
Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates



Abstract
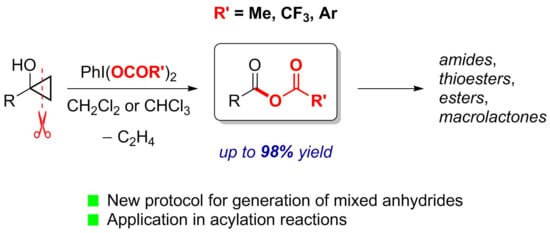
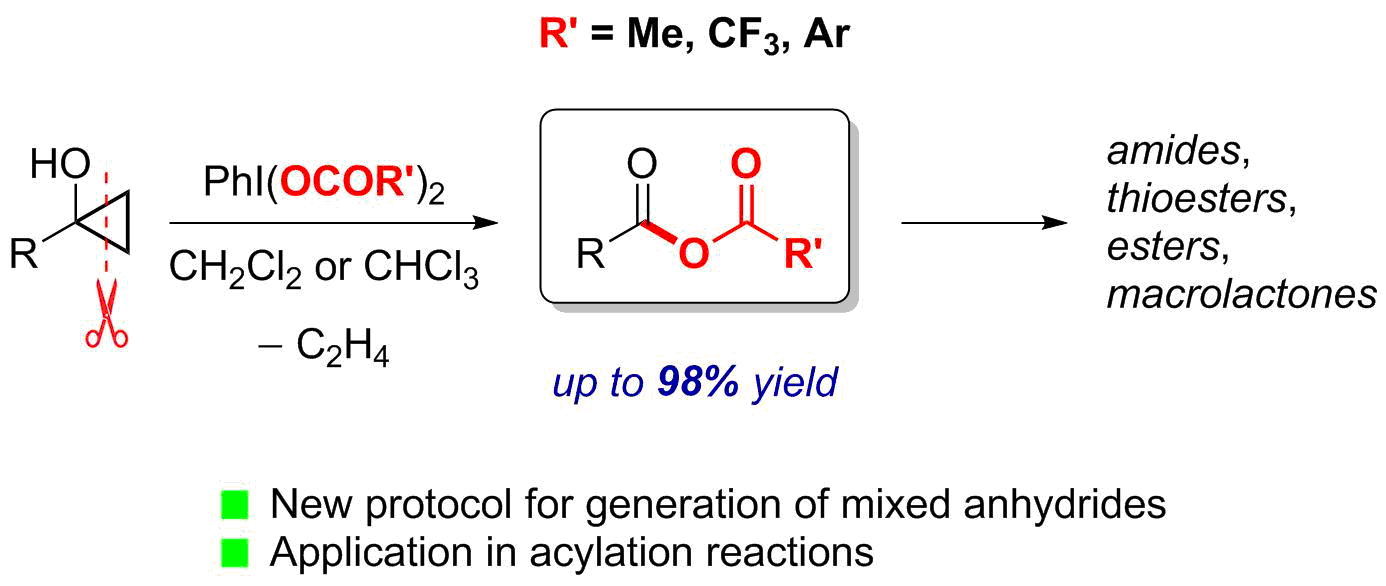
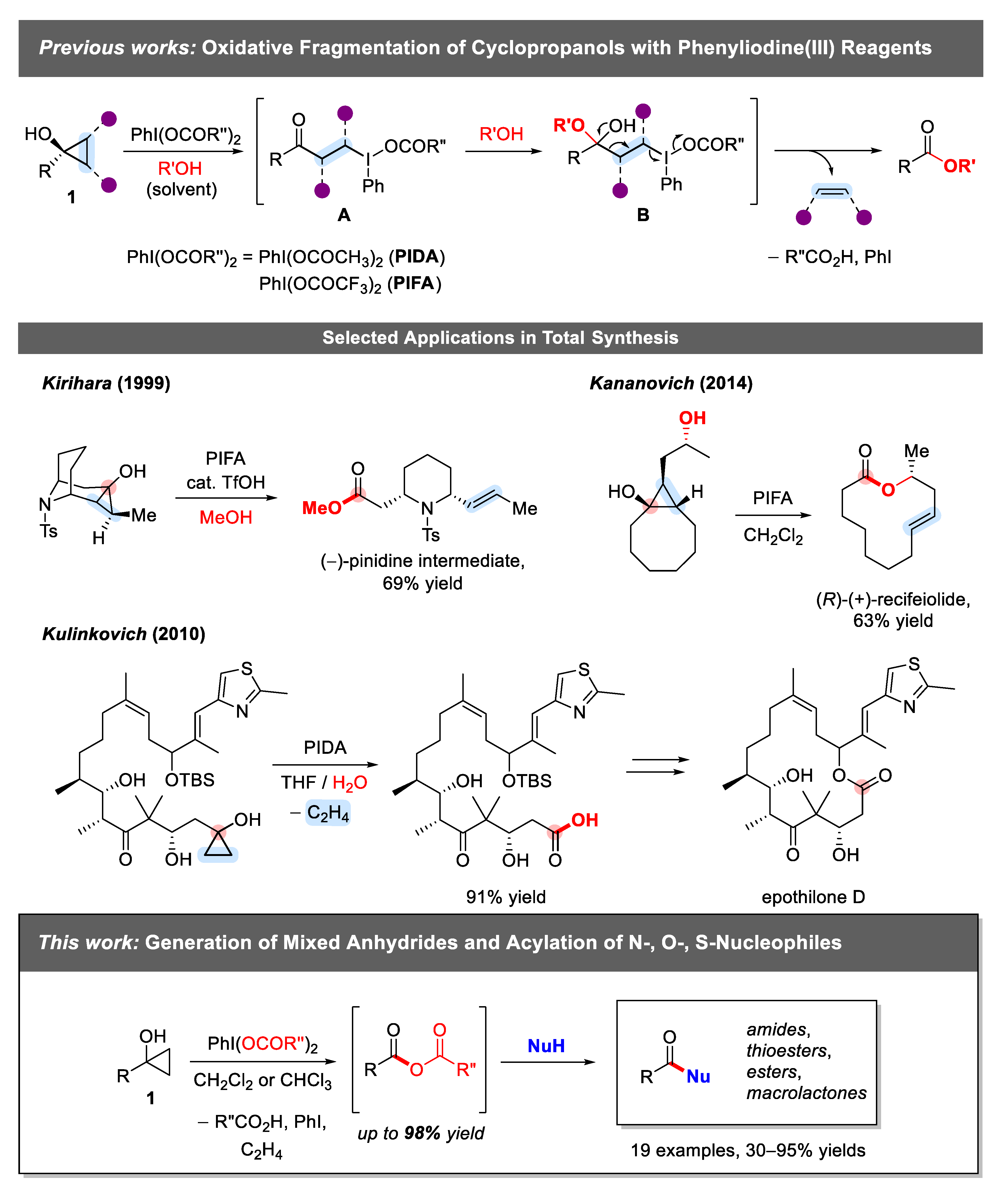
1. Introduction
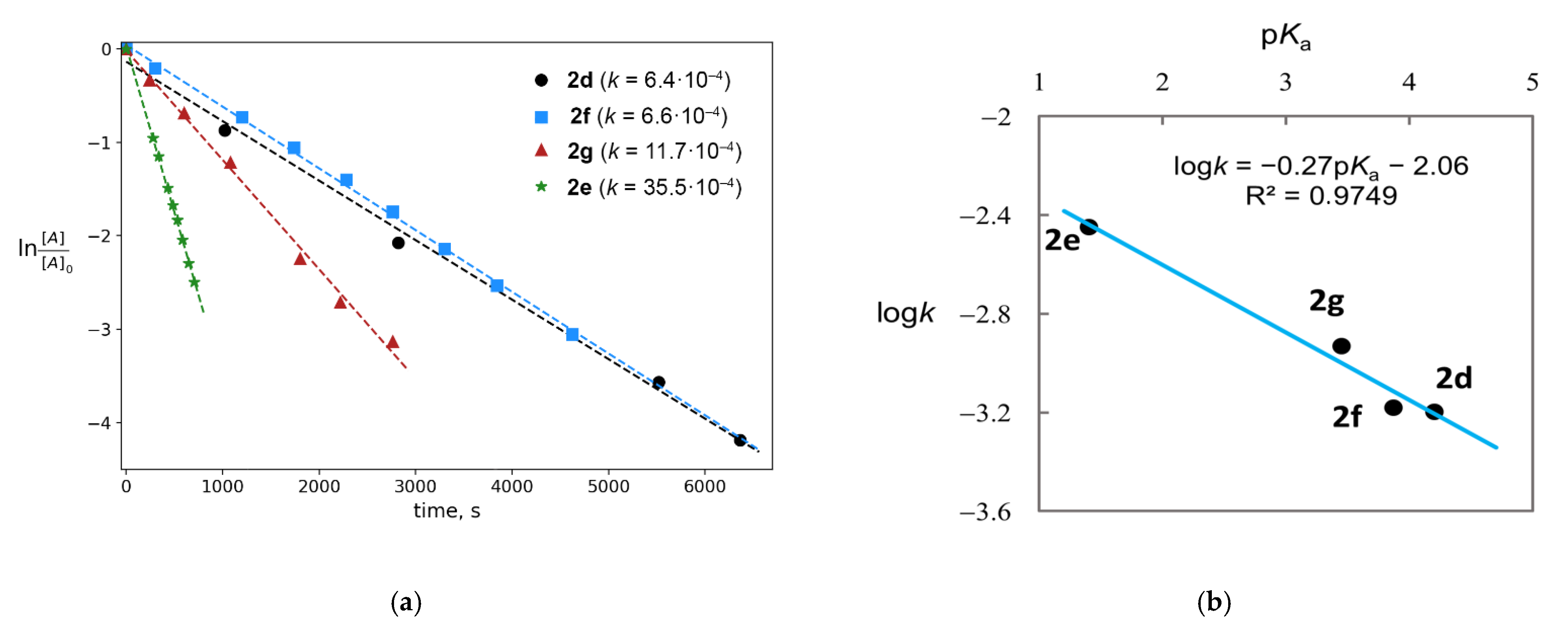
2. Results and Discussion
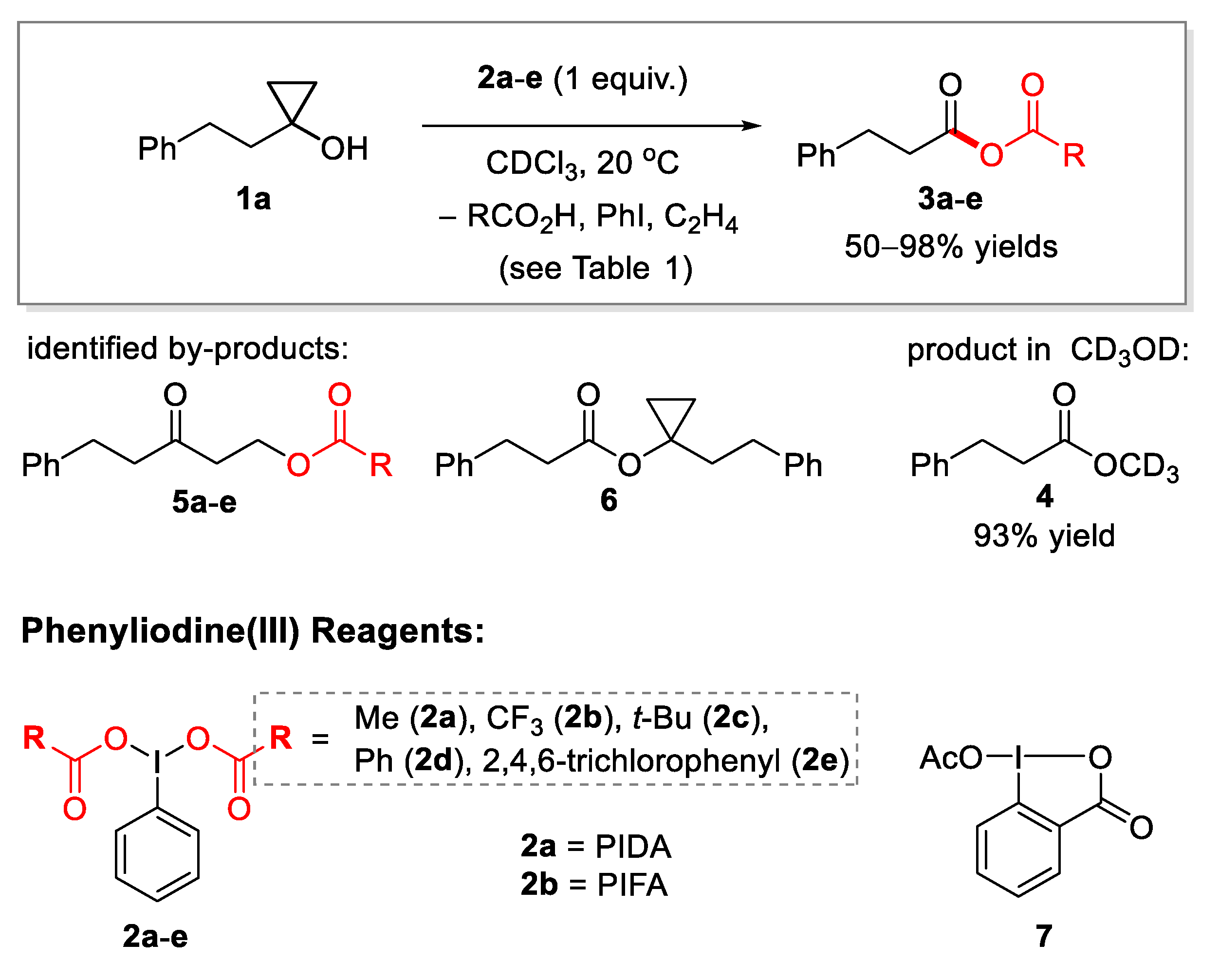
2.1. Generation of Mixed Anhydrides via Ring Scissoring of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates
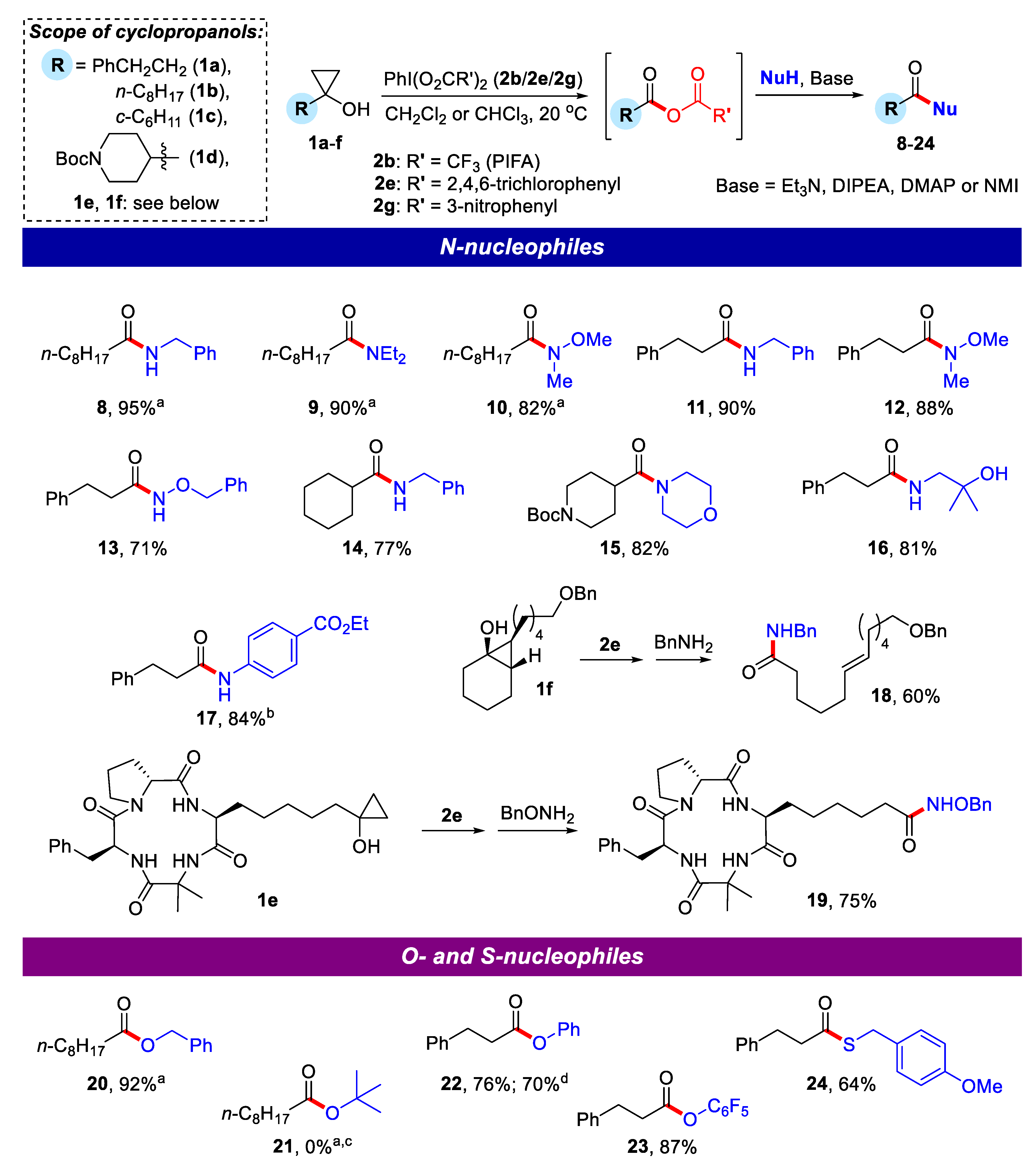
2.2. Application of Mixed Anhydrides for Acylation of N-, O-, S-Nucleophiles and in Macrolactonizations
3. Experimental Section
3.1. General Experimental Methods
3.2. Typical Procedure for Preparation of PhI(O2CAr)2 Reagents [for Ar = 2,4,6-trichlorophenyl, 2e]
3.3. General Procedure for Generation of Mixed Anhydrides from Cyclopropanols and Reagent 2e
3.3.1. N-Benzylnonanamide (8)
3.3.2. N,N-Diethylnonanamide (9)
3.3.3. N-Methoxy-N-methylnonanamide (10)
3.3.4. N-Benzyl-3-phenylpropanamide (11)
3.3.5. N-Methoxy-N-methyl-3-phenylpropanamide (12)
3.3.6. N-(Benzyloxy)-3-phenylpropanamide (13)
3.3.7. N-Benzylcyclohexanecarboxamide (14)
3.3.8. tert-Butyl 4-(morpholine-4-carbonyl)piperidine-1-carboxylate (15)
3.3.9. N-(2-Hydroxy-2-methylpropyl)-3-phenylpropanamide (16)
3.3.10. Ethyl 4-(3-phenylpropanamido)benzoate (17)
3.3.11. (E)-N-Benzyl-12-(benzyloxy)dodec-6-enamide (18)
3.3.12. 6-((3S,9S,14aR)-9-Benzyl-6,6-dimethyl-1,4,7,10-tetraoxotetradecahydropyrrolo[1,2-a][1,4,7,10]tetraazacyclododecin-3-yl)-N-(benzyloxy)hexanamide (cyclopeptide 19)
3.3.13. Benzyl nonanoate (20)
3.3.14. Phenyl 3-phenylpropanoate (22)
3.3.15. Pentafluorophenyl 3-phenylpropanoate (23)
3.3.16. S-(4-Methoxybenzyl) 3-phenylpropanethioate (24)
3.4. General Procedure for the Synthesis of Macrolactones 25a,b
3.4.1. Oxacycloundecan-2-one (25a)
3.4.2. Oxacyclotridecan-2-one (25b)
3.5. Preparation of (E)-oxacyclotridec-7-en-2-one (28)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kulinkovich, O.G. The Chemistry of Cyclopropanols. Chem. Rev. 2003, 103, 2597–2632. [Google Scholar] [CrossRef]
- Kulinkovich, O.G. Cyclopropanes in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Haym, I.; Brimble, M.A. The Kulinkovich hydroxycyclopropanation reaction in natural product synthesis. Org. Biomol. Chem. 2012, 10, 7649–7665. [Google Scholar] [CrossRef]
- Cai, X.; Liang, W.; Dai, M. Total Syntheses via Cyclopropanols. Tetrahedron 2019, 75, 193–208. [Google Scholar] [CrossRef]
- Wolan, A.; Six, Y. Synthetic transformations mediated by the combination of titanium(iv) alkoxides and grignard reagents: Selectivity issues and recent applications. Part 1: Reactions of carbonyl derivatives and nitriles. Tetrahedron 2010, 66, 15–61. [Google Scholar] [CrossRef]
