
Strategies for Natural Products Discovery from Uncultured Microorganisms


Abstract
1. Introduction
1.1. Natural Products and Uncultured/Uncharacterized Microbes
1.2. Culture-Independent Tools for the Study of Uncultured Microorganisms Related to NP Accumulation
1.2.1. Evaluation of Microbial Heterogeneity and Abundance in Environmental Samples
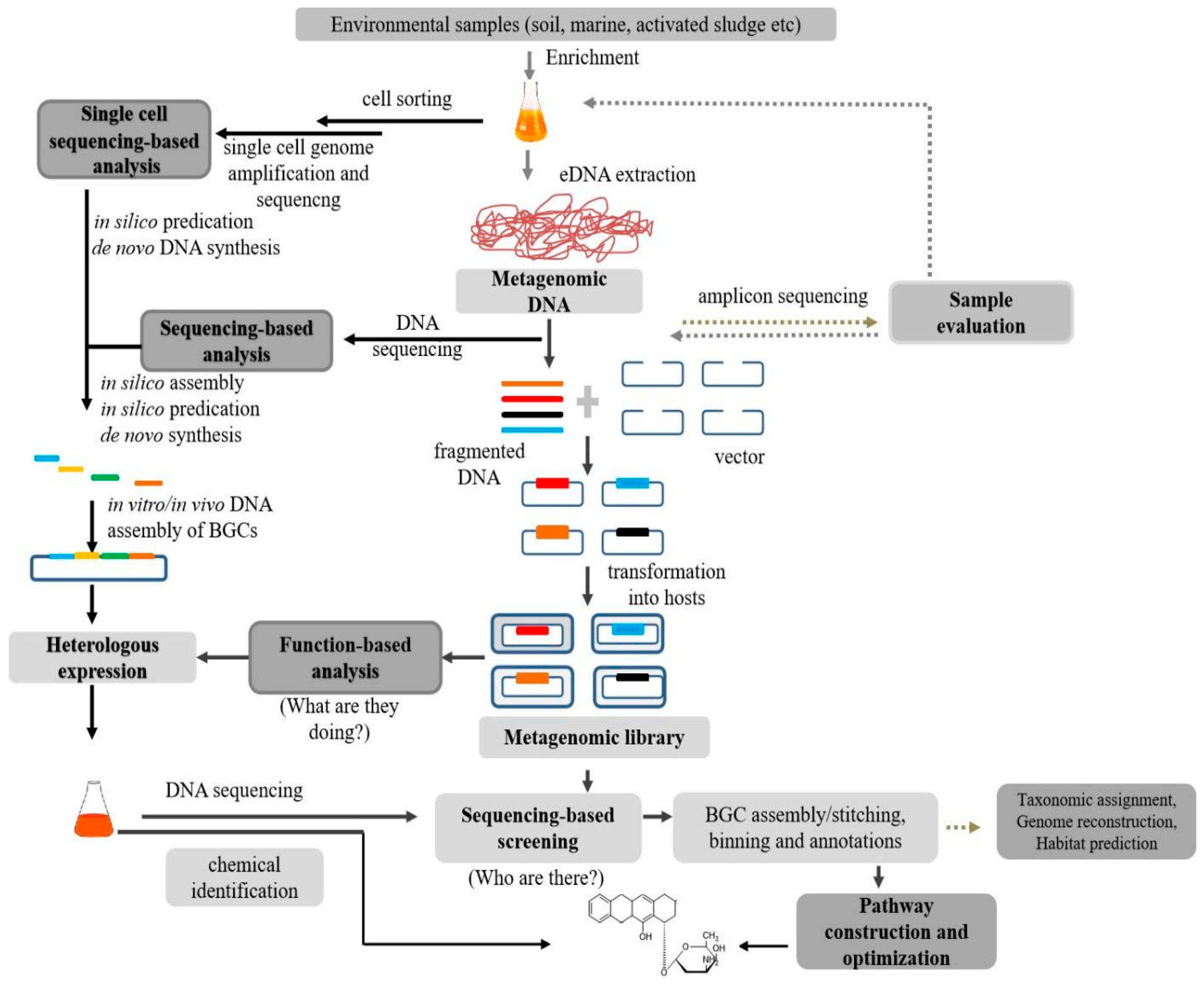
1.2.2. Metagenomics-Related Approaches
2. Function-Based Metagenomics for Exploration of NPs from Different Environmental Samples
2.1. Sample Preparation and Evaluation, Library Construction, and Heterologous Expression
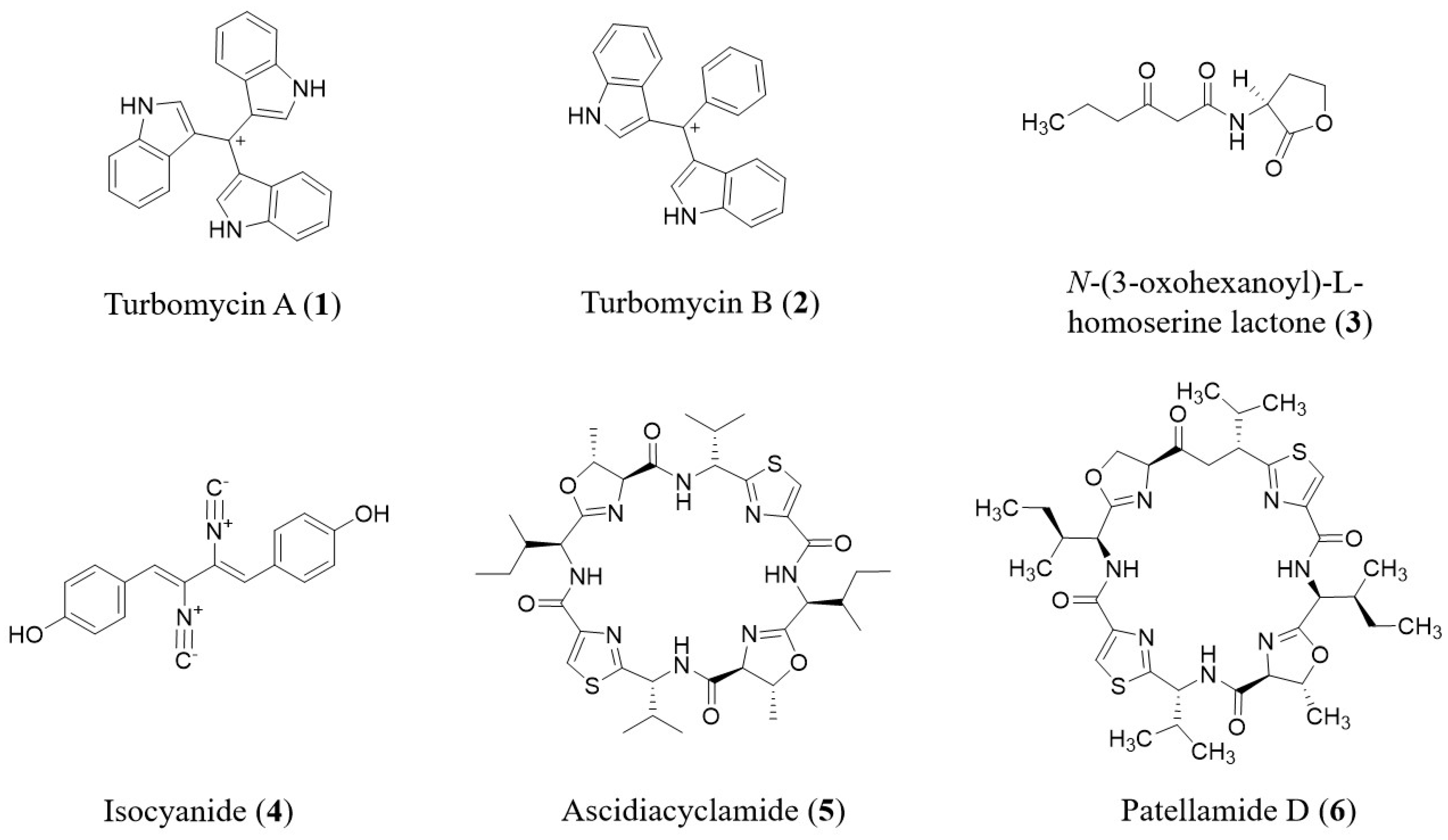
2.2. Function-Based Screening
2.2.1. Approaches for Function-Based Screening
2.2.2. Limitations of Function-Based Screening
3. Sequencing-Based Metagenomics

{kind=link}
{kind=link}
{kind=link}
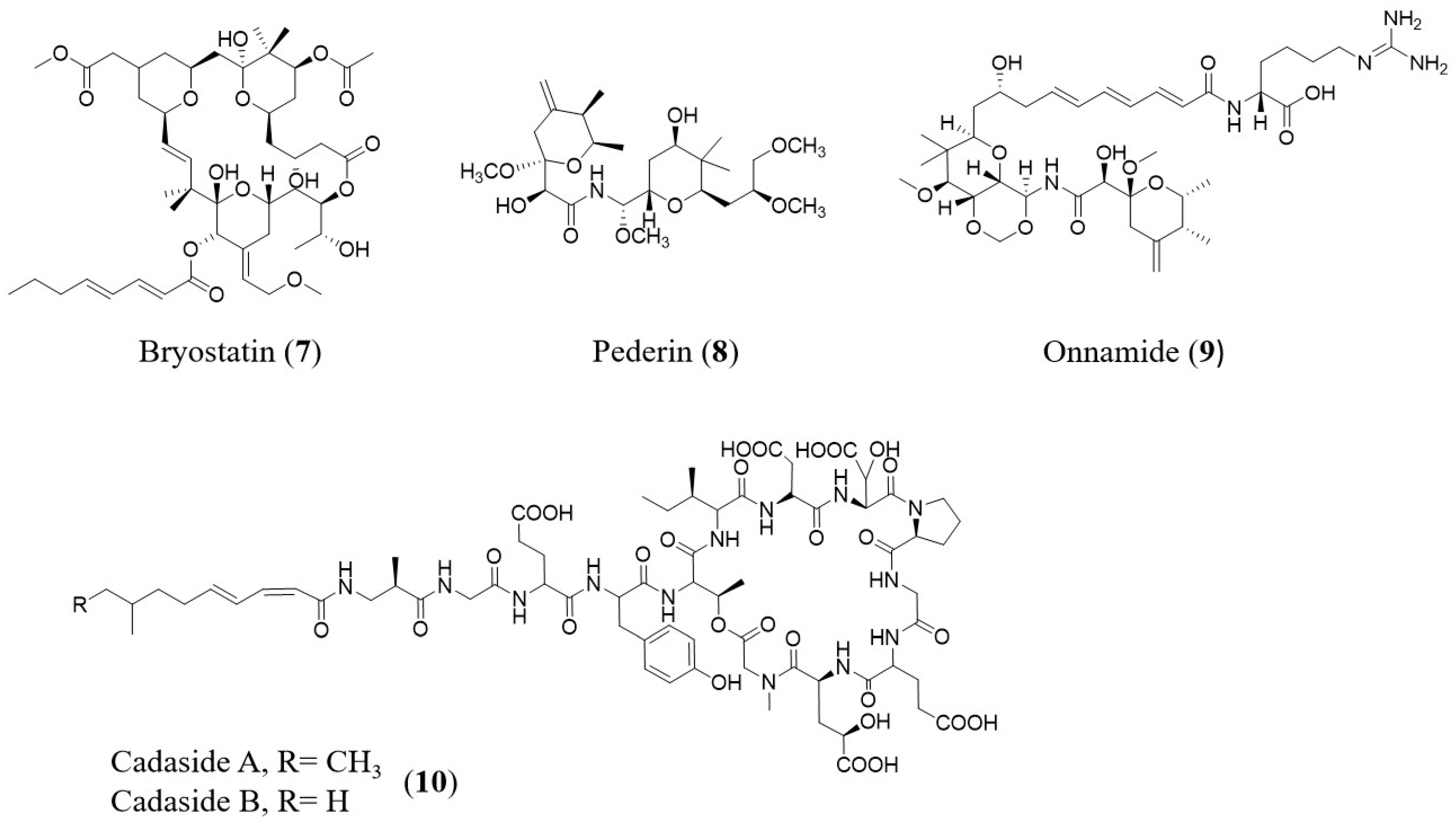
| Compound | Sources | Mode of Action | Reference |
|---|---|---|---|
| Bryostatin # | Bugula neritina | Antitumor | [76] |
| Pederin # | Paederus fuscipes | Antibacterial | [77,78] |
| Onnamide # | Theonella swinhoei | Antitumor | [79] |
| Cadasides | Soil sample | Antibacterial | [72] |
4. Single-Cell Metagenomics
4.1. Cell Sorting and Single-Cell Sequencing
4.2. Screening of NP Gene Clusters in Single-Cell Metagenomics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BGC | Biosynthetic Gene Cluster |
| NP | Natural Products |
| eDNA | Environmental DNA |
| BAC | Bacterial Artificial Chromosome |
| SAG | Single Amplified Genomes |
| NPST | Natural Product Sequence Tags |
| CP | Carrier Protein |
| eSNaPD | Environmental Surveyor of Natural Product Diversity |
| NaPDoS | Natural Product Domain Search |
References
- Kalkreuter, E.; Pan, G.; Cepeda, A.J.; Shen, B. Targeting bacterial genomes for natural product discovery. Trends Pharmacol. Sci. 2020, 41, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Shen, B. A new golden age of natural products drug discovery. Cell 2015, 163, 1297–1300. [Google Scholar] [CrossRef]
- Kim, E.; Moore, B.S.; Yoon, Y.J. Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat. Chem. Biol. 2015, 11, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Smanski, M.J.; Zhou, H.; Claesen, J.; Shen, B.; Fischbach, M.A.; Voigt, C.A. Synthetic biology to access and expand nature’s chemical diversity. Nat. Rev. Microbiol. 2016, 14, 135–149. [Google Scholar] [CrossRef]
- Xu, F.; Wu, Y.; Zhang, C.; Davis, K.M.; Moon, K.; Bushin, L.B.; Seyedsayamdost, M.R. A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat. Chem. Biol. 2019, 15, 161–168. [Google Scholar] [CrossRef]
- Baltz, R.H. Gifted microbes for genome mining and natural product discovery. J. Ind. Microbiol. Biotechnol. 2017, 44, 573–588. [Google Scholar] [CrossRef]
- Wilson, M.C.; Piel, J. Metagenomic approaches for exploiting uncultivated bacteria as a resource for novel biosynthetic enzymology. Chem. Biol. 2013, 20, 636–647. [Google Scholar] [CrossRef]
