
Preparation of the Key Dolutegravir Intermediate via MgBr2-Promoted Cyclization

Abstract
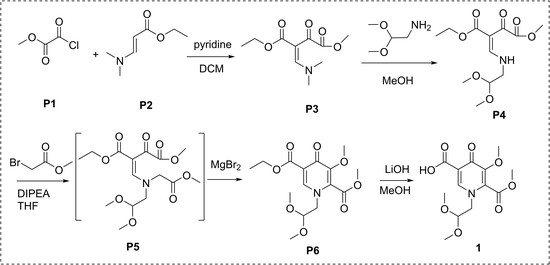
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Instrumentation
3.2. Syntheses
3.2.1. Synthesis of P3
3.2.2. Synthesis of P4
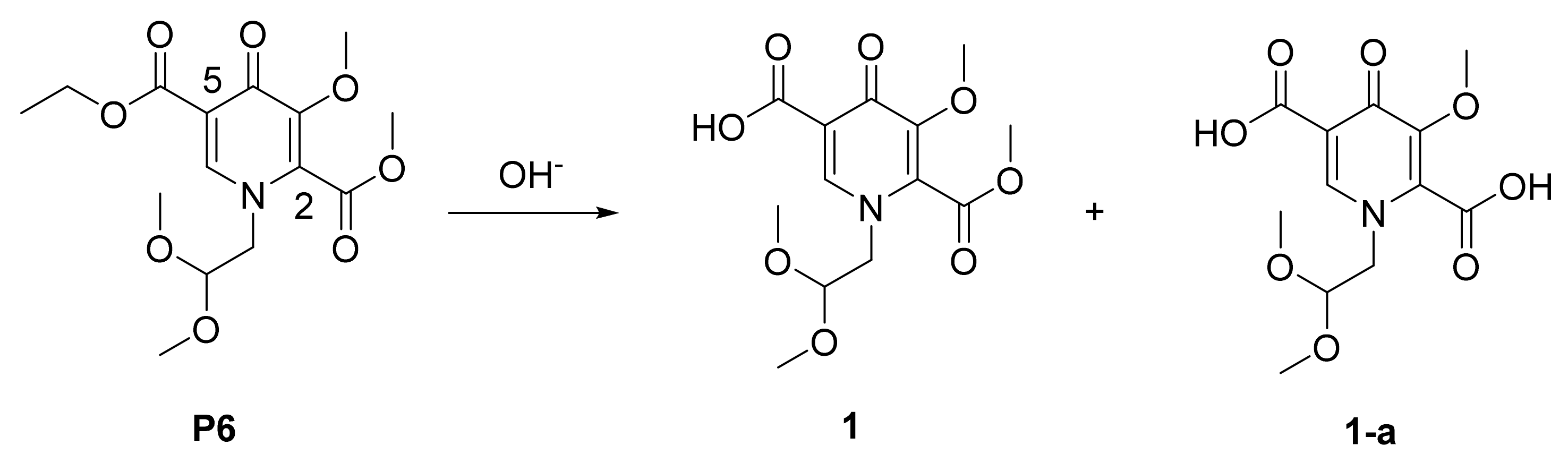
3.2.3. Synthesis of P6
3.2.4. Synthesis of 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vitoria, M.; Hill, A.M.; Ford, N.P.; Doherty, M.; Khoo, S.H.; Pozniak, A.L. Choice of antiretroviral drugs for continued treatment scale-up in a public health approach: What more do we need to know? J. Int. AIDS Soc. 2016, 19, 20504–20512. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.; Wiebe, N.; Smith, N.; Keiser, P.; Naicker, S.; Tonelli, M. Systematic review and meta-analysis: Renal safety of tenofovir disoproxil fumarate in HIV-infected patients. Clin. Infect. Dis. 2010, 51, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.; Andrade-Villanueva, J.; Chetchotisakd, P.; DeJesus, E.; Antunes, F.; Arastah, K. Cobicistat versus ritonavir as a pharmacoenhancer of atazanavir plus emticitabine/tenofovir disoproxil fumarate in treatment-naive HIV type 1-infected patients: Week 48 results. J. Infect. Dis. 2013, 20, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Abuduaini, T.; Roy, V.; Marlet, J.; Gaudy-Graffin, C.; Brand, D.; Baronti, C.; Touret, F.; Coutard, B.; McBrayer, T.R.; Schinazi, R.F.; et al. Synthesis and Antiviral Evaluation of (1,4-Disubstituted-1,2,3-Triazol)-(E)-2-Methyl-but-2-Enyl Nucleoside Phosphonate Prodrugs. Molecules 2021, 26, 1493. [Google Scholar] [CrossRef] [PubMed]
- Harjivan, S.G.; Charneira, C.; Martins, I.L.; Pereira, S.A.; Espadas, G.; Sabidó, E.; Beland, F.A.; Marques, M.M.; Antunes, A.M.M. Covalent Histone Modification by an Electrophilic Derivative of the Anti-HIV Drug Nevirapine. Molecules 2021, 26, 1349. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.; German, P.; Murray, B.; Wei, L.; Jain, A.; West, S.; Warren, D.; Hui, J.; Kearney, B.P. Pharmacokinetics and pharmacodynamics of GS-9350: A novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010, 87, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Johns, B.A.; Kawasuji, T.; Taishi, T.; Taoda, Y. Polycyclic Carbamoylpyridone Derivative Having HIV Integrase Inhibitory Activity. World Patent WO2006/116764A1, 28 April 2006. [Google Scholar]
- Kawasuji, T.; Johns, B.A.; Yoshida, H.; Taishi, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.; Yoshinaga, T.; Seki, T.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 1. Molecular Design and Establishment of an Advanced Two-Metal Binding Pharmacophore. J. Med. Chem. 2012, 55, 8735–8744. [Google Scholar] [CrossRef]