- McDonald, T.R.; Mills, L.R.; West, M.S.; Rousseaux, S.A.L. Selective Carbon–Carbon Bond Cleavage of Cyclopropanols. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pirenne, V.; Muriel, B.; Waser, J. Catalytic Enantioselective Ring-Opening Reactions of Cyclopropanes. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; You, B.; Xie, G.; Wang, X. Developments in the construction of cyclopropanols. Org. Biomol. Chem. 2020, 18, 191–204. [Google Scholar] [CrossRef]
- Cha, J.K.; Kulinkovich, O.G. The Kulinkovich Cyclopropanation of Carboxylic Acid Derivatives. Org. React. 2012, 77, 1–160. [Google Scholar] [CrossRef]
- Kulinkovich, O.G.; de Meijere, A. 1,n-Dicarbanionic Titanium Intermediates from Monocarbanionic Organometallics and Their Application in Organic Synthesis. Chem. Rev. 2000, 100, 2789–2834. [Google Scholar] [CrossRef]
- Du, H.; Long, J.; Shi, Y. Catalytic Asymmetric Simmons−Smith Cyclopropanation of Silyl Enol Ethers. Efficient Synthesis of Optically Active Cyclopropanol Derivatives. Org. Lett. 2006, 8, 2827–2829. [Google Scholar] [CrossRef]
- Cheng, K.; Carroll, P.J.; Walsh, P.J. Diasteroselective Preparation of Cyclopropanols Using Methylene Bis(iodozinc). Org. Lett. 2011, 13, 2346–2349. [Google Scholar] [CrossRef] [PubMed]
- Kananovich, D.G.; Zubrytski, D.M.; Kulinkovich, O.G. A Convenient Transformation of 2-Alkylidenecycloalkanones into Alkyl-Substituted Bicyclo[n.1.0]alkan-1-ols: Application to the Synthesis of Capsaicin. Synlett 2010, 7, 1043–1046. [Google Scholar] [CrossRef]
- Kananovich, D.G.; Kulinkovich, O.G. Studies on the origin of cis-diastereoselectivity of the titanium-mediated cyclopropanation of carboxylic esters with Grignard reagents. Stereochemistry of the intramolecular cyclization of β-metalloketones. Tetrahedron 2008, 64, 1536–1547. [Google Scholar] [CrossRef]
- Rivera, R.M.; Jang, Y.; Poteat, C.M.; Lindsay, V.N.G. General Synthesis of Cyclopropanols via Organometallic Addition to 1-Sulfonylcyclopropanols as Cyclopropanone Precursors. Org. Lett. 2020, 22, 6510–6515. [Google Scholar] [CrossRef]
- Yu, Z.; Mendoza, A. Enantioselective Assembly of Congested Cyclopropanes Using Redox-Active Aryldiazoacetates. ACS Catal. 2019, 9, 7870–7875. [Google Scholar] [CrossRef]
- Montesinos-Magraner, M.; Costantini, M.; Ramírez-Contreras, R.; Muratore, M.E.; Johansson, M.J.; Mendoza, A. General Cyclopropane Assembly via Enantioselective Transfer of a Redox-Active Carbene to Aliphatic Olefins. Angew. Chem. Int. Ed. 2019, 58, 5930–5935. [Google Scholar] [CrossRef]
- Simaan, M.; Marek, I. Asymmetric Catalytic Preparation of Polysubstituted Cyclopropanol and Cyclopropylamine Derivatives. Angew. Chem. Int. Ed. 2018, 57, 1543–1546. [Google Scholar] [CrossRef]
- Benoit, G.; Charette, A.B. Diastereoselective Borocyclopropanation of Allylic Ethers Using a Boromethylzinc Carbenoid. J. Am. Chem. Soc. 2017, 139, 1364–1367. [Google Scholar] [CrossRef]
