Iron Chelation in Local Infection




Abstract
1. Introduction
2. Iron Homeostasis
3. Iron in Infection
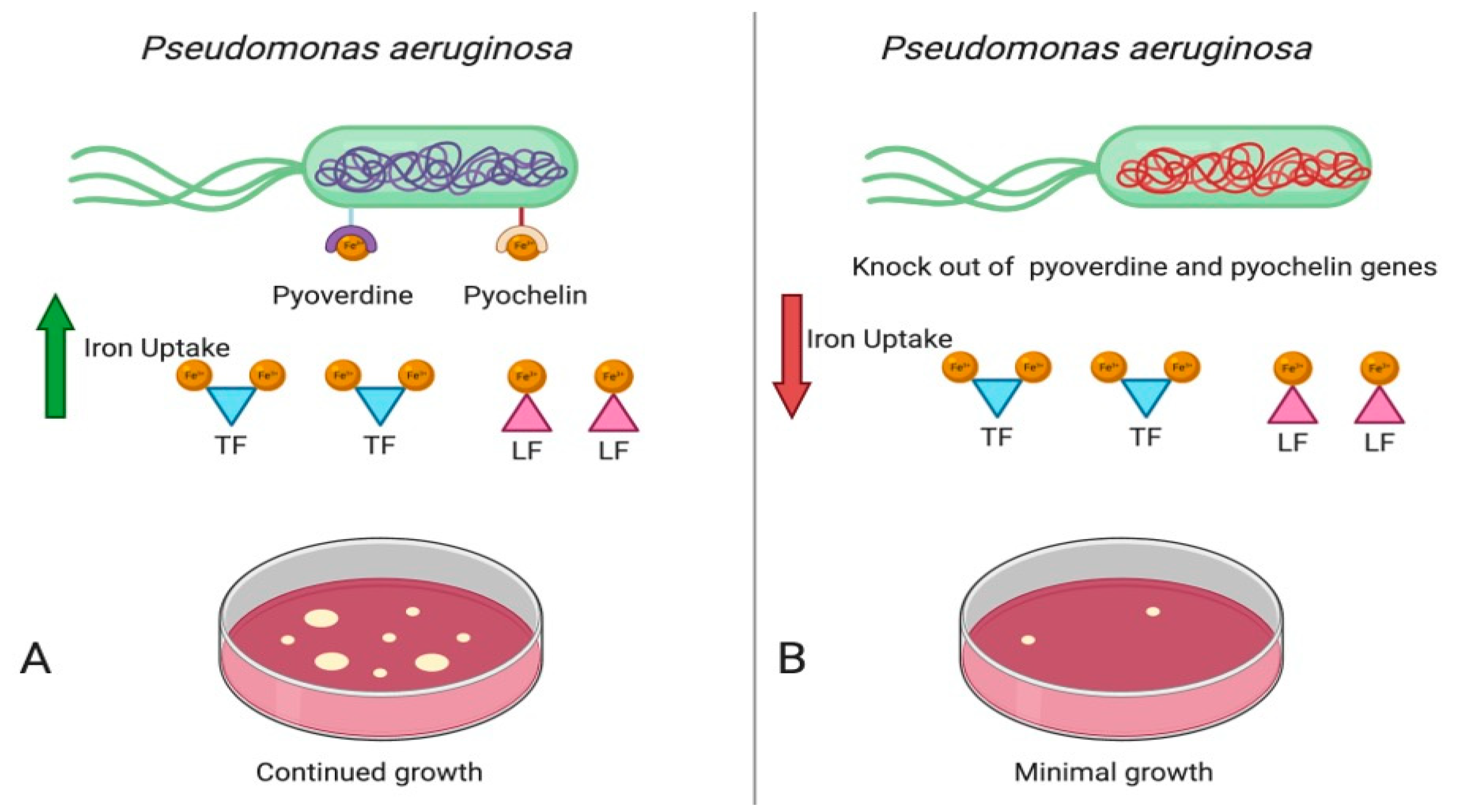
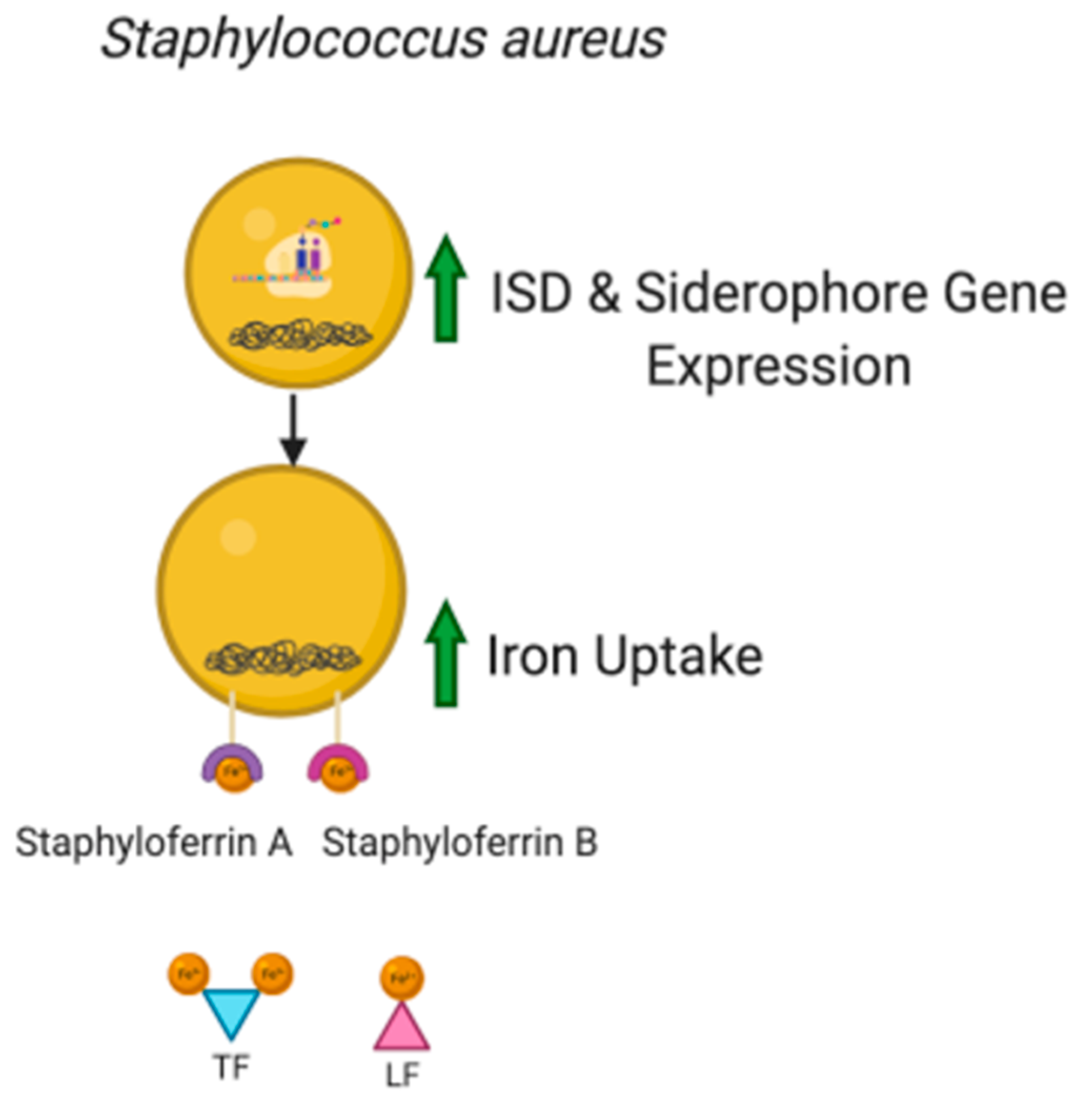
3.1. Role of Iron in Microorganism Pathogenicity
3.2. Siderophores
3.3. Synthetic Iron Chelators
4. Local Infection
4.1. Infections of the Eye
4.2. Skin Wound Infections
4.3. Bacterial Cystitis
4.4. Medical Device-Associated Infections
4.5. Iron in Viral Infection
5. Therapeutic Applications
5.1. Routs of Administration
5.2. Limitations
6. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Chifman, J.; Laubenbacher, R.; Torti, S.V. A Systems Biology Approach to Iron Metabolism. Adv. Exp. Med. Biol. 2014, 844, 201–225. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. An Int. Rev. J. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Shah, A.; Zhang, L. Heme, an essential nutrient from dietary proteins, critically impacts diverse physiological and pathological processes. Nutrients 2014, 6, 1080–1102. [Google Scholar] [CrossRef] [PubMed]
- Chua, A.C.G.; Graham, R.M.; Trinder, D.; Olynyk, J.K. The regulation of cellular iron metabolism. Crit. Rev. Clin. Lab. Sci. 2007, 44, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.C. Disorders of Iron Metabolism. N. Engl. J. Med. 1999, 341, 1986–1995. [Google Scholar] [CrossRef]
- Ganz, T. Hepcidin-a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract. Res. Clin. Haematol. 2005, 18, 171–182. [Google Scholar] [CrossRef]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Datta, V.; Lauth, X.; Johnson, R.S.; Nizet, V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 2006, 107, 3727–3732. [Google Scholar] [CrossRef]
- Theurl, I.; Theurl, M.; Seifert, M.; Mair, S.; Nairz, M.; Rumpold, H.; Zoller, H.; Bellmann-Weiler, R.; Niederegger, H.; Talasz, H.; et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 2008, 111, 2392–2399. [Google Scholar] [CrossRef]
- Guida, C.; Altamura, S.; Klein, F.A.; Galy, B.; Boutros, M.; Ulmer, A.J.; Hentze, M.W.; Muckenthaler, M.U. A novel inflammatory pathway mediating rapid hepcidin-independent hypoferremia. Blood 2015. [Google Scholar] [CrossRef] [PubMed]
- Frazier, M.D.; Mamo, L.B.; Ghio, A.J.; Turi, J.L. Hepcidin expression in human airway epithelial cells is regulated by interferon-γ. Respir. Res. 2011, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.L.; Heremans, J.F.; Schonne, E. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J. Exp. Med. 1969, 130, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Duck, K.A.; Connor, J.R. Iron uptake and transport across physiological barriers. BioMetals 2016, 29, 573–591. