
Sunscreen-Assisted Selective Photochemical Transformations


Abstract
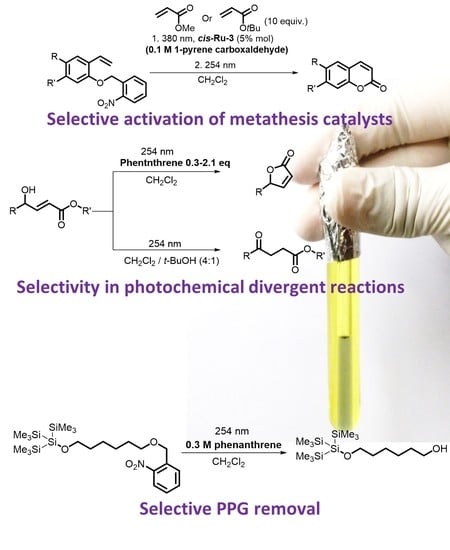
{kind=link}
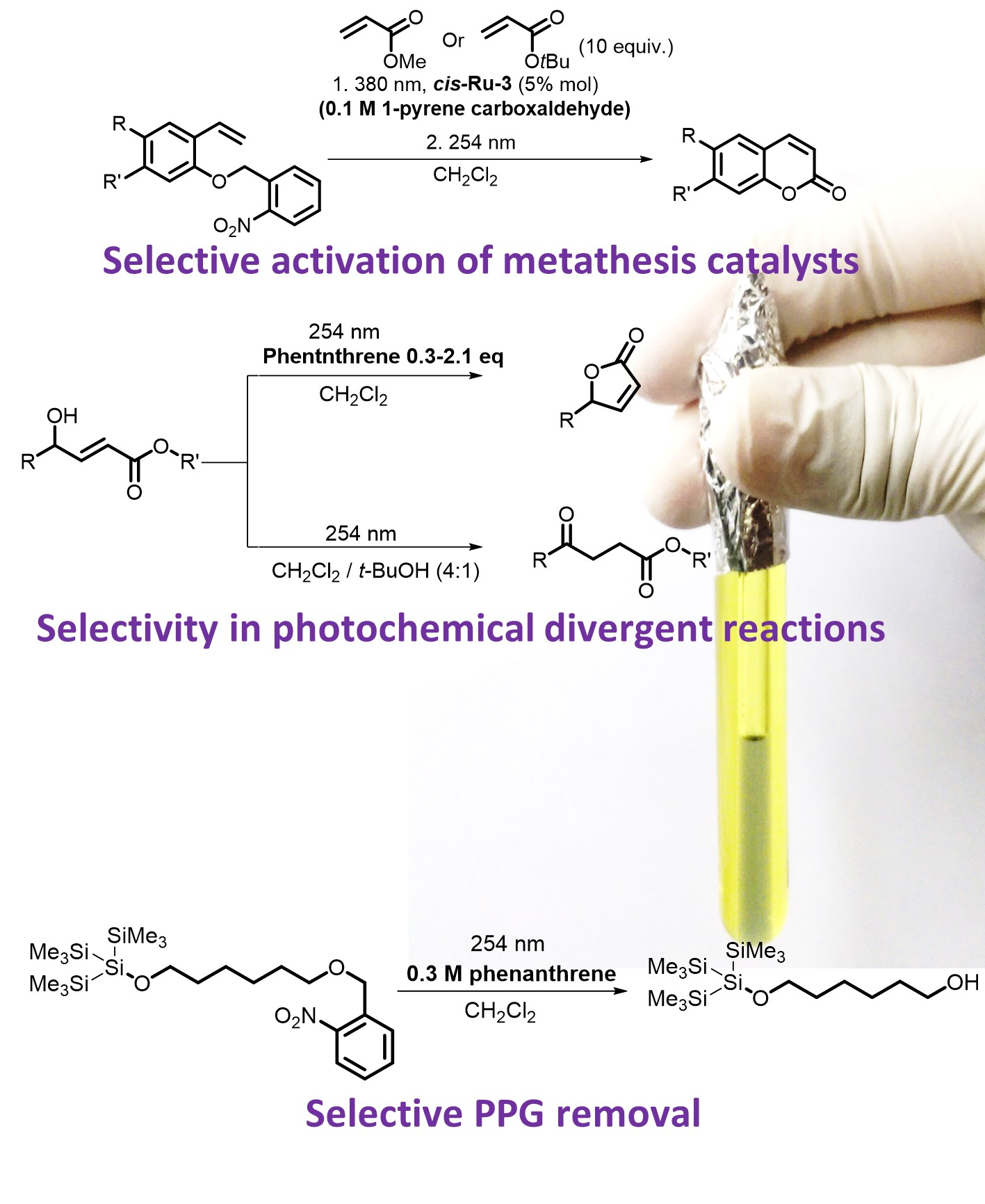
{kind=link}
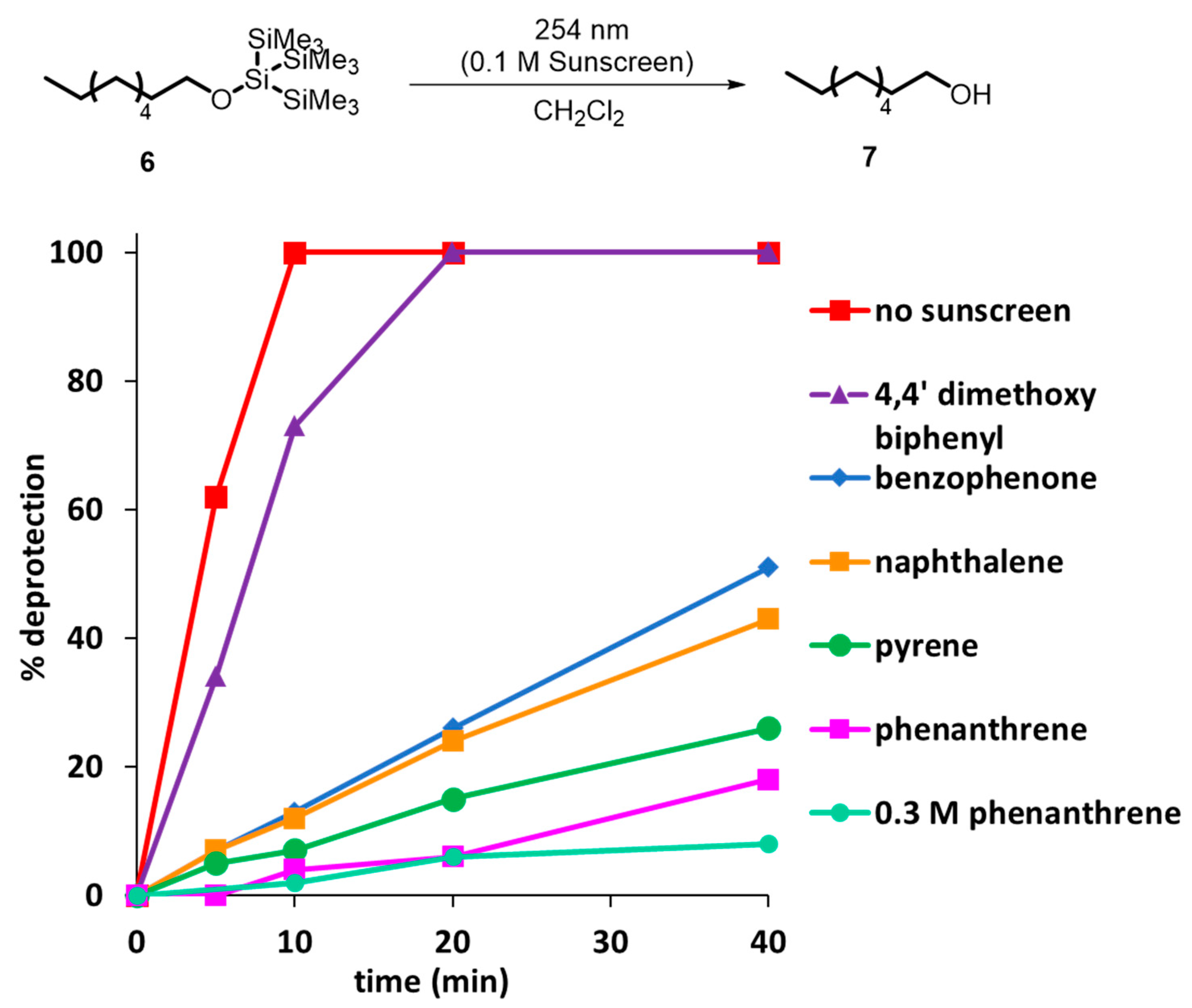
{kind=link}
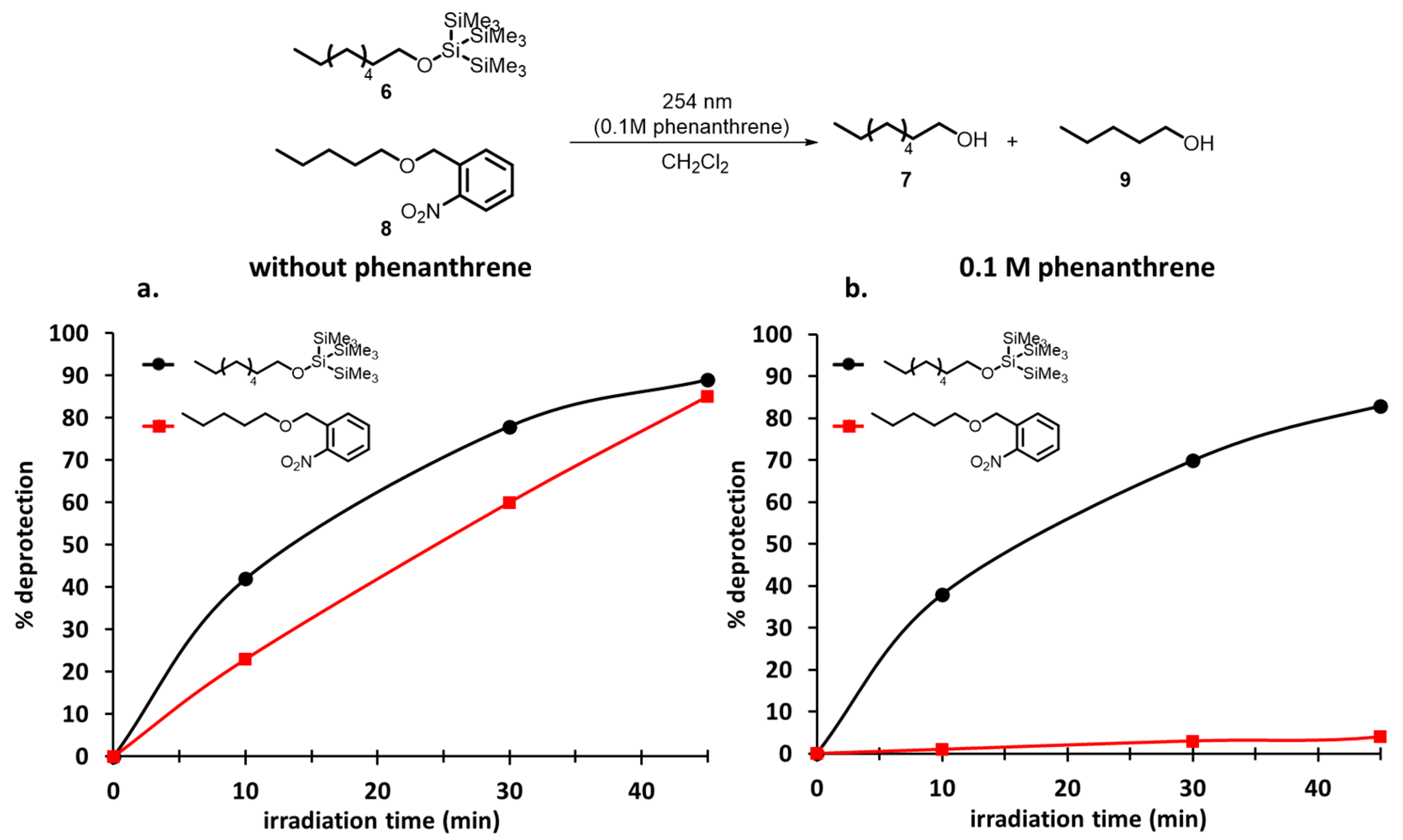
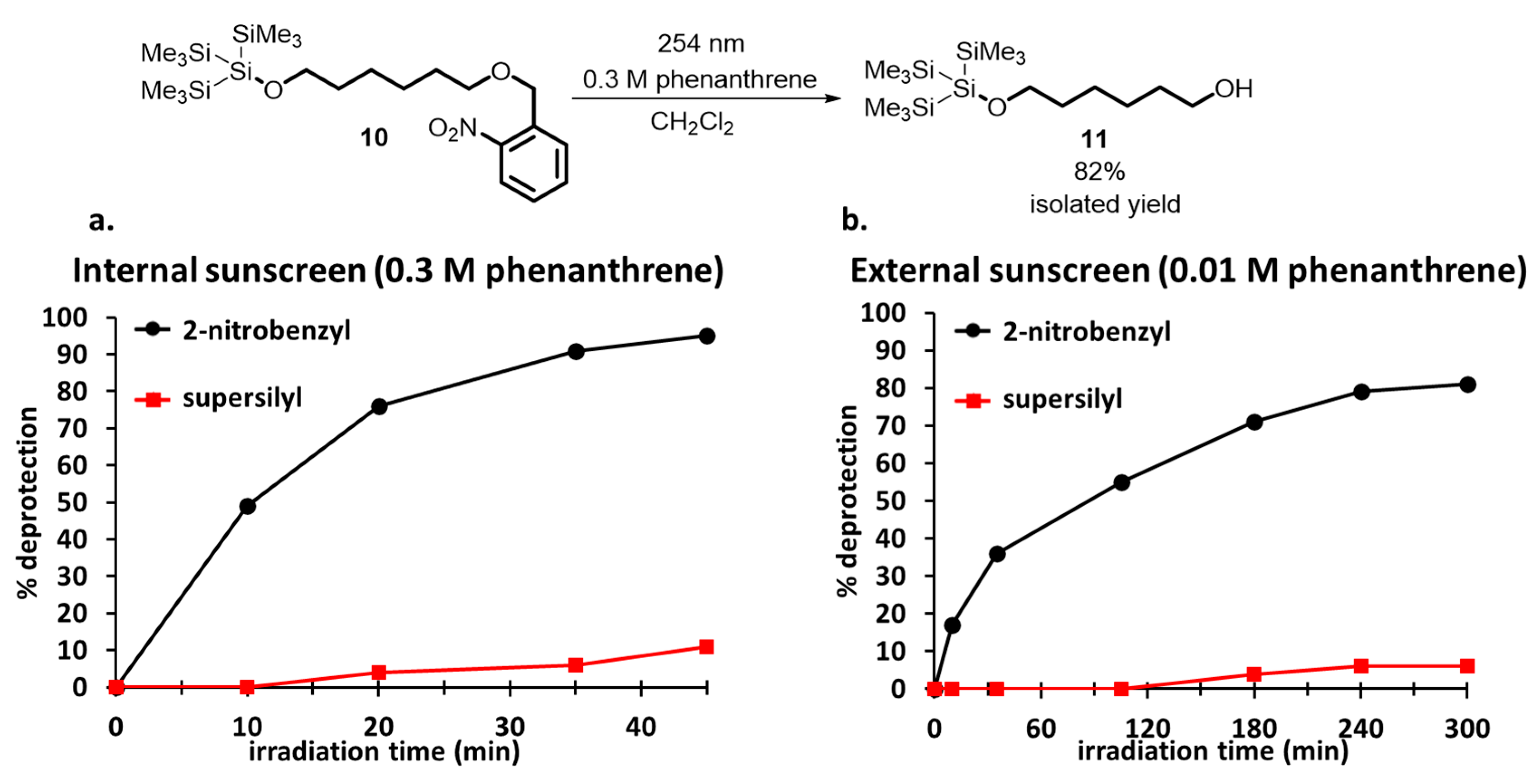{kind=link}
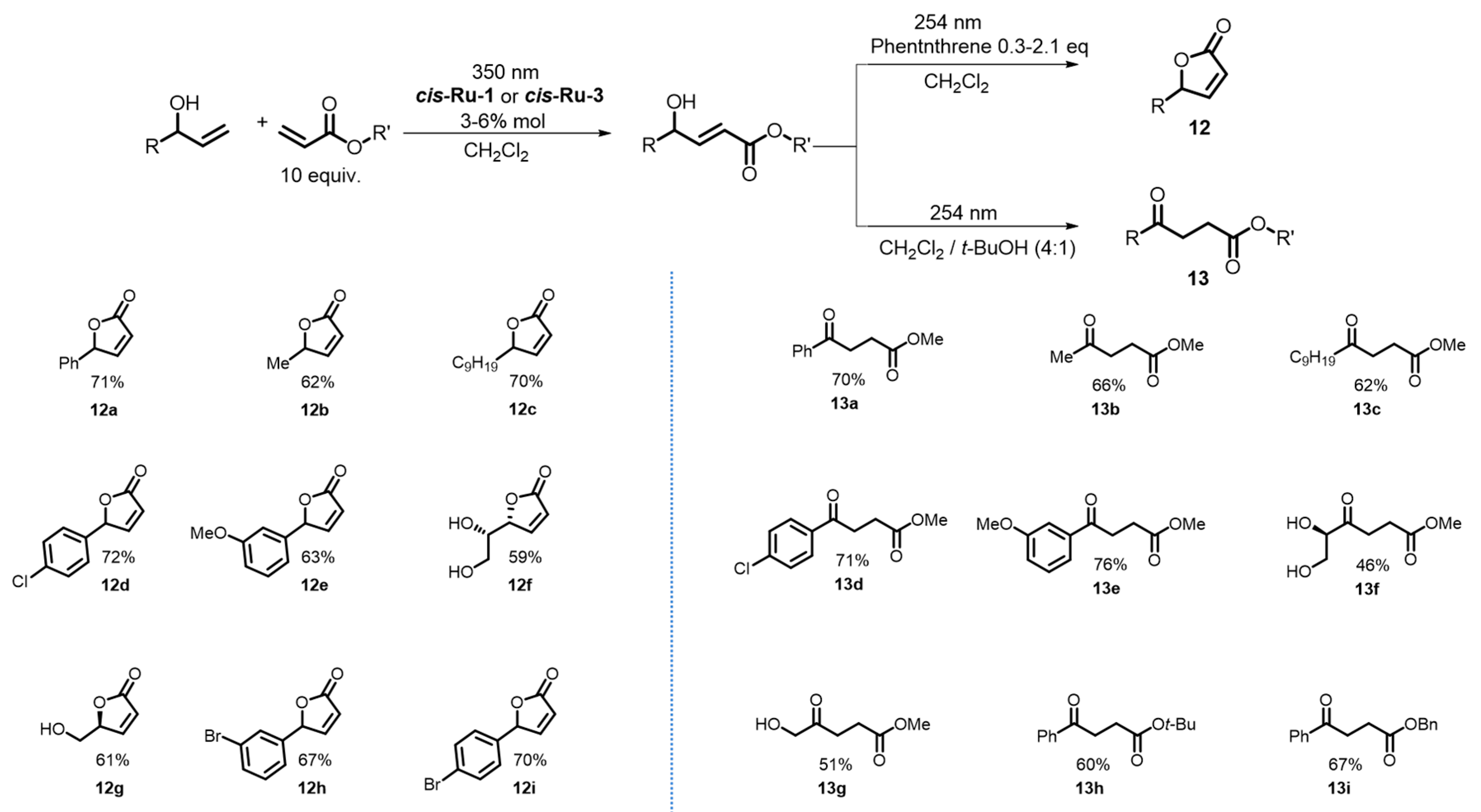{kind=link}
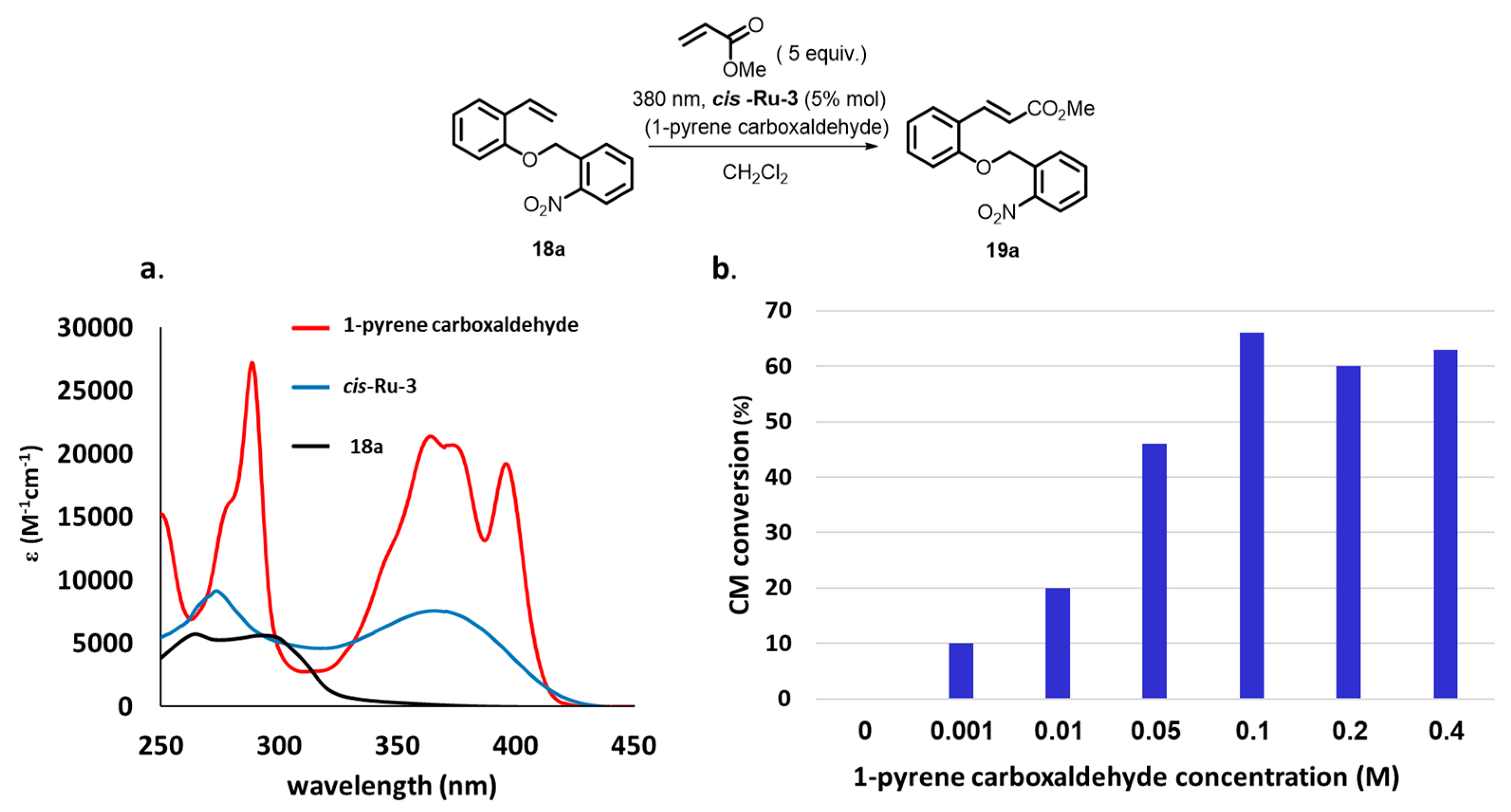{kind=link}
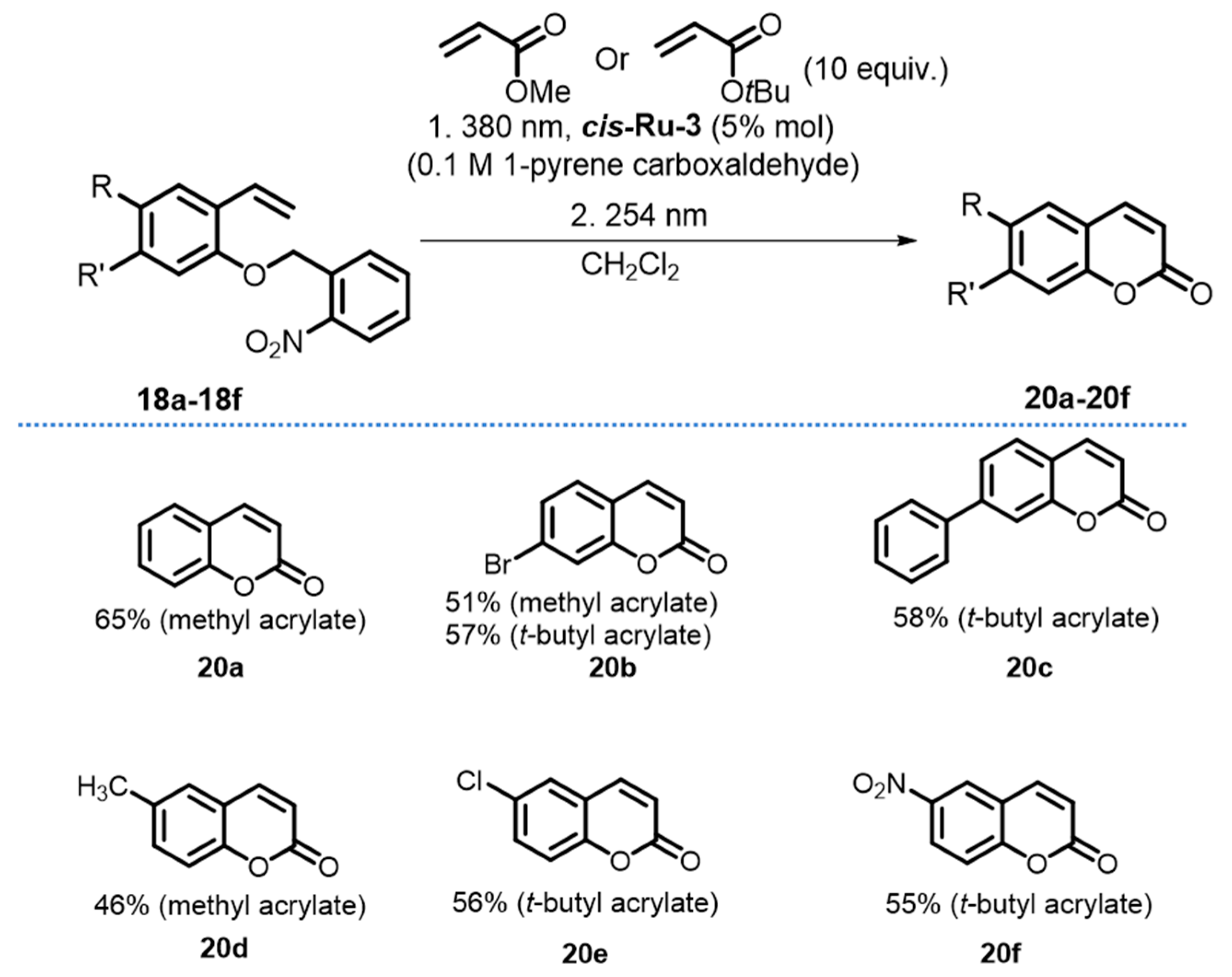{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
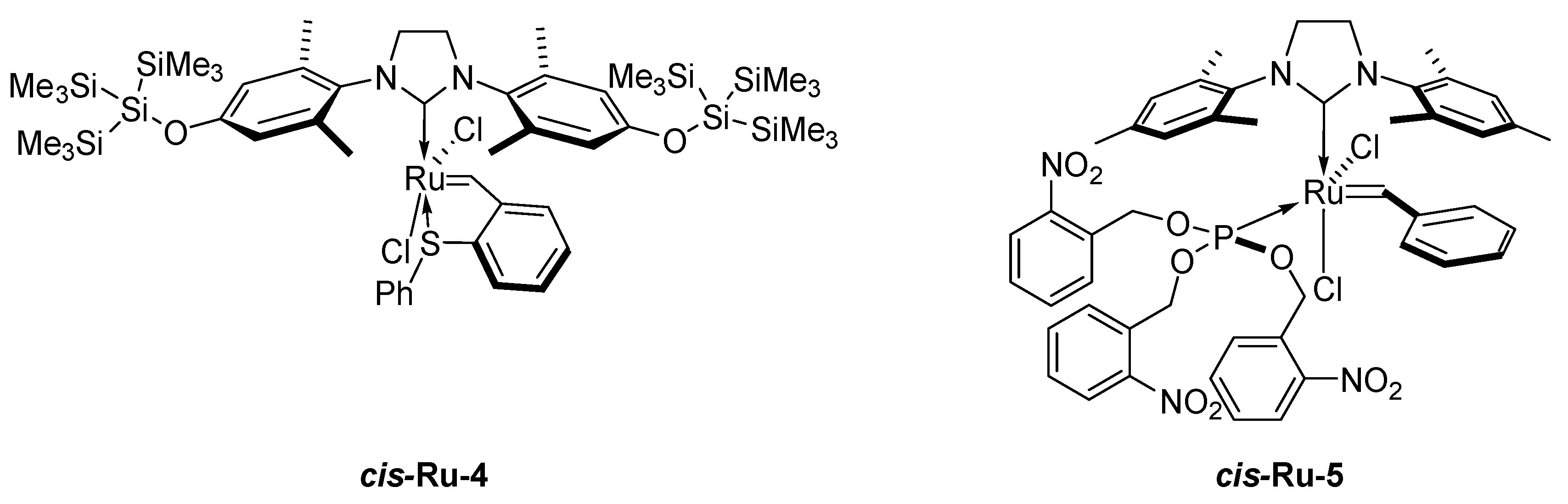
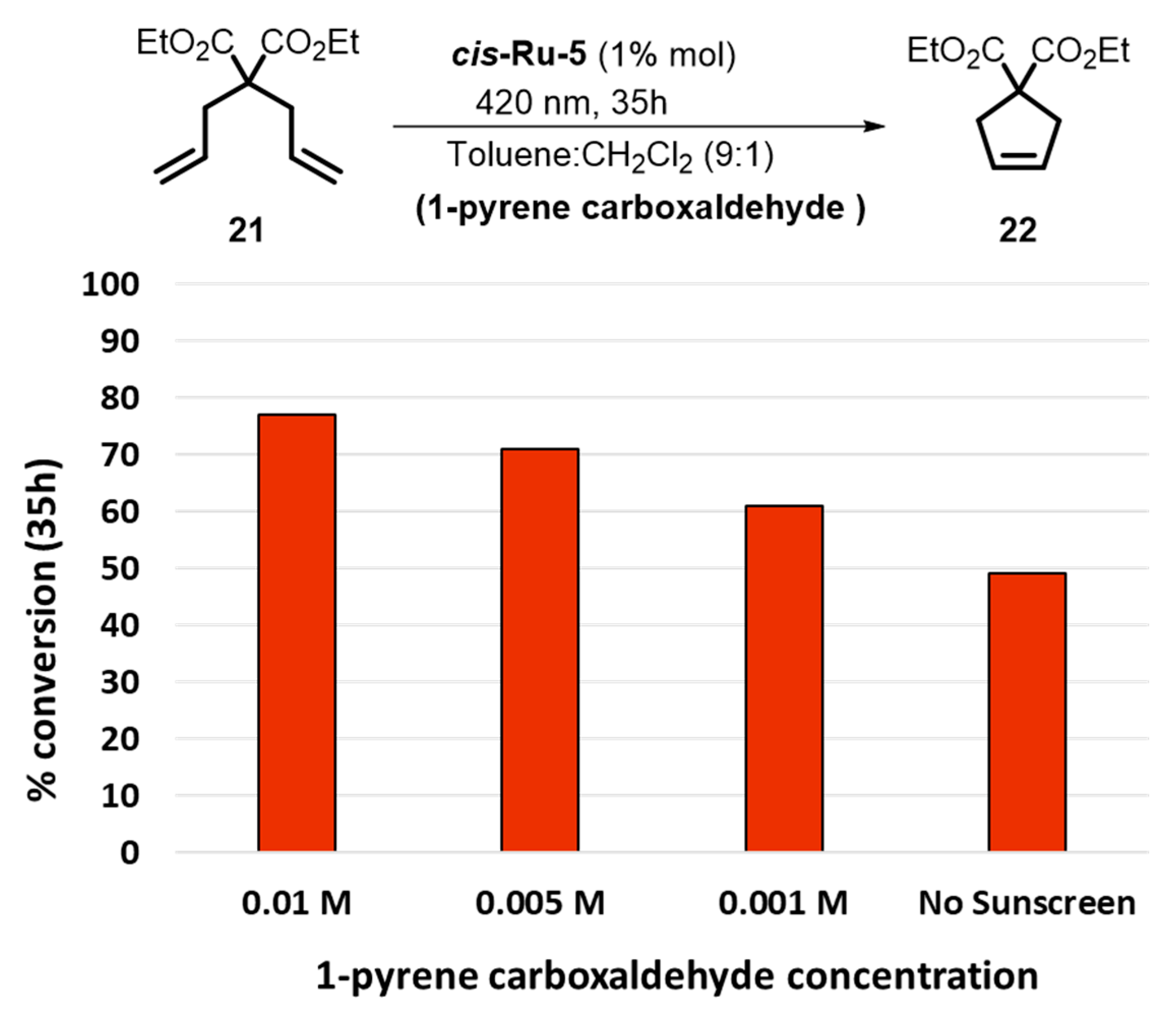
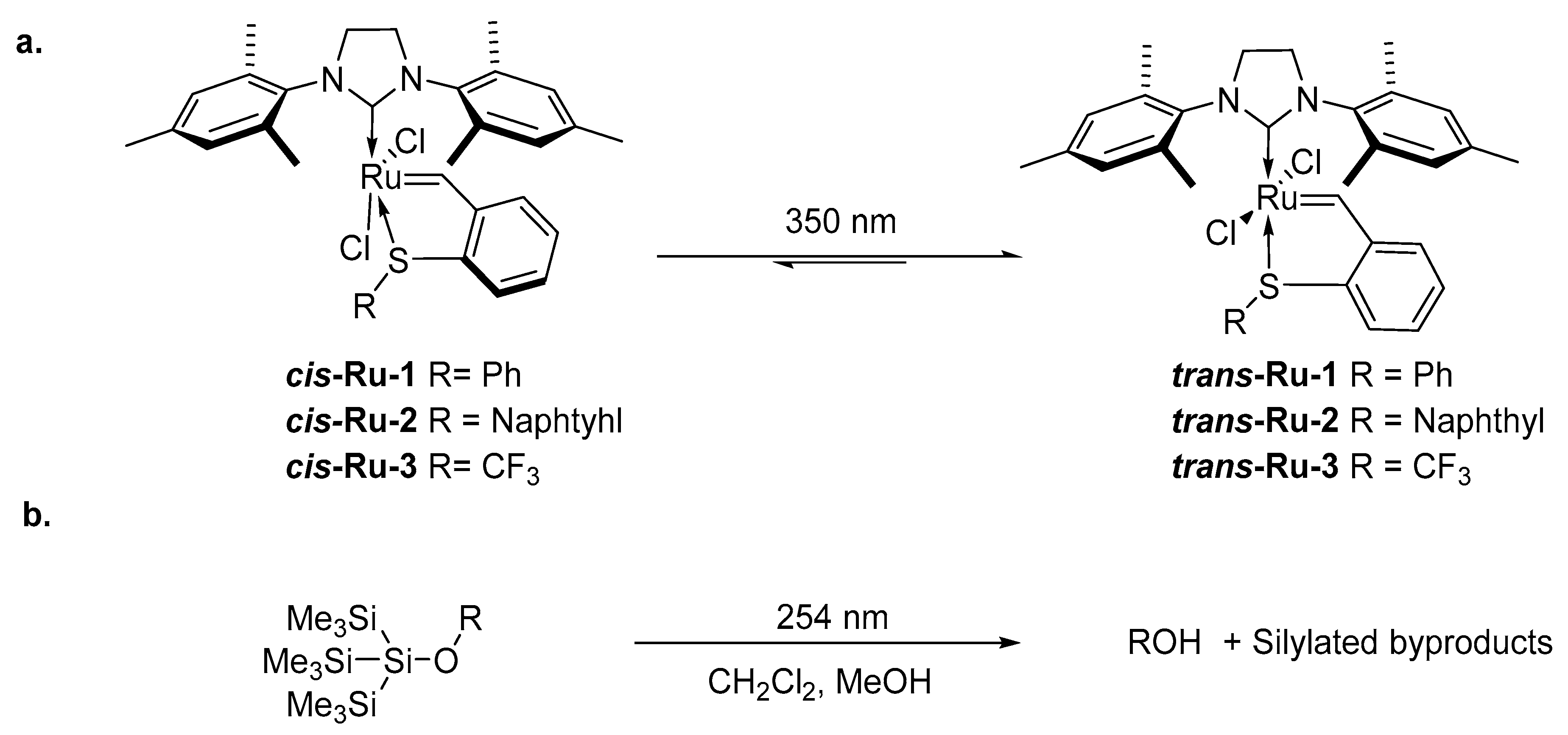
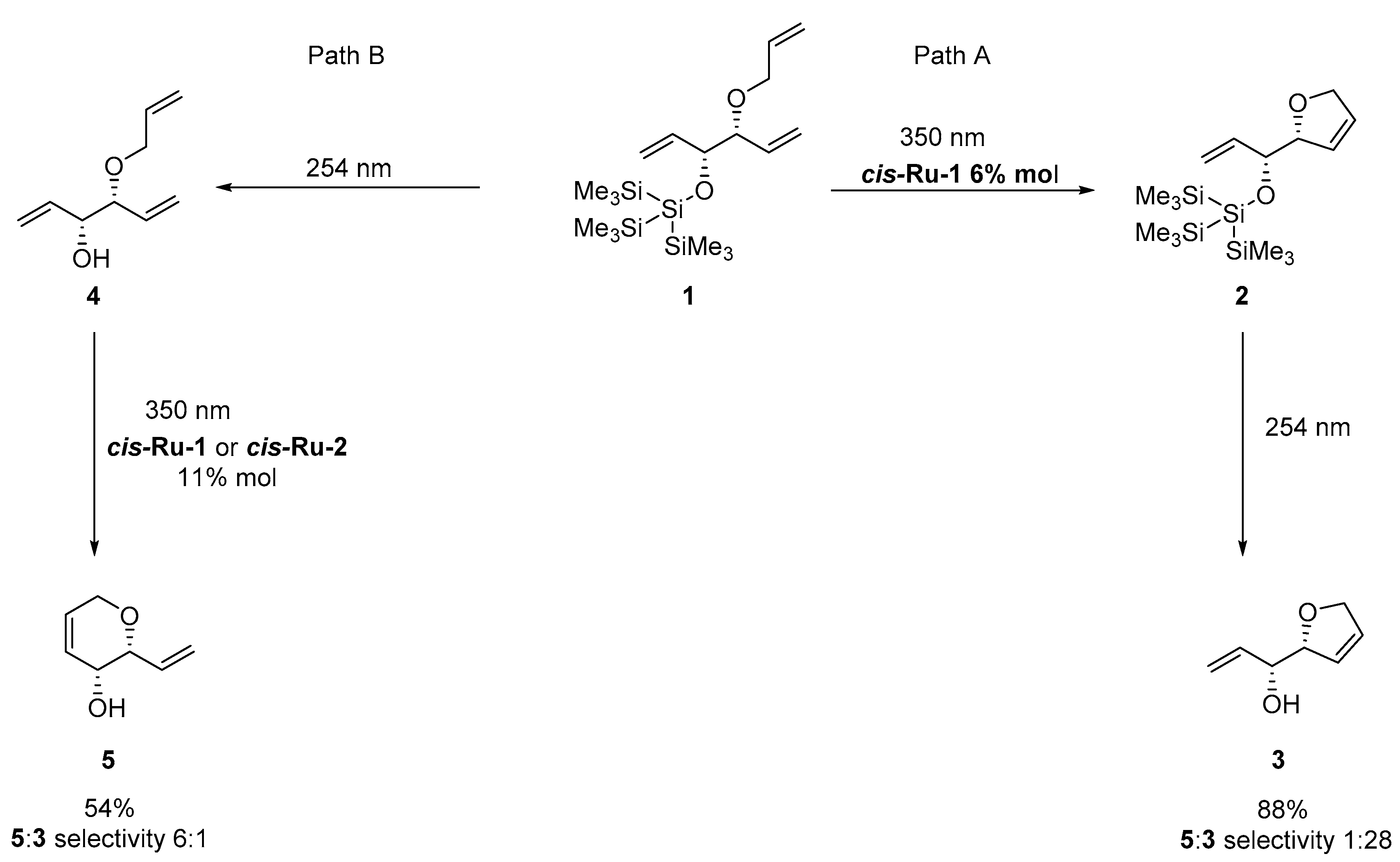
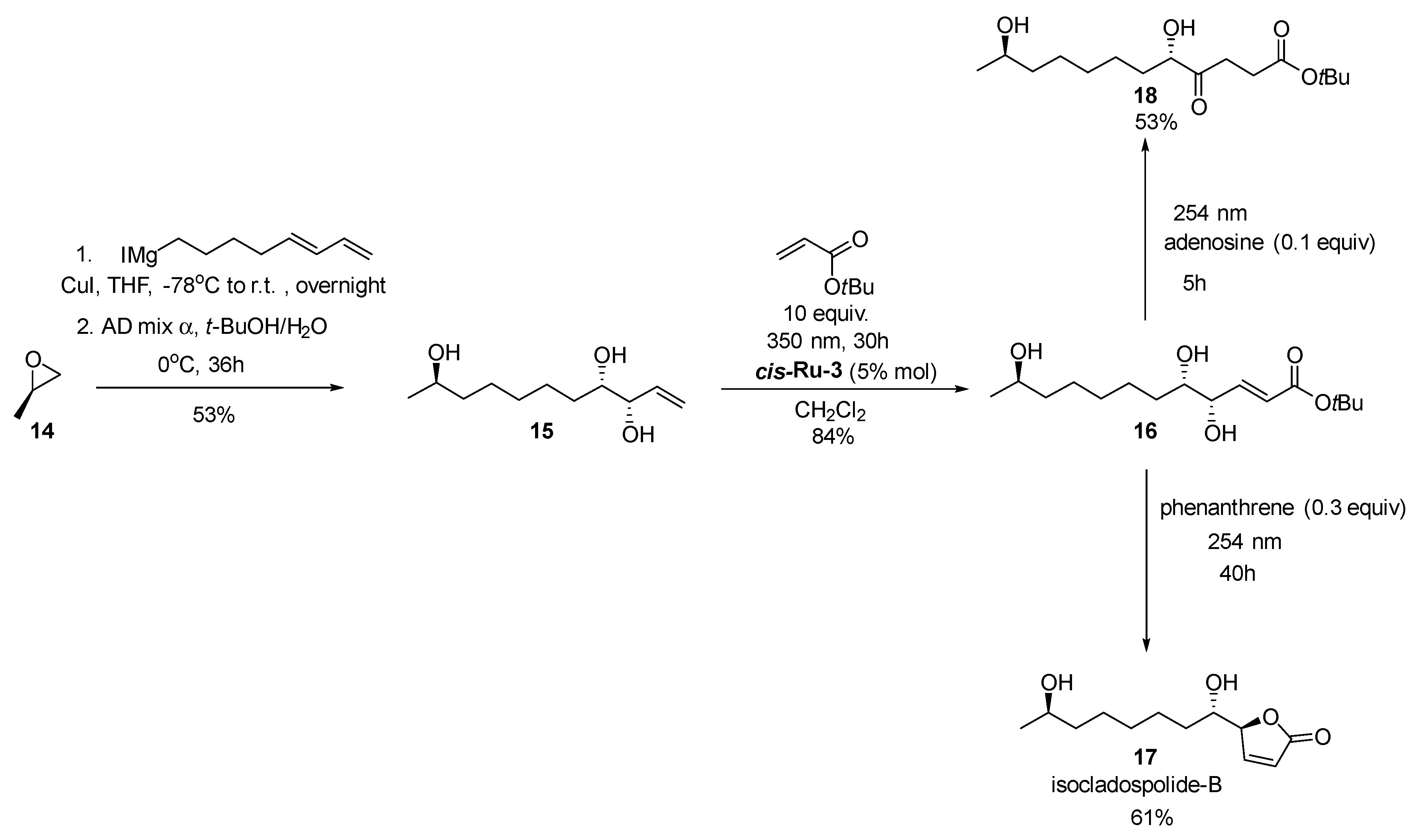
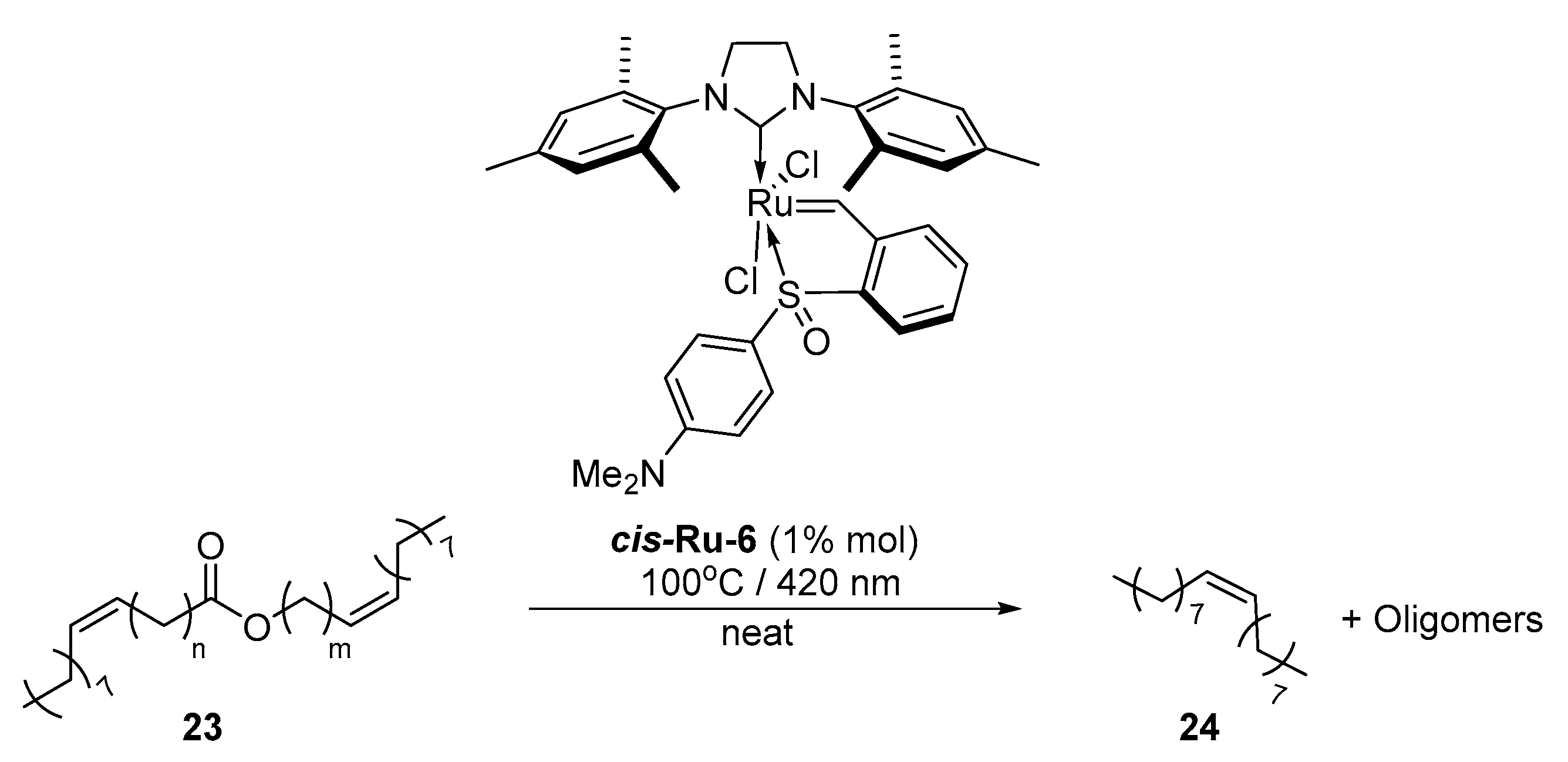
2. Development of the Method
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brieke, C.; Rohrbach, F.; Gottschalk, A.; Mayer, G.; Heckel, A. Light-Controlled Tools. Angew. Chem. Int. Ed. 2012, 51, 8446–8476. [Google Scholar] [CrossRef]
- Gruendling, T.; Oehlenschlaeger, K.K.; Frick, E.; Glassner, M.; Schmid, C.; Barner-Kowollik, C. Rapid UV Light-Triggered Macromolecular Click Conjugations via the Use of o-Quinodimethanes. Macromol. Rapid Commun. 2011, 32, 807–812. [Google Scholar] [CrossRef]
- Pauloehrl, T.; Delaittre, G.; Winkler, V.; Welle, A.; Bruns, M.; Börner, H.G.; Greiner, A.M.; Bastmeyer, M.; Barner-Kowollik, C. Adding Spatial Control to Click Chemistry: Phototriggered Diels-Alder Surface (Bio)functionalization at Ambient Temperature. Angew. Chem. Int. Ed. 2011, 51, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Remy, R.; Bochet, C.G. Arene–Alkene Cycloaddition. Chem. Rev. 2016, 116, 9816–9849. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Twilton, J.; Macmillan, D.W.C. Photoredox Catalysis in Organic Chemistry. J. Org. Chem. 2016, 81, 6898–6926. [Google Scholar] [CrossRef]
- Twilton, J.; Le, C.; Zhang, P.; Shaw, M.H.; Evans, R.W.; Macmillan, D.W.C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 52. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soulé, J.-F. Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef]
- Strieth-Kalthoff, F.; James, M.; Teders, M.; Pitzer, L.; Glorius, F. Energy transfer catalysis mediated by visible light: Principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. [Google Scholar] [CrossRef]
- Ligon, S.C.; Liska, R.; Stampfl, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef] [PubMed]
- Boydston, A.J.; Cao, B.; Nelson, A.; Ono, R.J.; Saha, A.; Schwartz, J.J.; Thrasher, C.J. Additive manufacturing with stimuli-responsive materials. J. Mater. Chem. A 2018, 6, 20621–20645. [Google Scholar] [CrossRef]
- Gilmore, K.; Seeberger, P.H. Continuous Flow Photochemistry. Chem. Rec. 2014, 14, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Beatty, J.W.; Douglas, J.J.; Miller, R.; McAtee, R.C.; Cole, K.P.; Stephenson, C.R.J. Photochemical Perfluoroalkylation with Pyridine N-Oxides: Mechanistic Insights and Performance on a Kilogram Scale. Chem 2016, 1, 456–472. [Google Scholar] [CrossRef]