- Chaudhary, D.K.; Khulan, A.; Kim, J. Development of a novel cultivation technique for uncultured soil bacteria. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of ‘unculturable’bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar]
- Katz, M.; Hover, B.M.; Brady, S.F. Culture-independent discovery of natural products from soil metagenomes. J. Ind. Microbiol. Biotechnol. 2016, 43, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, F. Single-cell metagenomics: Challenges and applications. Protein Cell 2018, 9, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Van Dorst, J.M.; Hince, G.; Snape, I.; Ferrari, B.C. Novel culturing techniques select for heterotrophs and hydrocarbon degraders in a subantarctic soil. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Shi, Z.J.; Seshadri, R.; Pollard, K.S.; Kyrpides, N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature 2019, 568, 505–510. [Google Scholar] [CrossRef]
- Scott, T.A.; Piel, J. The hidden enzymology of bacterial natural product biosynthesis. Nat. Rev. Chem. 2019, 3, 404–425. [Google Scholar] [CrossRef]
- Garrity, G.M. A new genomics-driven taxonomy of Bacteria and Archaea: Are we there yet? J. Clin. Microbiol. 2016, 54, 1956–1963. [Google Scholar] [CrossRef]
- Krishnani, K.K.; Kathiravan, V.; Natarajan, M.; Kailasam, M.; Pillai, S.M. Diversity of sulfur-oxidizing bacteria in greenwater system of coastal aquaculture. Appl. Biochem. Biotechnol. 2010, 162, 1225–1237. [Google Scholar] [CrossRef]
- Hedlund, B.P.; Dodsworth, J.A.; Murugapiran, S.K.; Rinke, C.; Woyke, T. Impact of single-cell genomics and metagenomics on the emerging view of extremophile “microbial dark matter”. Extremophiles 2014, 18, 865–875. [Google Scholar] [CrossRef]
- Madhavan, A.; Sindhu, R.; Parameswaran, B.; Sukumaran, R.K.; Pandey, A. Metagenome analysis: A powerful tool for enzyme bioprospecting. Appl. Biochem. Biotechnol. 2017, 183, 636–651. [Google Scholar] [CrossRef]
- Wei, Y.; Zhang, L.; Zhou, Z.; Yan, X. Diversity of gene clusters for polyketide and nonribosomal peptide biosynthesis revealed by metagenomic analysis of the yellow sea sediment. Front. Microbiol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N. Novel Biological Resources Screened from Uncultured Bacteria by a Metagenomic Method; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128134030. [Google Scholar]
- Lam, K.N.; Cheng, J.; Engel, K.; Neufeld, J.D.; Charles, T.C. Current and future resources for functional metagenomics. Front. Microbiol. 2015, 6, 1196. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Zepeda, A.; Vera-Ponce de León, A.; Sanchez-Flores, A. The road to metagenomics: From microbiology to DNA sequencing technologies and bioinformatics. Front. Genet. 2015, 6, 348. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R. Single cell genomics: An individual look at microbes. Curr. Opin. Microbiol. 2012, 15, 613–620. [Google Scholar] [CrossRef]
- Blainey, P.C. The future is now: Single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 2013, 37, 407–427. [Google Scholar] [CrossRef]