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Sheldon, J.R.; Laakso, H.A.; Heinrichs, D.E. Iron Acquisition Strategies of Bacterial Pathogens. Microbiol. Spectr. 2016, 4, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Habib, R.; Houle, S.; Caza, M.; Lépine, F.; Daigle, F.; Massé, E.; Dozois, C.M. The small RNA RyhB contributes to siderophore production and virulence of uropathogenic Escherichia coli. Infect. Immun. 2014, 82, 5056–5068. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, A.A.; Powell, D.A.; Nguyen, A.T.; O’Neill, M.; Djapgne, L.; Wilks, A.; Ernst, R.K.; Oglesby-Sherrouse, A.G. The prrF-encoded small regulatory RNAs are required for iron homeostasis and virulence of Pseudomonas aeruginosa. Infect. Immun. 2015, 83, 863–875. [Google Scholar] [CrossRef]
- Reinhart, A.A.; Oglesby-Sherrouse, A.G. Regulation of pseudomonas aeruginosa virulence by distinct iron sources. Genes (Basel) 2016, 7, 126. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Skaar, E.P.; Gaspar, A.H.; Humayun, M.; Gornicki, P.; Jelenska, J.; Joachmiak, A.; Missiakas, D.M.; Schneewind, O. Passage of Heme-Iron Across the Envelope of Staphylococcus aureus. Science 2003, 299, 906–910. [Google Scholar] [CrossRef]
- Pishchany, G.; Sheldon, J.R.; Dickson, C.F.; Alam, M.T.; Read, T.D.; Gell, D.A.; Heinrichs, D.E.; Skaar, E.P. IsdB-dependent hemoglobin binding is required for acquisition of heme by Staphylococcus aureus. J. Infect. Dis. 2014. [Google Scholar] [CrossRef]
- Wilson, B.R.; Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends Mol. Med. 2016, 22, 1077–1090. [Google Scholar] [CrossRef]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial siderophores in community and host interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, nature and utility of universal iron chelator-Siderophore: A review. Microbiol. Res. 2018, 212–213, 103–111. [Google Scholar] [CrossRef]
- Franchini, M.; Veneri, D. Iron-chelation therapy: An update. Hematol. J. 2004, 5, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Hoffbrand, A.V. The role of deferiprone in iron chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Katharina, R.; Thomas, N.; Claeys, J.; McGuane, J.; Prestidge, C.A.; Coenye, T.; Wormald, P.-J.; Vreugde, S. A Topical Hydrogel with Deferiprone and Gallium-Protoporphyrin Targets Bacterial Iron Metabolism and Has Antibiofilm Activity. Antimicrob. Agents Chemother. 2017, 61, e00481-17. [Google Scholar]