- Kawasuji, T.; Johns, B.A.; Yoshida, H.; Weatherhead, J.G.; Akiyama, T.; Taishi, T.; Taoda, Y.; Mikamiyama-Iwata, M.; Murai, H.; Kiyama, R.; et al. Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles. J. Med. Chem. 2013, 56, 1124–1135. [Google Scholar] [CrossRef]
- Yasunori, A.; Toshikazu, H.; Yuki, F.; Daisuke, Y.; Takao, O.; Yutaka, N.; Shoji, S.; Masahiko, N.; Naoki, M.; Yoshiyuki, T.; et al. Practical and Scalable Synthetic Method for Preparation of Dolutegravir Sodium: Improvement of a Synthetic Route for Large-Scale Synthesis. Org. Process. Res. Dev. 2019, 23, 558–564. [Google Scholar]
- Wang, H.; Kowalski, M.D.; Lakdawala, A.S.; Vogt, F.G.; Wu, L. An Efficient and Highly Diastereoselective Synthesis of GSK1265744, a Potent HIV Integrase Inhibitor. Org. Lett. 2015, 17, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, R.E.; Desai, B.K.; Jee, J.-A.; Gupton, B.F.; Roper, T.D.; Jamison, T.F. 7-Step Flow Synthesis of the HIV Integrase Inhibitor Dolutegravir. Angew. Chem. Int. Ed. 2018, 57, 7181–7185. [Google Scholar] [CrossRef] [PubMed]
- Tatsuro, Y.; Moriyasu, M.; Fumiya, I.; Kazuya, O.; Takanori, K.; Masahiko, N.; Yoshihide, S.; Naoki, M.; Shinichiro, H.; You, A.; et al. Practical Synthetic Method for the Preparation of Pyrone Diesters: An Efficient Synthetic Route for the Synthesis of Dolutegravir Sodium. Org. Process. Res. Dev. 2019, 23, 565–570. [Google Scholar]
- Ren, L. Synthesis Method of Dolutegravir Intermediate, and Related Substance Detection Method Thereof. Chinese Patent CN108101838A, 18 December 2017. [Google Scholar]
- Sumino, Y.; Masui, M.; Yamada, D.; Ikarashi, F.; Okamoto, K. Process for Preparing Compound Having HIV Integrase Inhibitory Activity. U.S. Scheme1 Change Colum Column. Patent Application 20140011995 A1, 9 January 2014. [Google Scholar]
- Sankareswaran, S.; Mannam, M.; Chakka, V.; Mandapati, S.R.; Kumar, P. Identification and Control of Critical Process Impurities: An Improved Process for the Preparation of Dolutegravir Sodium. Org. Process. Res. Dev. 2016, 20, 1461–1468. [Google Scholar] [CrossRef]
- Dandala, R. Process for the Preparation of Dolutegravir and Intermediates Thereof. World Patent WO2015/019310A1, 2 February 2015. [Google Scholar]
- David, L.H. Review of Synthetic Routes and Final Forms of Integrase Inhibitors Dolutegravir, Cabotegravir, and Bictegravir. Org. Process. Res. Dev. 2019, 23, 716–729. [Google Scholar]
- Wang, H. Post-Treatment Method for Continuous Synthesis of Methyl 4-Chloroacetoacetate. Chinese Patent CN111978182A, 31 August 2020. [Google Scholar]
- Kurz, G.; Camacho, G.J. Preparation of Novel Dihydropyridine Derivatives as Androgen Receptor and Glucocorticoid Receptor Modulators. Patent Application WO2019086720, 6 November 2017. [Google Scholar]