- Cambie, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noel, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Plutschack, M.B.; Pieber, B.; Gilmore, K.; Seeberger, P.H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. [Google Scholar] [CrossRef]
- Noel, T. A personal perspective on the future of flow photochemistry. J. Flow Chem. 2017, 7, 87–93. [Google Scholar] [CrossRef]
- Hansen, M.J.; Velema, W.A.; Lerch, M.M.; Szymanski, W.; Feringa, B.L. Wavelength-selective cleavage of photoprotecting groups: Strategies and applications in dynamic systems. Chem. Soc. Rev. 2015, 44, 3358–3377. [Google Scholar] [CrossRef]
- Wong, C.-H.; Zimmerman, S.C. Orthogonality in organic, polymer, and supramolecular chemistry: From Merrifield to click chemistry. Chem. Commun. 2013, 49, 1679–1695. [Google Scholar] [CrossRef]
- Miguel, V.S.; Bochet, C.G.; Del Campo, A. Wavelength-Selective Caged Surfaces: How Many Functional Levels Are Possible? J. Am. Chem. Soc. 2011, 133, 5380–5388. [Google Scholar] [CrossRef]
- Blanc, A.; Bochet, C.G. Wavelength-Controlled Orthogonal Photolysis of Protecting Groups. J. Org. Chem. 2002, 67, 5567–5577. [Google Scholar] [CrossRef] [PubMed]
- Blanc, A.; Bochet, C.G. Isotope Effects in Photochemistry: Application to Chromatic Orthogonality. Org. Lett. 2007, 9, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Debieux, J.-L.; Bochet, C.G. The all-photochemical synthesis of an OGP(10–14) precursor. Chem. Sci. 2012, 3, 405–406. [Google Scholar] [CrossRef]