- Stumpf, J.L.; Bonk, M.E. Deferasirox. Am. J. Health Pharm. 2007, 64, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Kumar, R.; Rojas, I.G.; Salvatori, O.; Edgertonb, M. Iron Chelator Deferasirox Reduces Candida albicans Invasion. Antimicrob. Agents Chemother. 2019, 63, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, T.; Aali, M.; Kostek, L.; LeTourneau-Paci, C.; Colp, P.; Zhou, J.; Holbein, B.; Hoskin, D.; Lehmann, C. Anti-inflammatory effects of a novel iron chelator, DIBI, in experimental sepsis. Clin. Hemorheol. Microcirc. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Del Carmen Parquet, M.; Savage, K.A.; Allan, D.S.; Ang, M.T.C.; Chen, W.; Logan, S.M.; Holbein, B.E. Antibiotic resistant Acinetobacter baumannii is susceptible to the novel iron-sequestering anti-infective DIBI in vitro and in experimental pneumonia in mice. Antimicrob. Agents Chemother. 2019, 63, e00855-19. [Google Scholar] [CrossRef]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar] [CrossRef]
- Gritz, D.; Wong, I.G. Incidence and prevalence of uveitis in Northern California The Northern California Epidemiology of Uveitis Study. Ophthalmology 2004, 111, 491–500. [Google Scholar] [CrossRef]
- Arora, N.; Caldwell, A.; Wafa, K.; Szczesniak, A.; Caldwell, M.; Al-Banna, N.; Sharawy, N.; Islam, S.; Zhou, J.; Holbein, B.; et al. DIBI, a polymeric hydroxypyridinone iron chelator, reduces ocular inflammation in local and systemic endotoxin-induced uveitis. Clin. Hemorheol. Microcirc. 2018, 69, 153–164. [Google Scholar] [CrossRef]
- Rao, N.A.; Romero, J.L.; Fernandez, M.A.S.; Sevanian, A.; Marak, G.E. Effect of Iron Chelation on Severity of Ocular Inflammation in an Animal Model. Arch. Ophthalmol. 1986, 104, 1369–1371. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.V.; Ivanova, M.V.; Levitsky, D.O. Echinochrome, a naturally occurring iron chelator and free radical scavenger in artificial and natural membrane systems. Life Sci. 2005, 76, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Lennikov, A.; Kitaichi, N.; Noda, K.; Mizuuchi, K.; Ando, R.; Dong, Z.; Fukuhara, J.; Kinoshita, S.; Namba, K.; Ohno, S.; et al. Amelioration of endotoxin-induced uveitis treated with the sea urchin pigment echinochrome in rats. Mol. Vis. 2014, 20, 171–177. [Google Scholar] [PubMed]
- Cheng, K.H.; Leung, S.L.; Hoekman, H.W.; Beekhuis, W.H.; Mulder, P.G.; Geerards, A.J.; Kijlstra, A. Incidence of contact-lens-associated microbial keratitis and its related morbidity. Lancet (Lond. Engl.) 1999, 354, 181–185. [Google Scholar] [CrossRef]