- Blainey, P.C.; Quake, S.R. Dissecting genomic diversity, one cell at a time. Nat. Methods 2014, 11, 19–21. [Google Scholar] [CrossRef]
- Nováková, J.; Farkašovský, M. Bioprospecting microbial metagenome for natural products. Biologia 2013, 68, 1079–1086. [Google Scholar] [CrossRef]
- Dias, R.; Silva, L.C.F.; Eller, M.R.; Oliveira, V.M.; De Paula, S.O.; Silva, C.C. Metagenomics: Library construction and screening methods. V Metagenom. Methods Appl. Perspect. 2014, 5, 28–34. [Google Scholar]
- Benbelgacem, F.F.; Salleh, H.M.; Noorbatcha, I.A. Construction of Metagenomic DNA Libraries and Enrichment Strategies. In Multifaceted Protocol in Biotechnology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 23–42. [Google Scholar]
- Craig, J.W.; Chang, F.-Y.; Kim, J.H.; Obiajulu, S.C.; Brady, S.F. Expanding small-molecule functional metagenomics through parallel screening of broad-host-range cosmid environmental DNA libraries in diverse proteobacteria. Appl. Environ. Microbiol. 2010, 76, 1633–1641. [Google Scholar] [CrossRef]
- Iqbal, H.A.; Low-Beinart, L.; Obiajulu, J.U.; Brady, S.F. Natural Product Discovery through Improved Functional Metagenomics in Streptomyces. J. Am. Chem. Soc. 2016, 138, 9341–9344. [Google Scholar] [CrossRef] [PubMed]
- Loganathachetti, D.S.; Muthuraman, S. Biomedical potential of natural products derived through metagenomic approaches. RSC Adv. 2015, 5, 101200–101213. [Google Scholar] [CrossRef]
- Gaida, S.M.; Sandoval, N.R.; Nicolaou, S.A.; Chen, Y.; Venkataramanan, K.P.; Papoutsakis, E.T. Expression of heterologous sigma factors enables functional screening of metagenomic and heterologous genomic libraries. Nat. Commun. 2015, 6, 7045. [Google Scholar] [CrossRef]
- Martinez, A.; Kolvek, S.J.; Yip, C.L.T.; Hopke, J.; Brown, K.A.; MacNeil, I.A.; Osburne, M.S. Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl. Environ. Microbiol. 2004, 70, 2452–2463. [Google Scholar] [CrossRef]
- Craig, J.W.; Chang, F.-Y.; Brady, S.F. Natural products from environmental DNA hosted in Ralstonia metallidurans. ACS Chem. Biol. 2009, 4, 23–28. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Mirete, S.; Morgante, V.; González-Pastor, J.E. Functional metagenomics of extreme environments. Curr. Opin. Biotechnol. 2016, 38, 143–149. [Google Scholar] [CrossRef]
- Feng, Z.; Chakraborty, D.; Dewell, S.B.; Reddy, B.V.B.; Brady, S.F. Environmental DNA-encoded antibiotics fasamycins A and B inhibit FabF in type II fatty acid biosynthesis. J. Am. Chem. Soc. 2012, 134, 2981–2987. [Google Scholar] [CrossRef]
- Lim, H.K.; Chung, E.J.; Kim, J.-C.; Choi, G.J.; Jang, K.S.; Chung, Y.R.; Cho, K.Y.; Lee, S.-W. Characterization of a forest soil metagenome clone that confers indirubin and indigo production on Escherichia coli. Appl. Environ. Microbiol. 2005, 71, 7768–7777. [Google Scholar] [CrossRef]
- Wang, G.-Y.-S.; Graziani, E.; Waters, B.; Pan, W.; Li, X.; McDermott, J.; Meurer, G.; Saxena, G.; Andersen, R.J.; Davies, J. Novel natural products from soil DNA libraries in a streptomycete host. Org. Lett. 2000, 2, 2401–2404. [Google Scholar] [CrossRef]
