Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units



Abstract
1. Introduction
2. Results and Discussion
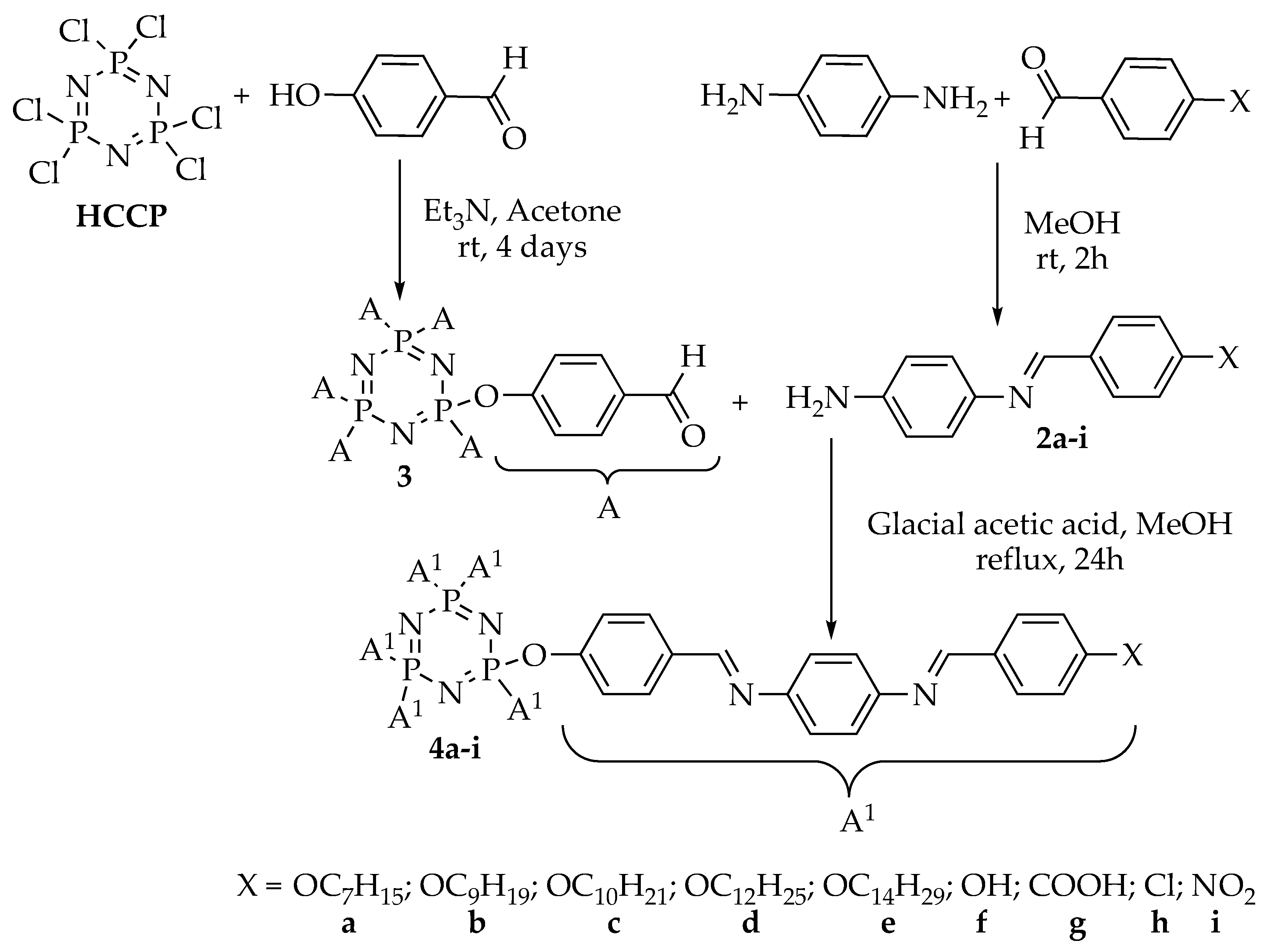
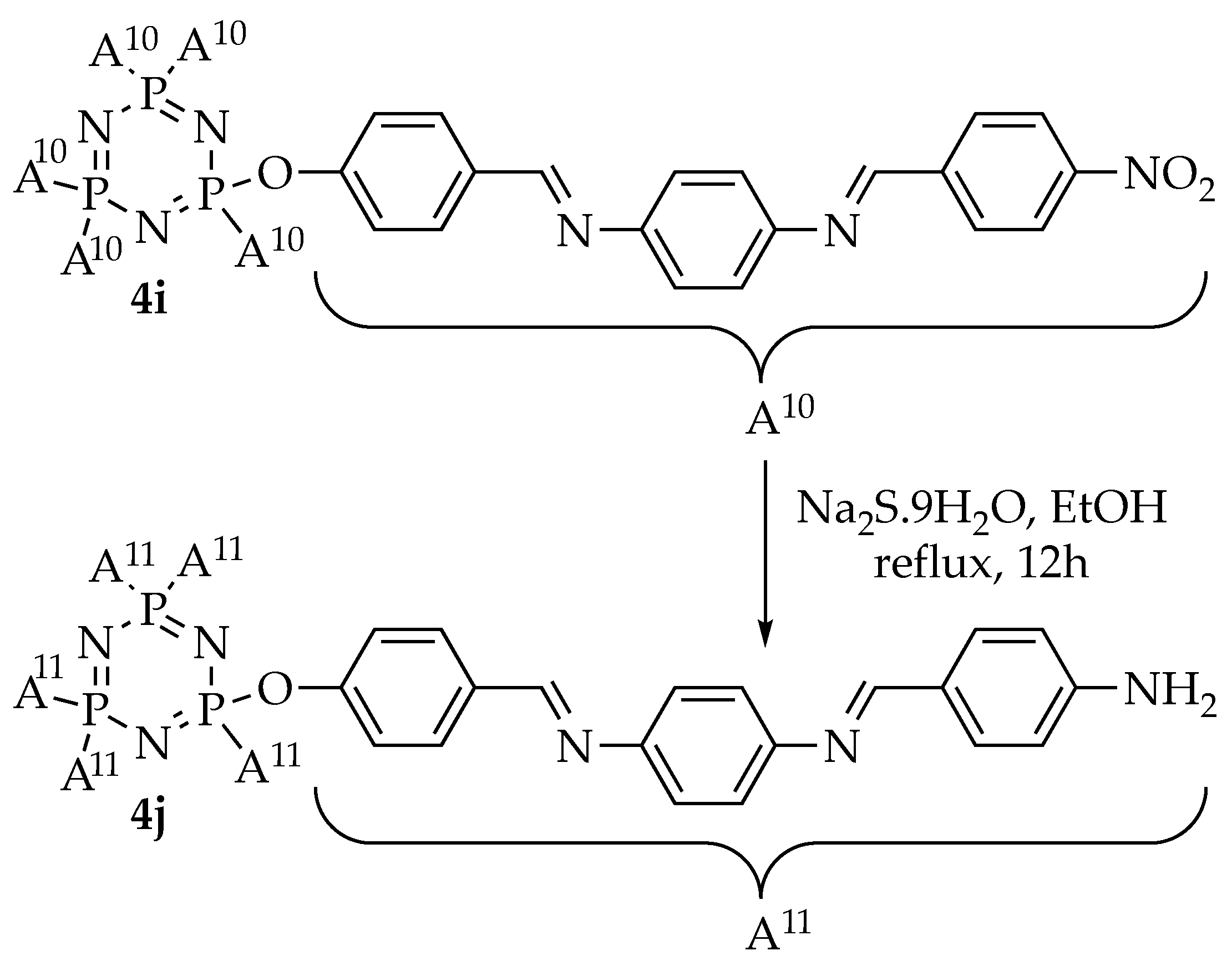
2.1. Synthesis of the