- McLaughlin-Borlace, L.; Stapleton, F.; Matheson, M.; Dart, J.K.G. Bacterial biofilm on contact lenses and lens storage cases in wearers with microbial keratitis. J. Appl. Microbiol. 1998, 84, 827–838. [Google Scholar] [CrossRef]
- Kang, D.; Kirienko, N.V. Interdependence between iron acquisition and biofilm formation in Pseudomonas aeruginosa. J. Microbiol. 2018, 56, 449–457. [Google Scholar] [CrossRef]
- Suzuki, T.; Gotoh, N.; Shiraishi, A. Role of pvdE Pyoverdine Synthesis in Pseudomonas. Cornea 2018, 37, 99–105. [Google Scholar] [CrossRef]
- Banin, E.; Vasil, M.L.; Greenberg, E.P. Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. USA 2005, 102, 11076–11081. [Google Scholar] [CrossRef]
- Banin, E.; Brady, K.M.; Greenberg, E.P. Chelator-induced dispersal and killing of Pseudomonas aeruginosa cells in a biofilm. Appl. Environ. Microbiol. 2006, 72, 2064–2069. [Google Scholar] [CrossRef]
- Cardona, A.F.; Wilson, S.E. Skin and Soft-Tissue Infections: A Critical Review and the Role of Telavancin in Their Treatment. Clin. Infect. Dis. 2015, 61, 69–78. [Google Scholar] [CrossRef]
- Ray, G.T.; Suaya, J.A.; Baxter, R. Microbiology of skin and soft tissue infections in the age of community-acquired methicillin-resistant Staphylococcus aureus. Diagn. Microbiol. Infect. Dis. 2013, 76, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Parquet, M. del C.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel Iron-Chelator DIBI Inhibits Staphylococcus aureus Growth, Suppresses Experimental MRSA Infection in Mice and Enhances the Activities of Diverse Antibiotics in vitro. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Horsburgh, M.J.; Ingham, E.; Foster, S.J. In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and Is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol. 2001, 183, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Escolar, L.; Pérez-Martín, J.; de Lorenzo, V. Opening the iron box: Transcriptional metalloregulation by the Fur protein. J. Bacteriol. 1999, 181, 6223–6229. [Google Scholar] [CrossRef]
- Hammer, N.D.; Skaar, E.P. Molecular Mechanisms of Staphylococcus aureus Iron Acquisition. Annu. Rev. Microbiol. 2010, 65, 129–147. [Google Scholar] [CrossRef]
- Foxman, B. Urinary tract infection syndromes. Occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect. Dis. Clin. North Am. 2014, 28, 1–13. [Google Scholar] [CrossRef]
- McLellan, L.K.; Hunstad, D.A. Urinary Tract Infection: Pathogenesis and Outlook. Trends Mol. Med. 2016, 22, 946–957. [Google Scholar] [CrossRef]