- Mills, L.R.; Rousseaux, S.A.L. Modern Developments in the Chemistry of Homoenolates. Eur. J. Org. Chem. 2019, 2019, 8–26. [Google Scholar] [CrossRef]
- Nikolaev, A.; Orellana, A. Transition-Metal-Catalyzed C–C and C–X Bond-Forming Reactions Using Cyclopropanols. Synthesis 2016, 48, 1741–1768. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.-L.; Chen, Z.; Zhou, C.-S.; Xiong, B.-Q.; Zhang, P.-L.; Yang, C.-A.; Zhou, Q. Oxidative radical ring-opening/cyclization of cyclopropane derivatives. Beilstein J. Org. Chem. 2019, 15, 256–278. [Google Scholar] [CrossRef] [PubMed]
- Rubottom, G.M.; Marrero, R.; Krueger, D.S.; Schreiner, J.L. The lead (IV) acetate oxidation of cyclopropane derivatives: A novel fragmentation reaction. Tetrahedron Lett. 1977, 18, 4013–4015. [Google Scholar] [CrossRef]
- Rubottom, G.M.; Beedle, E.C.; Kim, C.-W.; Mott, R.C. Stereochemistry of the Lead(IV) Acetate Fragmentation of 1-(Trimethylsiloxy)bicyclo[n.1.0]Alkanes. J. Am. Chem. Soc. 1985, 107, 4230–4233. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Kakuda, H.; Momose, T. Efficient Fragmentation of the Tertiary Cyclopropanol System: Oxidation of 1-(Trimethylsiloxy)bicyclo[n.1.0]alkanes and Analogues by Using Phenyliodine(III) Diacetate. Tetrahedron Lett. 1995, 36, 6907–6910. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Kakuda, H.; Momose, T. Hypervalent λn-Iodane-Mediated Fragmentation of Tertiary Cyclopropanol Systems. Tetrahedron 1998, 54, 13943–13954. [Google Scholar] [CrossRef]
- Zubrytski, D.M.; Kananovich, D.G.; Matiushenkov, E.A. Preparation of Stereochemically Pure E- and Z-Alkenoic Acids and Their Methyl Esters from Bicyclo[n.1.0]alkan-1-ols. Application in the Synthesis of Insect Pheromones. Russ. J. Org. Chem. 2017, 53, 813–823. [Google Scholar] [CrossRef]
- Momose, T.; Nishio, T.; Kirihara, M. Efficient Synthesis of an Enantiomeric Pair of Pinidine: An Illustration of Organochemical Carving on the Rigid Bridged System as the Stereochemical Tactics. Tetrahedron Lett. 1996, 37, 4987–4990. [Google Scholar] [CrossRef]
- Kirihara, M.; Nishio, T.; Yokoyama, S.; Kakuda, H.; Momose, T. Hypervalent λn-Iodane-Mediated Fragmentation of Tertiary Cyclopropanol Systems II: Application to Asymmetric Syntheses of Piperidine and Indolizidine Alkaloids. Tetrahedron 1999, 55, 2911–2926. [Google Scholar] [CrossRef]
- Zubrytski, D.M.; Kananovich, D.G.; Kulinkovich, O.G. A highly stereoselective route to medium-ring-sized trans-alkenolides via oxidative fragmentation of bicyclic oxycyclopropane precursors: Application to the synthesis of (+)-recifeiolide. Tetrahedron 2014, 70, 2944–2950. [Google Scholar] [CrossRef]
- Bekish, A.V.; Kulinkovich, O.G. Differentiation between the ethoxycarbonyl groups in diethyl malate via their titanium-catalyzed reductive cyclopropanation with ethylmagnesium bromide and subsequent site-selective three-carbon ring cleavage. Tetrahedron Lett. 2005, 46, 6975–6978. [Google Scholar] [CrossRef]