- Gillespie, D.E.; Brady, S.F.; Bettermann, A.D.; Cianciotto, N.P.; Liles, M.R.; Rondon, M.R.; Clardy, J.; Goodman, R.M.; Handelsman, J. Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Appl. Environ. Microbiol. 2002, 68, 4301–4306. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.F.; Chao, C.J.; Clardy, J. Long-chain N-acyltyrosine synthases from environmental DNA. Appl. Environ. Microbiol. 2004, 70, 6865–6870. [Google Scholar] [CrossRef]
- Brady, S.F.; Clardy, J. Palmitoylputrescine, an antibiotic isolated from the heterologous expression of DNA extracted from bromeliad tank water. J. Nat. Prod. 2004, 67, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Williamson, L.L.; Borlee, B.R.; Schloss, P.D.; Guan, C.; Allen, H.K.; Handelsman, J. Intracellular screen to identify metagenomic clones that induce or inhibit a quorum-sensing biosensor. Appl. Environ. Microbiol. 2005, 71, 6335–6344. [Google Scholar] [CrossRef]
- Uchiyama, T.; Abe, T.; Ikemura, T.; Watanabe, K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 2005, 23, 88–93. [Google Scholar] [CrossRef]
- Uchiyama, T.; Miyazaki, K. Product-induced gene expression, a product-responsive reporter assay used to screen metagenomic libraries for enzyme-encoding genes. Appl. Environ. Microbiol. 2010, 76, 7029–7035. [Google Scholar] [CrossRef]
- Podar, M.; Abulencia, C.B.; Walcher, M.; Hutchison, D.; Zengler, K.; Garcia, J.A.; Holland, T.; Cotton, D.; Hauser, L.; Keller, M. Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl. Environ. Microbiol. 2007, 73, 3205–3214. [Google Scholar] [CrossRef]
- Nasuno, E.; Kimura, N.; Fujita, M.J.; Nakatsu, C.H.; Kamagata, Y.; Hanada, S. Phylogenetically novel LuxI/LuxR-type quorum sensing systems isolated using a metagenomic approach. Appl. Environ. Microbiol. 2012, 78, 8067–8074. [Google Scholar] [CrossRef]
- Guan, C.; Ju, J.; Borlee, B.R.; Williamson, L.L.; Shen, B.; Raffa, K.F.; Handelsman, J. Signal mimics derived from a metagenomic analysis of the gypsy moth gut microbiota. Appl. Environ. Microbiol. 2007, 73, 3669–3676. [Google Scholar] [CrossRef]
- Meier, M.J.; Paterson, E.S.; Lambert, I.B. Metagenomic analysis of an aromatic hydrocarbon contaminated soil using substrate-induced gene expression. Appl. Environ. Microbiol. 2015. [Google Scholar] [CrossRef]
- Ufarté, L.; Potocki-Veronese, G.; Laville, E. Discovery of new protein families and functions: New challenges in functional metagenomics for biotechnologies and microbial ecology. Front. Microbiol. 2015, 6, 563. [Google Scholar] [CrossRef]
- Colin, P.-Y.; Kintses, B.; Gielen, F.; Miton, C.M.; Fischer, G.; Mohamed, M.F.; Hyvönen, M.; Morgavi, D.P.; Janssen, D.B.; Hollfelder, F. Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics. Nat. Commun. 2015, 6, 10008. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, T.C.; Dostal, S.M.; Griswold, K.E. A high-throughput screen for antibiotic drug discovery. Biotechnol. Bioeng. 