- Nicolle, L.E.; Bradley, S.; Colgan, R.; Rice, J.C.; Schaeffer, A.; Hooton, T.M. Infectious Diseases Society of America Guidelines for the Diagnosis and Treatment of Asymptomatic Bacteriuria in Adults. Clin. Infect. Dis. 2005, 40, 643–654. [Google Scholar] [CrossRef]
- Bauckman, K.A.; Mysorekar, I.U. Ferritinophagy drives uropathogenic Escherichia coli persistence in bladder epithelial cells. Autophagy 2016, 12, 850–863. [Google Scholar] [CrossRef]
- Yu, X.; Gan, Z.; Wang, Z.; Tang, X.; Guo, B.; Du, H. Increased Iron Availability Aggravates the Infection of Escherichia coli O157:H7 in Mice. Biol. Trace Elem. Res. 2019, 190, 457–465. [Google Scholar] [CrossRef]
- Bauckman, K.A.; Matsuda, R.; Higgins, C.B.; DeBosch, B.J.; Wang, C.; Mysorekar, I.U. Dietary restriction of iron availability attenuates UPEC pathogenesis in a mouse model of urinary tract infection. Am. J. Physiol. Ren. Physiol. 2019, 316, F814–F822. [Google Scholar] [CrossRef] [PubMed]
- Hagan, E.C.; Lloyd, A.L.; Rasko, D.A.; Faerber, G.J.; Mobley, H.L.T. Escherichia coli global gene expression in urine from women with urinary tract infection. PLoS Pathog. 2010, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- VanEpps, J.S.; Younger, J.G. Implantable Device Related Infection. Shock 2016, 46, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Vestby, L.K.; Grønseth, T.; Simm, R.; Nesse, L.L. Bacterial biofilm and its role in the pathogenesis of disease. Antibiotics 2020, 9, 59. [Google Scholar] [CrossRef]
- Darouiche, R.O. Device-associated infections: A macroproblem that starts with microadherence. Clin. Infect. Dis. 2001, 33, 1567–1572. [Google Scholar] [CrossRef]
- Natsuhara, K.M.; Shelton, T.J.; Meehan, J.P.; Lum, Z.C. Mortality During Total Hip Periprosthetic Joint Infection. J. Arthroplasty 2019, 34, S337–S342. [Google Scholar] [CrossRef]
- Nicolle, L.E. Catheter associated urinary tract infections. Antimicrob. Resist. Infect. Control 2014, 3, 1–8. [Google Scholar] [CrossRef]
- Zarb, P.; Coignard, B.; Griskeviciene, J.; Muller, A.; Vankerckhoven, V.; Weist, K.; Goossens, M.M.; Vaerenberg, S.; Hopkins, S.; Catry, B.; et al. The european centre for disease prevention and control (ECDC) pilot point prevalence survey of healthcare-associated infections and antimicrobial use. Eurosurveillance 2012, 17, 1–16. [Google Scholar] [CrossRef]
- Grigoryan, L.; Abers, M.S.; Kizilbash, Q.F.; Petersen, N.J.; Trautner, B.W. A comparison of the microbiologic profile of indwelling versus external urinary catheters. Am. J. Infect. Control 2014, 42, 682–684. [Google Scholar] [CrossRef]