Intermediates and Final Compounds
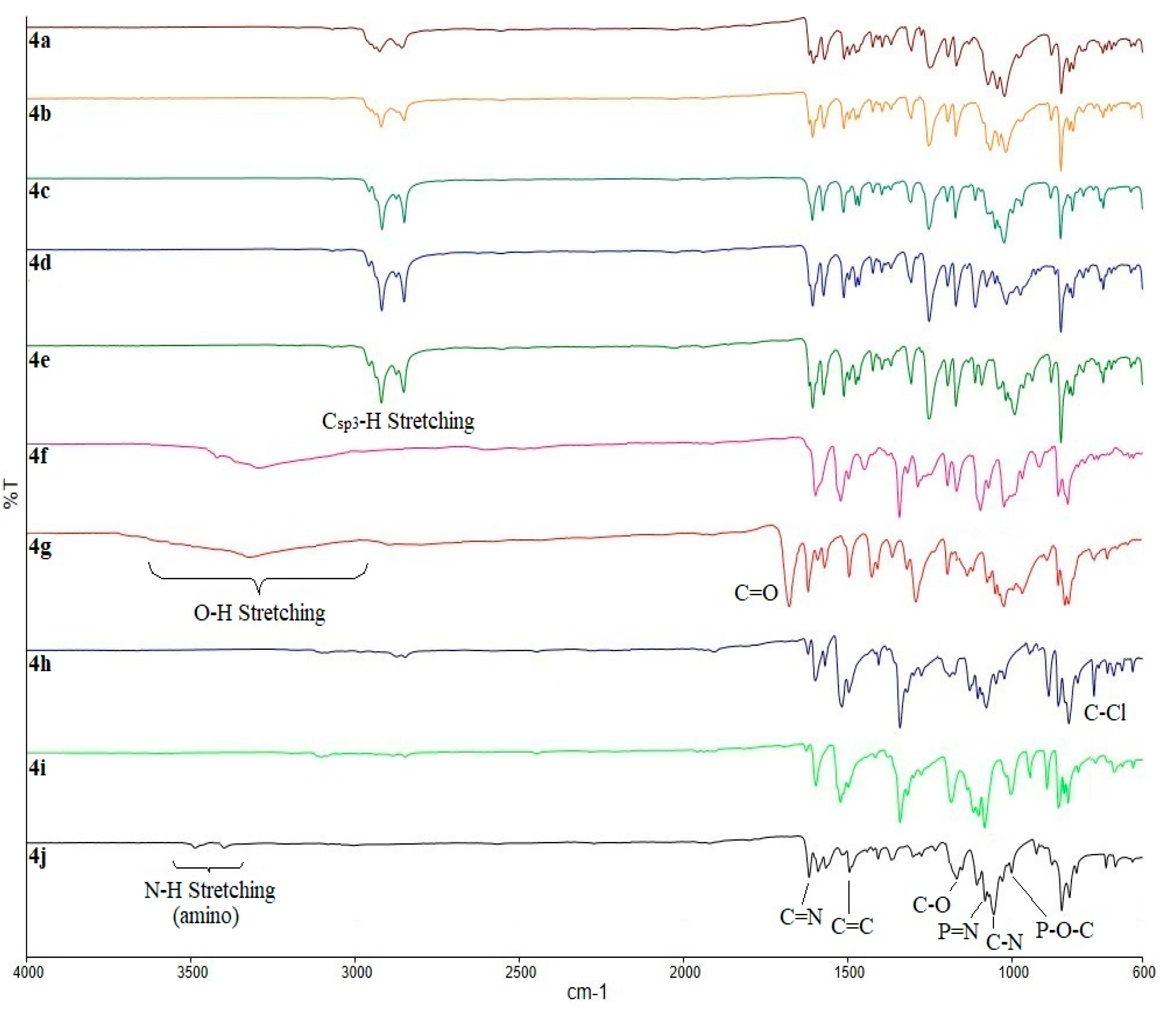
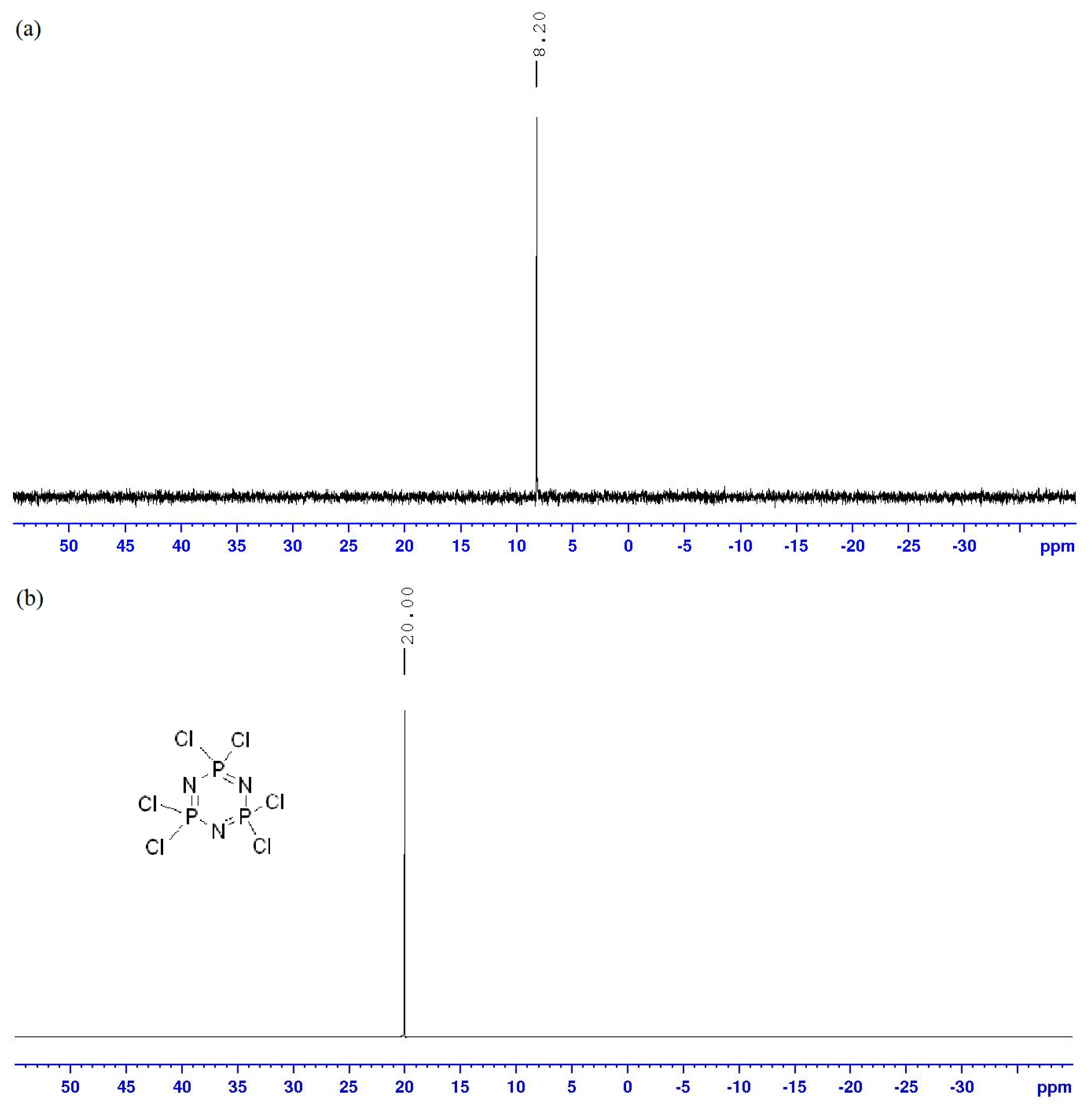