- Bekish, A.V.; Isakov, V.E.; Kulinkovich, O.G. A cyclopropanol approach to the synthesis of the C13–C21 fragment of epothilones from diethyl (S)-malate. Tetrahedron Lett. 2005, 46, 6979–6981. [Google Scholar] [CrossRef]
- Kovalenko, V.N.; Masalov, N.V.; Kulinkovich, O.G. Synthesis of (+)-Disparlure from Diethyl (−)-Malate via Opening and Fragmentation of the Three-Membered Ring in Tertiary Cyclopropanols. Russ. J. Org. Chem. 2009, 45, 1318–1324. [Google Scholar] [CrossRef]
- Shklyaruck, D.G.; Fedarkevich, A.N.; Kozyrkov, Y.Y. The First Synthesis of Spirocyclic Sulfates from Tertiary Cyclopropanols and Their Reaction with Normant Homocuprates. Synlett 2014, 25, 1855–1858. [Google Scholar] [CrossRef][Green Version]
- Shklyaruck, D.G. A cyclopropanol-based approach to synthesis of Unit A of the cryptophycins. Tetrahedron Asymmetry 2014, 25, 644–649. [Google Scholar] [CrossRef]
- Shklyaruck, D.G. New Synthesis of syn-Stereodiad Building Block for Polyketides. Formal Synthesis of Arenamides A and C. Russ. J. Org. Chem. 2015, 51, 582–590. [Google Scholar] [CrossRef]
- Hurski, A.L.; Kulinkovich, O.G. Total synthesis of epothilone D by sixfold ring cleavage of cyclopropanol intermediates. Tetrahedron Lett. 2010, 51, 3497–3500. [Google Scholar] [CrossRef]
- Hurski, A.L.; Kulinkovich, O.G. Synthesis of Epothilone D with the Forced Application of Oxycyclopropane Intermediates. Russ. J. Org. Chem. 2011, 47, 1653–1674. [Google Scholar] [CrossRef]
- Elek, G.Z.; Koppel, K.; Zubrytski, D.M.; Konrad, N.; Järving, I.; Lopp, M.; Kananovich, D.G. Divergent Access to Histone Deacetylase Inhibitory Cyclopeptides via a Late-Stage Cyclopropane Ring Cleavage Strategy. Short Synthesis of Chlamydocin. Org. Lett. 2019, 21, 8473–8478. [Google Scholar] [CrossRef]
- Nishino, N.; Jose, B.; Shinta, R.; Kato, T.; Komatsu, Y.; Yoshida, M. Chlamydocin–hydroxamic acid analogues as histone deacetylase inhibitors. Bioorg. Med. Chem. 2004, 12, 5777–5784. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Wei, Y.; Xiu, Z.; Nishino, N. Discovery of Potent HDAC Inhibitors Based on Chlamydocin with Inhibitory Effects on Cell Migration. ChemMedChem 2014, 9, 627–637. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Kitir, B.; Maolanon, A.R.; Ohm, R.G.; Colaço, A.R.; Fristrup, P.; Madsen, A.S.; Olsen, C.A. Chemical Editing of Macrocyclic Natural Products and Kinetic Profiling Reveal Slow, Tight-Binding HDAC Inhibitors with Picomolar Affinities. Biochemistry 2017, 56, 5134–5146. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Brown, D.G.; Young, R.J.; Keserü, G.M. Expanding the medicinal chemistry synthetic toolbox. Nat. Rev. Drug Discov. 2018, 17, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
