.; Clement, S.C. A discontinuous energy-density functional. J. Chem. Phys. 1982, 77, 3140–3147. [Google Scholar] [CrossRef]
- Politzer, P.; Sjoberg, P. A formula for calculating molecular energy differences from electrostatic potentials at nuclei. J. Chem. Phys. 1983, 78, 7008–7009. [Google Scholar] [CrossRef]
- Levy, M.; Tal, Y. Atomic binding energies from fundamental theorems involving the electron density, <r−1>, and the Z-1 perturbation expansion. J. Chem. Phys. 1980, 72, 3416–3417. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Politzer, P.; Murray, J.S. The Neglected Nuclei. Molecules 2021, 26, 2982. https://doi.org/10.3390/molecules26102982
Politzer P, Murray JS. The Neglected Nuclei. Molecules. 2021; 26(10):2982. https://doi.org/10.3390/molecules26102982
Chicago/Turabian StylePolitzer, Peter, and Jane S. Murray. 2021. "The Neglected Nuclei" Molecules 26, no. 10: 2982. https://doi.org/10.3390/molecules26102982
APA StylePolitzer, P., & Murray, J. S. (2021). The Neglected Nuclei. Molecules, 26(10), 2982. https://doi.org/10.3390/molecules26102982