- Hiltebrandt, K.; Pauloehrl, T.; Blinco, J.P.; Linkert, K.; Börner, H.G.; Barner-Kowollik, C. λ-Orthogonal Pericyclic Macromolecular Photoligation. Angew. Chem. Int. Ed. 2015, 54, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Hiltebrandt, K.; Kaupp, M.; Molle, E.; Menzel, J.P.; Blinco, J.P.; Barner-Kowollik, C. Star polymer synthesis via ?-orthogonal photochemistry. Chem. Commun. 2016, 52, 9426–9429. [Google Scholar] [CrossRef]
- Menzel, J.P.; Noble, B.; Lauer, A.; Coote, M.L.; Blinco, J.P.; Barner-Kowollik, C. Wavelength Dependence of Light-Induced Cycloadditions. J. Am. Chem. Soc. 2017, 139, 15812–15820. [Google Scholar] [CrossRef]
- Menzel, J.P.; Feist, F.; Tuten, B.; Weil, T.; Blinco, J.P.; Barner-Kowollik, C. Light-Controlled Orthogonal Covalent Bond Formation at Two Different Wavelengths. Angew. Chem. Int. Ed. 2019, 58, 7470–7474. [Google Scholar] [CrossRef]
- Marschner, D.E.; Franck, C.O.; Abt, D.; Mutlu, H.; Barner-Kowollik, C. Fully independent photochemical reactivity in one molecule. Chem. Commun. 2019, 55, 9877–9880. [Google Scholar] [CrossRef]
- Kessler, M.; Glatthar, R.; Giese, B.; Bochet, C.G. Sequentially Photocleavable Protecting Groups in Solid-Phase Synthesis. Org. Lett. 2003, 5, 1179–1181. [Google Scholar] [CrossRef]
- Piloto, A.M.; Costa, S.P.G.; Gonçalves, M.S.T. Wavelength-selective cleavage of o-nitrobenzyl and polyheteroaromatic benzyl protecting groups. Tetrahedron 2014, 70, 650–657. [Google Scholar] [CrossRef]