- Sharma, P.; Dube, D.; Sinha, M.; Yadav, S.; Kaur, P.; Sharma, S.; Singh, T.P. Structural Insights into the Dual Strategy of Recognition by Peptidoglycan Recognition Protein, PGRP-S: Structure of the Ternary Complex of PGRP-S with Lipopolysaccharide and Stearic Acid. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef]
- Mettrick, K.; Hassan, K.; Lamont, I.; Reid, D. The Iron chelator, N,N’-bis (2-hydroxybenzyl) Ethylenediamine-N,N’-diacetic acid is an Effective Colistin Adjunct against Clinical Strains of Biofilm-Dwelling Pseudomonas aeruginosa. Antibiotics 2020, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Noelting, J.; Jurewitsch, B.; Allard, J.P. Non-antibiotic antimicrobial catheter lock solutions in patients on home parenteral nutrition. Nutrients 2018, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Ong, K.; Lau, E.; Mowat, F.; Halpern, M. Projections of primary and revision hip and knee arthroplasty in the United States from 2005 to 2030. J. Bone Jt. Surg.-Ser. A 2007, 89, 780–785. [Google Scholar] [CrossRef]
- Gbejuade, H.O.; Lovering, A.M.; Webb, J.C. The role of microbial biofilms in prosthetic joint infections: A review. Acta Orthop. 2015, 86, 147–158. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Stoodley, P.; Kathju, S.; Høiby, N.; Moser, C.; William Costerton, J.; Moter, A.; Bjarnsholt, T. Towards diagnostic guidelines for biofilm-associated infections. FEMS Immunol. Med. Microbiol. 2012, 65, 127–145. [Google Scholar] [CrossRef]
- Coraça-Huber, D.C.; Dichtl, S.; Steixner, S.; Nogler, M.; Weiss, G. Iron chelation destabilizes bacterial biofilms and potentiates the antimicrobial activity of antibiotics against coagulase-negative Staphylococci. Pathog. Dis. 2018, 76, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Kraus, F.; Popella, P.; Baykal, A.; Guttroff, C.; François, P.; Sass, P.; Plietker, B.; Götz, F. The polycyclic polyprenylated acylphloroglucinol antibiotic PPAP 23 targets the membrane and iron metabolism in Staphylococcus aureus. Front. Microbiol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Crossing the iron gate: Why and how transferrin receptors mediate viral entry. Annu. Rev. Nutr. 2018, 38, 431–458. [Google Scholar] [CrossRef]
- Parker, J.S.L.; Murphy, W.J.; Wang, D.; O’Brien, S.J.; Parrish, C.R. Canine and Feline Parvoviruses Can Use Human or Feline Transferrin Receptors To Bind, Enter, and Infect Cells. J. Virol. 2001. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef]