- Massolo, E.; Pirola, M.; Benaglia, M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4651. [Google Scholar] [CrossRef]
- de Figueiredo, R.M.; Suppo, J.-S.; Campagne, J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef]
- Papadopoulos, G.N.; Kokotos, C.G. Photoorganocatalytic One-Pot Synthesis of Hydroxamic Acids from Aldehydes. Chem. Eur. J. 2016, 22, 6964–6967. [Google Scholar] [CrossRef]
- Montalbetti, C.A.G.N.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Kirihara, M.; Kakuda, H. Hypervalent Iodine-Mediated Fragmentation of Tertiary Cyclopropanol Systems. J. Synth. Org. Chem. Jpn. 2004, 62, 919–928. [Google Scholar] [CrossRef]
- Kirihara, M.; Yokoyama, S.; Momose, T. A Significant Effect of Triflic Acid on the Hypervalent λn-Iodane-Mediated Fragmentation of the Tertiary Cyclopropanol System. Synth. Commun. 1998, 28, 1947–1956. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Zefirov, N.S.; Zhdankin, V.V.; Dan’kov, Y.V.; Sorokin, V.D.; Semerikov, V.N.; Koz’min, A.S.; Caple, R.; Berglund, B.A. Novel Reagents Containing Hypervalent Iodine and Their Use for Electrophilic Additions to Olefins. Tetrahedron Lett. 1986, 27, 3971–3974. [Google Scholar] [CrossRef]
- Tóth, B.L.; Béke, F.; Egyed, O.; Bényei, A.; Stirling, A.; Novák, Z. Synthesis of Multifunctional Aryl(Trifloxyalkenyl)iodonium Triflate Salts. ACS Omega 2019, 4, 9188–9197. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, L.-N.; Wang, H.; Duan, X.-H. Alkynylation of Tertiary Cycloalkanols via Radical C–C Bond Cleavage: A Route to Distal Alkynylated Ketones. Org. Lett. 2015, 17, 4798–4801. [Google Scholar] [CrossRef]
- Jia, K.; Zhang, F.; Huang, H.; Chen, Y. Visible-Light-Induced Alkoxyl Radical Generation Enables Selective C(sp3)–C(sp3) Bond Cleavage and Functionalizations. J. Am. Chem. Soc. 2016, 138, 1514–1517. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Shu, Y.-J.; Cao, P.; Xu, T.; Xu, H.-J. Copper(I)-catalyzed ring-opening cyanation of cyclopropanols. Org. Biomol. Chem. 2017, 15, 3590–3593. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Bellman, T.M.; Chi, T.; Dai, M. Efficient Synthesis of β-CF3/SCF3-Substituted Carbonyls via Copper-Catalyzed Electrophilic Ring-Opening Cross-Coupling of Cyclopropanols. Org. Lett. 2015, 17, 2186–2189. [Google Scholar] [CrossRef]
- Kananovich, D.G.; Konik, Y.A.; Zubrytski, D.M.; Järving, I.; Lopp, M. A Simple Access to β-Trifluoromethyl-Substituted Ketones via Copper-Catalyzed Ring-Opening Trifluoromethylation of Substituted Cyclopropanols. Chem. Commun. 2015, 51, 8349–8352. [Google Scholar] [CrossRef]
- Stang, P.J.; Boehshar, M.; Wingert, H.; Kitamura, T. Acetylenic Esters. Preparation and Characterization of Alkynyl Carboxylates via Polyvalent Iodonium Species. J. Am. Chem. Soc. 1988, 110, 3272–3278. [Google Scholar] [CrossRef]