- Rodrigues-Correia, A.; Knapp-Bühle, D.; Engels, J.W.; Heckel, A. Selective Uncaging of DNA through Reaction Rate Selectivity. Org. Lett. 2014, 16, 5128–5131. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Qureishi, Z.; Thomas, S.W. Light-Controlled Selective Disruption, Multilevel Patterning, and Sequential Release with Polyelectrolyte Multilayer Films Incorporating Four Photocleavable Chromophores. Chem. Mater. 2017, 29, 2951–2960. [Google Scholar] [CrossRef]
- Levin, E.; Mavila, S.; Eivgi, O.; Tzur, E.; Lemcoff, N.G. Regioselective Chromatic Orthogonality with Light-Activated Metathesis Catalysts. Angew. Chem. Int. Ed. 2015, 54, 12384–12388. [Google Scholar] [CrossRef] [PubMed]
- Ben-Asuly, A.; Aharoni, A.; Diesendruck, C.E.; Vidavsky, Y.; Goldberg, I.; Straub, B.F.; Lemcoff, N.G. Photoactivation of Ruthenium Olefin Metathesis Initiators. Organometallics 2009, 28, 4652–4655. [Google Scholar] [CrossRef]
- Brook, M.; Balduzzi, S.; Mohamed, M.; Gottardo, C. The photolytic and hydrolytic lability of sisyl (Si(SiMe3)3) ethers, an alcohol protecting group. Tetrahedron 1999, 55, 10027–10040. [Google Scholar] [CrossRef]
- Brook, M.; Gottardo, C.; Balduzzi, S.; Mohamed, M. The sisyl (tris(trimethylsilyl)silyl) group: A fluoride resistant, photolabile alcohol protecting group. Tetrahedron Lett. 1997, 38, 6997–7000. [Google Scholar] [CrossRef]
- Schmidt, B.; Nave, S. Synthesis of Dihydrofurans and Dihydropyrans with Unsaturated Side Chains Based on Ring Size-Selective Ring-Closing Metathesis. Adv. Synth. Catal. 2007, 349, 215–230. [Google Scholar] [CrossRef]