- Zou, D.M.; Sun, W.L. Relationship between hepatitis C virus infection and iron overload. Chin. Med. J. (Engl). 2017, 130, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rong, L.; Li, Y.P. Flaviviridae Viruses and Oxidative Stress: Implications for Viral Pathogenesis. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Ikeda, M.; Abe, K.I.; Dansako, H.; Ohkoshi, S.; Aoyagi, Y.; Kato, N. Comprehensive analysis of the effects of ordinary nutrients on hepatitis C virus RNA replication in cell culture. Antimicrob. Agents Chemother. 2007. [Google Scholar] [CrossRef] [PubMed]
- Minaiyan, M.; Mostaghel, E.; Mahzouni, P. Preventive therapy of experimental colitis with selected iron chelators and anti-oxidants. Int. J. Prev. Med. 2012, 3, S162–S169. [Google Scholar] [PubMed]
- Vermylen, C. What is new in iron overload? Eur. J. Pediatr. 2008, 167, 377–381. [Google Scholar] [CrossRef]
- Li, L.; Frei, B. Iron chelation inhibits NF-κB-mediated adhesion molecule expression by inhibiting p22phox protein expression and NADPH oxidase activity. Arterioscler. Thromb. Vasc. Biol. 2006. [Google Scholar] [CrossRef]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Act -Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Desferrioxamine (DFO) | Deferasirox (DFX) | Deferiprone (DFP) |
|---|---|---|---|
| Binding capacity (chelator: iron) | Hexadentate (1:1) | Bidentate (2:1) | Tridentate (3:1) |
| Route of administration | Subcutaneous, intravenous | Oral tablet | Oral tablet |
| Side effects | Growth retardation Local skin reaction Ophthalmological Auditory Allergic reaction Pulmonary at high doses Neurological at high doses | Rash Rise in creatinine Auditory Gastrointestinal Ophthalmological | Gastrointestinal Zinc deficiency Agranulocytosis Musculoskeletal and joint pains |
| Half-life | 47–134 min | 8–16 h | 3–4 h |
| Structure |  |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scott, C.; Arora, G.; Dickson, K.; Lehmann, C. Iron Chelation in Local Infection. Molecules 2021, 26, 189. https://doi.org/10.3390/molecules26010189
Scott C, Arora G, Dickson K, Lehmann C. Iron Chelation in Local Infection. Molecules. 2021; 26(1):189. https://doi.org/10.3390/molecules26010189
Chicago/Turabian StyleScott, Cassidy, Gaurav Arora, Kayle Dickson, and Christian Lehmann. 2021. "Iron Chelation in Local Infection" Molecules 26, no. 1: 189. https://doi.org/10.3390/molecules26010189
APA StyleScott, C., Arora, G., Dickson, K., & Lehmann, C. (2021). Iron Chelation in Local Infection. Molecules, 26(1), 189. https://doi.org/10.3390/molecules26010189