- Koposov, A.Y.; Boyarskikh, V.V.; Zhdankin, V.V. Amino Acid-Derived Iodobenzene Dicarboxylates: Reagents for Oxidative Conversion of Alkenes to Amino Acid Esters. Org. Lett. 2004, 6, 3613–3615. [Google Scholar] [CrossRef]
- Lundblad, R.L.; Macdonald, F.M. Handbook of Biochemistry and Molecular Biology, 4th ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 1–1098. [Google Scholar]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q. TCFH–NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-A.; Han, G.; Kim, H.-J.; Kim, H.-M.; Cho, S.-D. Chemopreventive and chemotherapeutic effect of a novel histone deacetylase inhibitor, by specificity protein 1 in MDA-MB-231 human breast cancer cells. Eur. J. Cancer Prev. 2014, 23, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Smith, S.; Meza-Reyes, S.; Viñas-Bravo, O.; Fernández-Herrera, M.A.; Martínez-Pascual, R.; Sandoval-Ramírez, J.; Fuente, A.; Reyes, M.; Ruiz, J.A. In situ preparation of mixed anhydrides containing the trifluoroacetyl moiety. Application to the esterification of cholesterol and phenol. Arkivoc 2005, 2005, 127–135. [Google Scholar] [CrossRef]
- Shiina, I. Total Synthesis of Natural 8- and 9-Membered Lactones: Recent Advancements in Medium-Sized Ring Formation. Chem. Rev. 2007, 107, 239–273. [Google Scholar] [CrossRef]
- Parenty, A.; Moreau, X.; Niel, G.; Campagne, J.-M. Update 1 of: Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2013, 113, PR1–PR40. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef]
- Shiina, I.; Mukaiyama, T. A Novel Method for the Preparation of Macrolides from ω-Hydroxycarboxylic Acids. Chem. Lett. 1994, 23, 677–680. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. An Effective Use of Benzoic Anhydride and Its Derivatives for the Synthesis of Carboxylic Esters and Lactones: A Powerful and Convenient Mixed Anhydride Method Promoted by Basic Catalysts. J. Org. Chem. 2004, 69, 1822–1830. [Google Scholar] [CrossRef]
- Ishihara, K.; Kubota, M.; Kurihara, H.; Yamamoto, H. Scandium Trifluoromethanesulfonate as an Extremely Active Lewis Acid Catalyst in Acylation of Alcohols with Acid Anhydrides and Mixed Anhydrides. J. Org. Chem. 1996, 61, 4560–4567. [Google Scholar] [CrossRef]
- Shiina, I. An effective method for the synthesis of carboxylic esters and lactones using substituted benzoic anhydrides with Lewis acid catalysts. Tetrahedron 2004, 60, 1587–1599. [Google Scholar] [CrossRef]
- de Léséleuc, M.; Collins, S.K. Direct synthesis of macrodiolides via hafnium(IV) catalysis. Chem. Commun. 2015, 51, 10471–10474. [Google Scholar] [CrossRef] [PubMed]
- de Léséleuc, M.; Collins, S.K. Direct Macrolactonization of Seco Acids via Hafnium(IV) Catalysis. ACS Catal. 2015, 5, 1462–1467. [Google Scholar] [CrossRef]