- Schmidt, B.; Nave, S. Control of ring size selectivity by substrate directable RCM. Chem. Commun. 2006, 2489–2491. [Google Scholar] [CrossRef]
- Eivgi, O.; Levin, E.; Lemcoff, N.G. Modulation of Photodeprotection by the Sunscreen Protocol. Org. Lett. 2015, 17, 740–743. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2012, 113, 119–191. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Plant, P.J.; Schofield, P. Photosensitive protective groups. Chem. Commun. 1966, 822. [Google Scholar] [CrossRef]
- Patchornik, A.; Amit, B.; Woodward, R.B. Photosensitive protecting groups. J. Am. Chem. Soc. 1970, 92, 6333–6335. [Google Scholar] [CrossRef]
- Šolomek, T.; Mercier, S.; Bally, T.; Bochet, C.G. Photolysis of ortho-nitrobenzylic derivatives: The importance of the leaving group. Photochem. Photobiol. Sci. 2012, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Šolomek, T.; Bochet, C.G.; Bally, T. The Primary Steps in Excited-State Hydrogen Transfer: The Phototautomerization of o- Nitrobenzyl Derivatives. Chem. A Eur. J. 2014, 20, 8062–8067. [Google Scholar] [CrossRef]
- Sutar, R.; Sen, S.; Eivgi, O.; Segalovich, G.; Schapiro, I.; Reany, O.; Lemcoff, N.G. Guiding a divergent reaction by photochemical control: Bichromatic selective access to levulinates and butenolides. Chem. Sci. 2017, 9, 1368–1374. [Google Scholar] [CrossRef]
- Sutar, R.; Levin, E.; Butilkov, D.; Goldberg, I.; Reany, O.; Lemcoff, N.G. A Light-Activated Olefin Metathesis Catalyst Equipped with a Chromatic Orthogonal Self-Destruct Function. Angew. Chem. Int. Ed. 2015, 55, 764–767. [Google Scholar] [CrossRef]