2014, 111, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, F.; Hugenholtz, P. Building on basic metagenomics with complementary technologies. Genome Biol. 2007, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Hoshino, Y.; Nishikawa, Y.; Hirose, T.; Yoon, D.H.; Mori, T.; Sekiguchi, T.; Shoji, S.; Takeyama, H. Droplet-based microfluidics for high-throughput screening of a metagenomic library for isolation of microbial enzymes. Biosens. Bioelectron. 2015, 67, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-J.; Chen, G.; Huang, J.; Huang, Q.; Jin, K.; Shen, P.-H.; Li, J.-F.; Wu, B. A novel β-glucosidase with lipolytic activity from a soil metagenome. Folia Microbiol. 2011, 56, 563–570. [Google Scholar] [CrossRef]
- Jiménez, D.J.; Montana, J.S.; Alvarez, D.; Baena, S. A novel cold active esterase derived from Colombian high Andean forest soil metagenome. World J. Microbiol. Biotechnol. 2012, 28, 361–370. [Google Scholar] [CrossRef]
- Bunterngsook, B.; Kanokratana, P.; Thongaram, T.; Tanapongpipat, S.; Uengwetwanit, T.; Rachdawong, S.; Vichitsoonthonkul, T.; Eurwilaichitr, L. Identification and characterization of lipolytic enzymes from a peat-swamp forest soil metagenome. Biosci. Biotechnol. Biochem. 2010, 74, 1848–1854. [Google Scholar] [CrossRef][Green Version]
- Brady, S.F.; Clardy, J. Cloning and heterologous expression of isocyanide biosynthetic genes from environmental DNA. Angew. Chem. Int. Ed. 2005, 44, 7063–7065. [Google Scholar]
- Long, P.F.; Dunlap, W.C.; Battershill, C.N.; Jaspars, M. Shotgun cloning and heterologous expression of the patellamide gene cluster as a strategy to achieving sustained metabolite production. ChemBioChem 2005, 6, 1760–1765. [Google Scholar] [CrossRef]
- Chung, E.J.; Lim, H.K.; Kim, J.-C.; Choi, G.J.; Park, E.J.; Lee, M.H.; Chung, Y.R.; Lee, S.-W. Forest soil metagenome gene cluster involved in antifungal activity expression in Escherichia coli. Appl. Environ. Microbiol. 2008, 74, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.P.; Sloan, W.T.; Scannell, J.W. Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 2002, 99, 10494–10499. [Google Scholar] [CrossRef]
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A new enzyme superfamily—the phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef]
- Pfeifer, B.A.; Admiraal, S.J.; Gramajo, H.; Cane, D.E.; Khosla, C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 2001, 291, 1790–1792. [Google Scholar] [CrossRef]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Brady, S.F. Chemical-biogeographic survey of secondary metabolism in soil. Proc. Natl. Acad. Sci. USA 2014, 111, 3757–3762. [Google Scholar] [CrossRef] [PubMed]
- Charlop-Powers, Z.; Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Guimarães, D.O.; de Frias, U.A.; Pupo, M.T.; Seepe, P.; Feng, Z.; Brady, S.F. Global biogeographic sampling of bacterial secondary metabolism. Elife 2015, 4, e05048. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.B.; Kallifidas, D.; Kim, J.H.; Charlop-Powers, Z.; Feng, Z.; Brady, S.F. Natural product biosynthetic gene diversity in geographically distinct soil microbiomes. Appl. Environ. Microbiol. 2012, 78, 3744–3752. [Google Scholar] [CrossRef]