- Mocci, F.; Uccheddu, G.; Frongia, A.; Cerioni, G. Solution Structure of Some λ3 Iodanes: An 17O NMR and DFT Study. J. Org. Chem. 2007, 72, 4163–4168. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, F.; Sugiura, R.; Murai, T. Direct Thionation and Selenation of Amides Using Elemental Sulfur and Selenium and Hydrochlorosilanes in the Presence of Amines. Org. Lett. 2009, 11, 3064–3067. [Google Scholar] [CrossRef]
- Fukuyama, T.; Nishitani, S.; Inouye, T.; Morimoto, K.; Ryu, I. Effective Acceleration of Atom Transfer Carbonylation of Alkyl Iodides by Metal Complexes. Application to the Synthesis of the Hinokinin Precursor and Dihydrocapsaicin. Org. Lett. 2006, 8, 1383–1386. [Google Scholar] [CrossRef]
- Lee, J.I. A Novel Synthesis of N-Methoxy-N-Methylamides from 4,6-Pyrimidyl Urethane and Grignard Reagents. Bull. Korean Chem. Soc. 2007, 28, 695–697. [Google Scholar] [CrossRef]
- Allen, C.L.; Davulcu, S.; Williams, J.M.J. Catalytic Acylation of Amines with Aldehydes or Aldoximes. Org. Lett. 2010, 12, 5096–5099. [Google Scholar] [CrossRef]
- Murphy, J.A.; Commeureuc, A.G.J.; Snaddon, T.N.; McGuire, T.M.; Khan, T.A.; Hisler, K.; Dewis, M.L.; Carling, R. Direct Conversion of N-Methoxy-N-Methylamides (Weinreb Amides) to Ketones via a Nonclassical Wittig Reaction. Org. Lett. 2005, 7, 1427–1429. [Google Scholar] [CrossRef]
- Gissot, A.; Volonterio, A.; Zanda, M. One-Step Synthesis of O-Benzyl Hydroxamates from Unactivated Aliphatic and Aromatic Esters. J. Org. Chem. 2005, 70, 6925–6928. [Google Scholar] [CrossRef]
- Maki, T.; Ishihara, K.; Yamamoto, H. 4,5,6,7-Tetrachlorobenzo[d][1,3,2]Dioxaborol-2-ol as an Effective Catalyst for the Amide Condensation of Sterically Demanding Carboxylic Acids. Org. Lett. 2006, 8, 1431–1434. [Google Scholar] [CrossRef]
- Besbes, N. Regiospecific ring opening of N-acylaziridines by neutral hydrolysis. Tetrahedron Lett. 1999, 40, 6569–6570. [Google Scholar] [CrossRef]
- Sheikh, M.C.; Takagi, S.; Ogasawara, A.; Ohira, M.; Miyatake, R.; Abe, H.; Yoshimura, T.; Morita, H. Studies on the Lossen-Type rearrangement of N-(3-phenylpropionyloxy)phthalimide and N-tosyloxy derivatives with several nucleophiles. Tetrahedron 2010, 66, 2132–2140. [Google Scholar] [CrossRef]
- Hon, Y.-S.; Wong, Y.-C.; Chang, C.-P.; Hsieh, C.-H. Tishchenko reactions of aldehydes promoted by diisobutylaluminum hydride and its application to the macrocyclic lactone formation. Tetrahedron 2007, 63, 11325–11340. [Google Scholar] [CrossRef]
- Muller, T.; Coowar, D.; Hanbali, M.; Heuschling, P.; Luu, B. Improved synthesis of tocopherol fatty alcohols and analogs: Microglial activation modulators. Tetrahedron 2006, 62, 12025–12040. [Google Scholar] [CrossRef]
- Chen, H.; Sun, S.; Liao, X. Nickel-Catalyzed Decarboxylative Alkenylation of Anhydrides with Vinyl Triflates or Halides. Org. Lett. 2019, 21, 3625–3630. [Google Scholar] [CrossRef]