- Dai, H.-Q.; Kang, Q.-J.; Li, G.-H.; Shen, Y.-M. Three New Polyketide Metabolites from the Endophytic Fungal StrainCladosporium tenuissimum LR463 ofMaytenus hookeri. Helvetica Chim. Acta 2006, 89, 527–531. [Google Scholar] [CrossRef]
- Edwards, J.T.; Merchant, R.R.; McClymont, K.S.; Knouse, K.W.; Qin, T.; Malins, L.R.; Vokits, B.; Shaw, S.A.; Bao, D.-H.; Wei, F.-L.; et al. Decarboxylative alkenylation. Nature 2017, 545, 213–218. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and Recyclable Monomeric and Dendritic Ru-Based Metathesis Catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J.; Hoveyda, A.H. A Recyclable Ru-Based Metathesis Catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Chatterjee, A.K.; Choi, T.-L.; Sanders, D.P.; Grubbs, R.H. A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. [Google Scholar] [CrossRef] [PubMed]
- Eivgi, O.; Sutar, R.; Reany, O.; Lemcoff, N.G. Bichromatic Photosynthesis of Coumarins by UV Filter-Enabled Olefin Metathesis. Adv. Synth. Catal. 2017, 359, 2352–2357. [Google Scholar] [CrossRef]
- Diesendruck, C.E.; Iliashevsky, O.; Ben-Asuly, A.; Goldberg, I.; Lemcoff, N.G. Latent and Switchable Olefin Metathesis Catalysts. Macromol. Symp. 2010, 293, 33–38. [Google Scholar] [CrossRef]
- Vidavsky, Y.; Lemcoff, N.G. Light-induced olefin metathesis. Beilstein J. Org. Chem. 2010, 6, 1106–1119. [Google Scholar] [CrossRef]
- Lemcoff, N.G.; Eivgi, O. Turning the Light On: Recent Developments in Photoinduced Olefin Metathesis. Synthesis 2017, 50, 49–63. [Google Scholar] [CrossRef]