- Reddy, B.V.B.; Milshteyn, A.; Charlop-powers, Z.; Brady, S.F. Resource eSNaPD: A Versatile, Web-Based Bioinformatics Platform for Surveying and Mining Natural Product Biosynthetic Diversity from Metagenomes. Chem. Biol. 2014, 21, 1023–1033. [Google Scholar] [CrossRef]
- Wu, C.; Shang, Z.; Lemetre, C.; Ternei, M.A.; Brady, S.F. Cadasides, calcium-dependent acidic lipopeptides from the soil metagenome that are active against multidrug-resistant bacteria. J. Am. Chem. Soc. 2019, 141, 3910–3919. [Google Scholar] [CrossRef]
- Stein, J.L.; Marsh, T.L.; Wu, K.Y.; Shizuya, H.; DeLong, E.F. Characterization of uncultivated prokaryotes: Isolation and analysis of a 40-kilobase-pair genome fragment from a planktonic marine archaeon. J. Bacteriol. 1996, 178, 591–599. [Google Scholar] [CrossRef]
- Weinstock, G.M. Genomic approaches to studying the human microbiota. Nature 2012, 489, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Waggoner, L.E.; Liu, H.; Sudek, S.; Allen, S.; Anderson, C.; Sherman, D.H.; Haygood, M. bryA: An unusual modular polyketide synthase gene from the uncultivated bacterial symbiont of the marine bryozoan Bugula neritina. Chem. Biol. 2004, 11, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [Google Scholar] [CrossRef] [PubMed]
- Kačar, D.; Schleissner, C.; Cañedo, L.M.; Rodríguez, P.; de la Calle, F.; Galán, B.; García, J.L. Genome of Labrenzia sp. PHM005 reveals a complete and active trans-AT PKS gene cluster for the biosynthesis of labrenzin. Front. Microbiol. 2019, 10, 2561. [Google Scholar] [CrossRef] [PubMed]
- Piel, J.; Hui, D.; Wen, G.; Butzke, D.; Platzer, M.; Fusetani, N.; Matsunaga, S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef]
- Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Charlop-Powers, Z.; Calle, P.Y.; Kim, J.H.; Brady, S.F. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc. Natl. Acad. Sci. USA 2013, 110, 11797–11802. [Google Scholar] [CrossRef]
- Owen, J.G.; Charlop-Powers, Z.; Smith, A.G.; Ternei, M.A.; Calle, P.Y.; Reddy, B.V.B.; Montiel, D.; Brady, S.F. Multiplexed metagenome mining using short DNA sequence tags facilitates targeted discovery of epoxyketone proteasome inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, 4221–4226. [Google Scholar] [CrossRef]
- Kang, H.; Brady, S.F. Arimetamycin A: Improving clinically relevant families of natural products through sequence-guided screening of soil metagenomes. Angew. Chem. Int. Ed. 2013, 52, 11063–11067. [Google Scholar] [CrossRef]
- Chang, F.-Y.; Ternei, M.A.; Calle, P.Y.; Brady, S.F. Targeted metagenomics: Finding rare tryptophan dimer natural products in the environment. J. Am. Chem. Soc. 2015, 137, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Zotchev, S.B. Marine actinomycetes as an emerging resource for the drug development pipelines. J. Biotechnol. 2012, 158, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Niu, G. Genomics-driven natural product discovery in actinomycetes. Trends Biotechnol. 2018, 36, 238–241. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Hover, B.M.; Kim, S.-H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.H. Discovering antibiotics through soil metagenomics. Nat. Rev. Drug Discov. 2018, 17, 241. [Google Scholar] [CrossRef]