- Pulikottil, F.T.; Pilli, R.; Suku, R.V.; Rasappan, R. Nickel-Catalyzed Cross-Coupling of Alkyl Carboxylic Acid Derivatives with Pyridinium Salts via C–N Bond Cleavage. Org. Lett. 2020, 22, 2902–2907. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
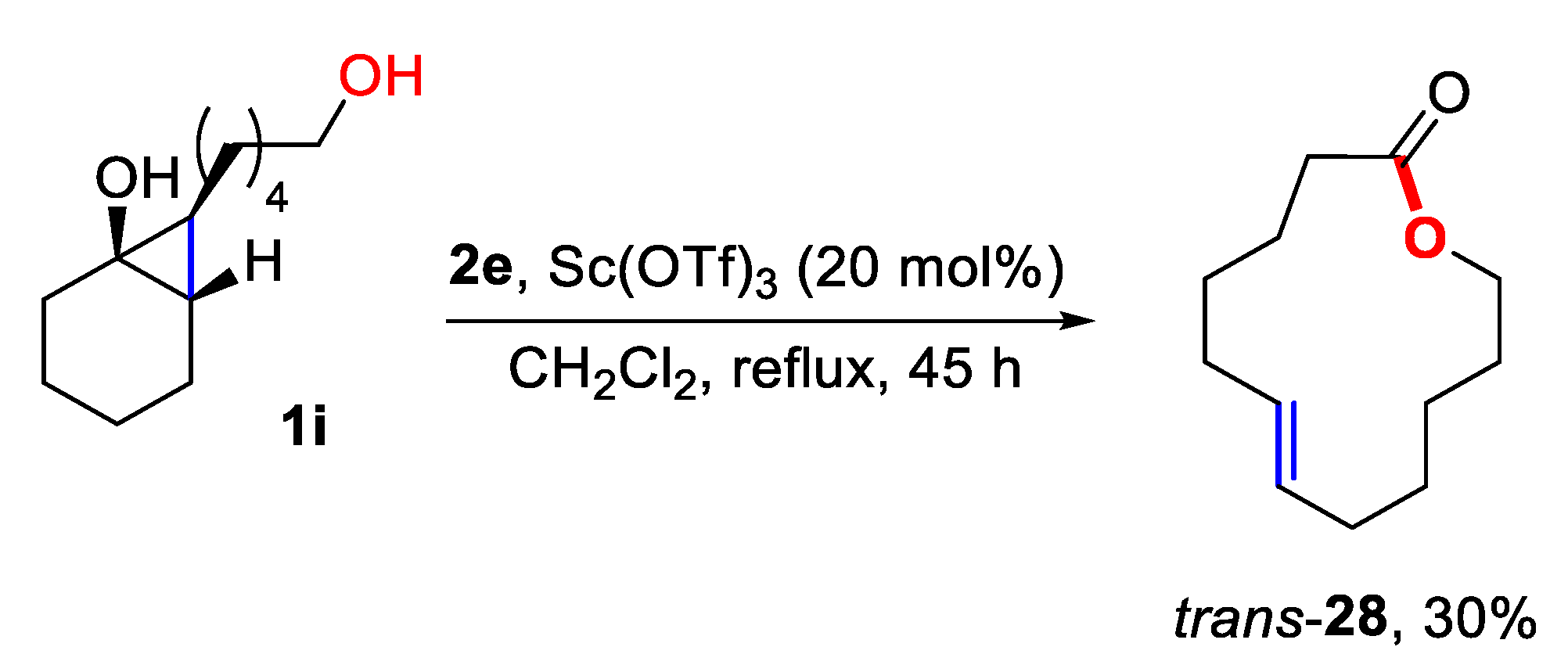{kind=link}
| Entry | Reagent | Solvent | Additive or Catalyst | Reaction Time, Min b | Main Product | Yield % c | Yield of 5, % c |
|---|---|---|---|---|---|---|---|
| 1 | 2a | CD3OD | – | <15 | 4 | 93 | – |
| 2 | 2a | CDCl3 | BnNH2 (3 equiv.) | 300 | no reaction | ||
| 3 | 2a | CDCl3 | – | 600 d | 3a | 50 | 12 |
| 4 | 2a | CDCl3 | TfOH (1 mol%) | <5 | 3a | 90 | 10 e |
| 5 | 2b | CDCl3 | – | 7 | 3b | 95 | Trace f |
| 6 | 2c | CDCl3 | – | 1500 | – g | – g | – g |
| 7 | 2c | CDCl3 | TfOH (1 mol%) | 1500 | – g | – g | – g |
| 8 | 2d | CDCl3 | – | 500 d | 3d | 46 | 8 |
| 9 | 2e | CDCl3 | – | 100 | 3e | 98 | 2 |
| 10 | 2e | CDCl3 | TfOH (1 mol%) | <5 | 3e | 70 | 30 e |
| 11 | 2e | CDCl3 | MsOH (1 mol%) | 30 | 3e | 95 | 4 e |
| 12 | 2e | CDCl3 | CF3CO2H (1 mol%) | 90 | 3e | 94 | 5 e |
| 13 | 2e | toluene-d8 | – | <15 | 3e | 90 | 4 |
| 14 | 7 | CD3ODor CDCl3 | – | 1500 | no reaction | ||

| Entry | Reagent 2a-k | R | Time, min b | Yield of 8, % c |
|---|---|---|---|---|
| 1 | 2a | Me | 120 | 22 e,d |
| 2 | 2h |  | 95 | 91 |
| 3 | 2e |  | 30 | 94 |
| 4 | 2i |  | <14 | 88 |
| 5 | 2g |  | 90 | 95 |
| 6 | 2j |  | – f | 58 |
| 7 | 2k |  | – f | – f |

| Entry | Substrate | Reagent | Catalyst | Solvent | Time, h | Yield of 25, % a | Yield of 26, % a |
|---|---|---|---|---|---|---|---|
| 1 | 1g (n = 9) | 2g | Sc(OTf)3 | CH3CN/THF | 20 | 0 b | 0 |
| 2 | 2g | Sc(OTf)3 | CH2Cl2/CH3CN c | 17 | 62 | 14 | |
| 3 | 1h (n = 11) | 2g | Sc(OTf)3 | CH2Cl2/CH3CN | 189 | 82 | 10 |
| 4 | 2e | Sc(OTf)3 | CH2Cl2/CH3CN | 187 | 91 | 8 | |
| 5 | 2g | Hf(OTf)4 | CH2Cl2/CH3CN | 178 | 93 | >7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubrytski, D.M.; Elek, G.Z.; Lopp, M.; Kananovich, D.G. Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules 2021, 26, 140. https://doi.org/10.3390/molecules26010140
Zubrytski DM, Elek GZ, Lopp M, Kananovich DG. Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules. 2021; 26(1):140. https://doi.org/10.3390/molecules26010140
Chicago/Turabian StyleZubrytski, Dzmitry M., Gábor Zoltán Elek, Margus Lopp, and Dzmitry G. Kananovich. 2021. "Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates" Molecules 26, no. 1: 140. https://doi.org/10.3390/molecules26010140
APA StyleZubrytski, D. M., Elek, G. Z., Lopp, M., & Kananovich, D. G. (2021). Generation of Mixed Anhydrides via Oxidative Fragmentation of Tertiary Cyclopropanols with Phenyliodine(III) Dicarboxylates. Molecules, 26(1), 140. https://doi.org/10.3390/molecules26010140