- Mercedes, T.; Salvador, G.; Margarita, P. New Synthetic Methods to 2-Pyridone Rings. Curr. Org. Chem. 2005, 9, 1757–1779. [Google Scholar]
- Gianluigi, A.; Laura, A.A. From Alkynes to Heterocycles through Metal-Promoted Silylformylation and Silylcarbocyclization Reactions. Catalysts 2020, 10, 1012. [Google Scholar]
- Preeti; Krishna, N.S. Metal-Free Multicomponent Reactions: A Benign Access to Monocyclic Six-membered N-Heterocycles. Org. Biomol. Chem. 2021, 19, 2622–2657. [Google Scholar] [CrossRef] [PubMed]
- Simonetta, B.; Romeo, R.; Carmela, D.R.; Giampiero, S.; Vinicio, Z. Mastering. beta.-Keto Esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Base | Temperature | Yield |
|---|---|---|---|
| 1 | TEA (1.2 equiv) | −5 °C | 56% |
| 2 | Pyridine (1.2 equiv) | −5 °C | 95% |
| 3 | Pyridine (1.2 equiv) | −10 °C | 90% |
| 4 | Pyridine (1.2 equiv) | 8 °C | 79% |
| 5 | NaOCH3 (1.2 equiv) | −5 °C | 36% |
| 6 | Mg(OCH3)2 (0.6 equiv) | −5 °C | 85% |
| Entry | Base | Additive | Yield a | |
|---|---|---|---|---|
| P6 | P6-Isos | |||
| 1 | TEA (0.5 equiv) | \ | 10% | 25% |
| 2 | DIPEA (1.0 equiv) | \ | 20% | 25% |
| 3 | sodium tert-butoxide (1.0 equiv) | \ | 2% | 2% |
| 4 | DIPEA (1.0 equiv) | MgBr2 | 50% | 10% |
| 5 | DIPEA (1.0 equiv) | MgCl2 | 38% | 15% |
| 6 | DIPEA (1.0 equiv) | MgI2 | 51% | 10% |
| 7 | DIPEA (1.0 equiv) | MgSO4 | 23% | 20% |
| 8 | Mg(OCH3)2 (0.5 equiv) | \ | 10% | 10% |
| Entry | Base | Temperature | Yield | |
|---|---|---|---|---|
| 1 | 1-a | |||
| 1 | NaOH (2.0 equiv) | 0 °C | 62% | 32% |
| 2 | KOH (2.0 equiv) | 0 °C | 59% | 35% |
| 3 | LiOH (2.0 equiv) | 0 °C | 90% | 8% |
| 4 * | LiOH (2.0 equiv) | −10 °C | 91% | 7% |
| 5 | LiOH (2.0 equiv) | 10 °C | 82% | 15% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, J.; Xia, H.; He, R.; Chen, H.; Yu, Y. Preparation of the Key Dolutegravir Intermediate via MgBr2-Promoted Cyclization. Molecules 2021, 26, 2850. https://doi.org/10.3390/molecules26102850
Kong J, Xia H, He R, Chen H, Yu Y. Preparation of the Key Dolutegravir Intermediate via MgBr2-Promoted Cyclization. Molecules. 2021; 26(10):2850. https://doi.org/10.3390/molecules26102850
Chicago/Turabian StyleKong, Jiahui, Haijian Xia, Renbao He, Hao Chen, and Yongping Yu. 2021. "Preparation of the Key Dolutegravir Intermediate via MgBr2-Promoted Cyclization" Molecules 26, no. 10: 2850. https://doi.org/10.3390/molecules26102850
APA StyleKong, J., Xia, H., He, R., Chen, H., & Yu, Y. (2021). Preparation of the Key Dolutegravir Intermediate via MgBr2-Promoted Cyclization. Molecules, 26(10), 2850. https://doi.org/10.3390/molecules26102850