- Nayfach, S.; Pollard, K.S. Toward accurate and quantitative comparative metagenomics. Cell 2016, 166, 1103–1116. [Google Scholar] [CrossRef]
- Abbasi, M.N.; Fu, J.; Bian, X.; Wang, H.; Zhang, Y.; Li, A. Recombineering for Genetic Engineering of Natural Product Biosynthetic Pathways. Trends Biotechnol. 2020, 38, 715–728. [Google Scholar] [CrossRef]
- Mardanov, A.V.; Kadnikov, V.V.; Ravin, N.V. Metagenomics: A paradigm shift in microbiology. In Metagenomics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–13. [Google Scholar]
- Ottesen, E.A.; Hong, J.W.; Quake, S.R.; Leadbetter, J.R. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 2006, 314, 1464–1467. [Google Scholar] [CrossRef]
- Binga, E.K.; Lasken, R.S.; Neufeld, J.D. Something from (almost) nothing: The impact of multiple displacement amplification on microbial ecology. ISME J. 2008, 2, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, T.; Niederkrüger, H.; Gernert, C.; Siegl, A.; Taudien, S.; Platzer, M.; Crews, P.; Hentschel, U.; Piel, J. Linking chemical and microbial diversity in marine sponges: Possible role for Poribacteria as producers of methyl-branched fatty acids. ChemBioChem 2010, 11, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Cahn, J.K.B.; Wilson, M.C.; Meoded, R.A.; Wiebach, V.; Martinez, A.F.C.; Helfrich, E.J.N.; Albersmeier, A.; Wibberg, D.; Dätwyler, S. Single-bacterial genomics validates rich and varied specialized metabolism of uncultivated Entotheonella sponge symbionts. Proc. Natl. Acad. Sci. USA 2018, 115, 1718–1723. [Google Scholar] [CrossRef] [PubMed]
- Siegl, A.; Hentschel, U. PKS and NRPS gene clusters from microbial symbiont cells of marine sponges by whole genome amplification. Environ. Microbiol. Rep. 2010, 2, 507–513. [Google Scholar] [CrossRef]
- Siegl, A.; Kamke, J.; Hochmuth, T.; Piel, J.; Richter, M.; Liang, C.; Dandekar, T.; Hentschel, U. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 2011, 5, 61–70. [Google Scholar] [CrossRef]
- Lu, S.; Zong, C.; Fan, W.; Yang, M.; Li, J.; Chapman, A.R.; Zhu, P.; Hu, X.; Xu, L.; Yan, L. Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 2012, 338, 1627–1630. [Google Scholar] [CrossRef]
- Sha, Y.; Sha, Y.; Ji, Z.; Ding, L.; Zhang, Q.; Ouyang, H.; Lin, S.; Wang, X.; Shao, L.; Shi, C. Comprehensive Genome Profiling of Single Sperm Cells by Multiple Annealing and Looping-Based Amplification Cycles and Next-Generation Sequencing from Carriers of Robertsonian Translocation. Ann. Hum. Genet. 2017, 81, 91–97. [Google Scholar] [CrossRef]
- Lasken, R.S. Single-cell genomic sequencing using multiple displacement amplification. Curr. Opin. Microbiol. 2007, 10, 510–516. [Google Scholar] [CrossRef]
- Piel, J.; Cahn, J. Opening up the Single-Cell Toolbox for Microbial Natural Products Research. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef]