- Eivgi, O.; Guidone, S.; Frenklah, A.; Kozuch, S.; Goldberg, I.; Lemcoff, N.G. Photoactivation of Ruthenium Phosphite Complexes for Olefin Metathesis. ACS Catal. 2018, 8, 6413–6418. [Google Scholar] [CrossRef]
- Eivgi, O.; Vaisman, A.; Nechmad, N.B.; Baranov, M.; Lemcoff, N.G. Latent Ruthenium Benzylidene Phosphite Complexes for Visible-Light-Induced Olefin Metathesis. ACS Catal. 2019, 10, 2033–2038. [Google Scholar] [CrossRef]
- Segalovich-Gerendash, G.; Rozenberg, I.; Alassad, N.; Nechmad, N.B.; Goldberg, I.; Kozuch, S.; Lemcoff, N.G. Imposing Latency in Ruthenium Sulfoxide-Chelated Benzylidenes: Expanding Opportunities for Thermal and Photoactivation in Olefin Metathesis. ACS Catal. 2020, 10, 4827–4834. [Google Scholar] [CrossRef]













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eivgi, O.; Lemcoff, N.G. Sunscreen-Assisted Selective Photochemical Transformations. Molecules 2020, 25, 2125. https://doi.org/10.3390/molecules25092125
Eivgi O, Lemcoff NG. Sunscreen-Assisted Selective Photochemical Transformations. Molecules. 2020; 25(9):2125. https://doi.org/10.3390/molecules25092125
Chicago/Turabian StyleEivgi, Or, and N. Gabriel Lemcoff. 2020. "Sunscreen-Assisted Selective Photochemical Transformations" Molecules 25, no. 9: 2125. https://doi.org/10.3390/molecules25092125
APA StyleEivgi, O., & Lemcoff, N. G. (2020). Sunscreen-Assisted Selective Photochemical Transformations. Molecules, 25(9), 2125. https://doi.org/10.3390/molecules25092125