- Fieseler, L.; Horn, M.; Wagner, M.; Hentschel, U. Discovery of the novel candidate phylum “Poribacteria” in marine sponges. Appl. Environ. Microbiol. 2004, 70, 3724–3732. [Google Scholar] [CrossRef]
- Woyke, T.; Tighe, D.; Mavromatis, K.; Clum, A.; Copeland, A.; Schackwitz, W.; Lapidus, A.; Wu, D.; McCutcheon, J.P.; McDonald, B.R. One bacterial cell, one complete genome. PLoS ONE 2010, 5, e10314. [Google Scholar] [CrossRef]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.-F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C. Single-cell genomics of microbial dark matter. In Microbiome Analysis; Springer: Berlin/Heidelberg, Germany, 2018; pp. 99–111. [Google Scholar]
- Wang, L.; Ravichandran, V.; Yin, Y.; Yin, J.; Zhang, Y. Natural products from mammalian gut microbiota. Trends Biotechnol. 2019, 37, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Leclerc, M.C.; Joossens, M.; Mondot, S.; Blottière, H.M.; Raes, J.; Ehrlich, D.; Doré, J. A metagenomic insight into our gut’s microbiome. Gut 2013, 62, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Sudarikov, K.; Tyakht, A.; Alexeev, D. Methods for the metagenomic data visualization and analysis. Curr. Issues Mol. Biol. 2017, 24, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Bitok, J.K.; Lemetre, C.; Ternei, M.A.; Brady, S.F. Identification of biosynthetic gene clusters from metagenomic libraries using PPTase complementation in a Streptomyces host. FEMS Microbiol. Lett. 2017, 364, 1–8. [Google Scholar] [CrossRef]
- Santana-Pereira, A.L.R.; Sandoval-Powers, M.; Monsma, S.; Zhou, J.; Santos, S.R.; Mead, D.A.; Liles, M.R. Discovery of novel biosynthetic gene cluster diversity from a soil metagenomic library. Front. Microbiol. 2020, 11, 585398. [Google Scholar] [CrossRef]
- Ghurye, J.S.; Cepeda-Espinoza, V.; Pop, M. Metagenomic Assembly: Overview, Challenges and Applications. Yale J. Biol. Med. 2016, 89, 353–362. [Google Scholar]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Bhushan, A.; Egli, P.J.; Peters, E.E.; Freeman, M.F.; Piel, J. Genome mining-and synthetic biology-enabled production of hypermodified peptides. Nat. Chem. 2019, 11, 931–939. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, K.; Abbasi, M.N.; Hao, J.; Zhang, Y.; Li, A. Strategies for Natural Products Discovery from Uncultured Microorganisms. Molecules 2021, 26, 2977. https://doi.org/10.3390/molecules26102977
Alam K, Abbasi MN, Hao J, Zhang Y, Li A. Strategies for Natural Products Discovery from Uncultured Microorganisms. Molecules. 2021; 26(10):2977. https://doi.org/10.3390/molecules26102977
Chicago/Turabian StyleAlam, Khorshed, Muhammad Nazeer Abbasi, Jinfang Hao, Youming Zhang, and Aiying Li. 2021. "Strategies for Natural Products Discovery from Uncultured Microorganisms" Molecules 26, no. 10: 2977. https://doi.org/10.3390/molecules26102977
APA StyleAlam, K., Abbasi, M. N., Hao, J., Zhang, Y., & Li, A. (2021). Strategies for Natural Products Discovery from Uncultured Microorganisms. Molecules, 26(10), 2977. https://doi.org/10.3390/molecules26102977