2.2. FTIR Spectral Data of the Intermediates and Final Compounds
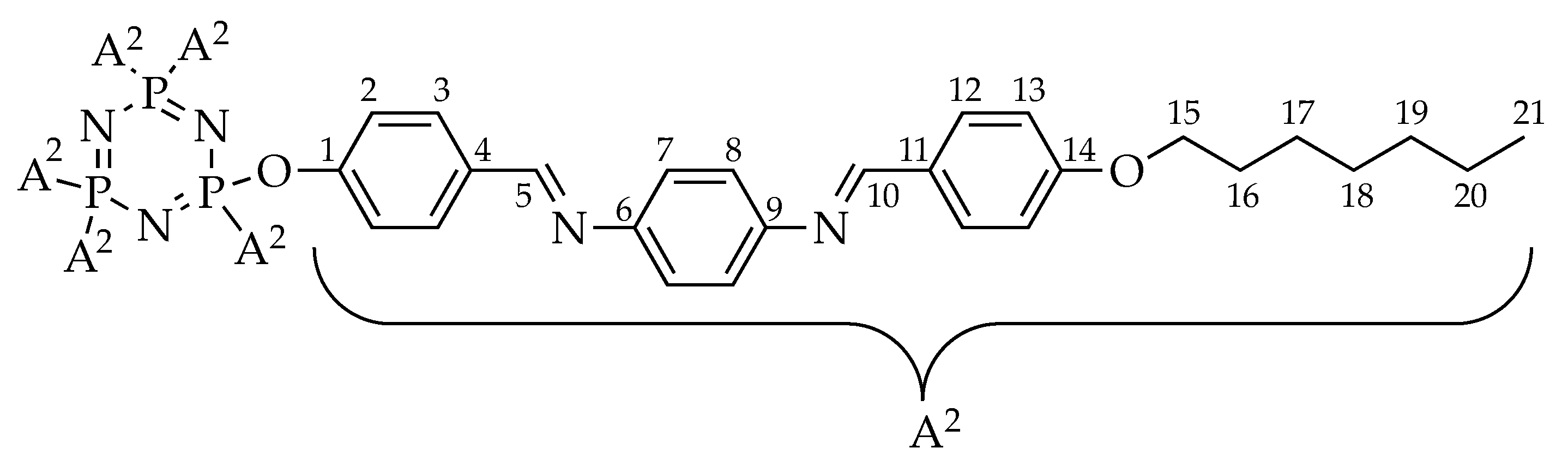
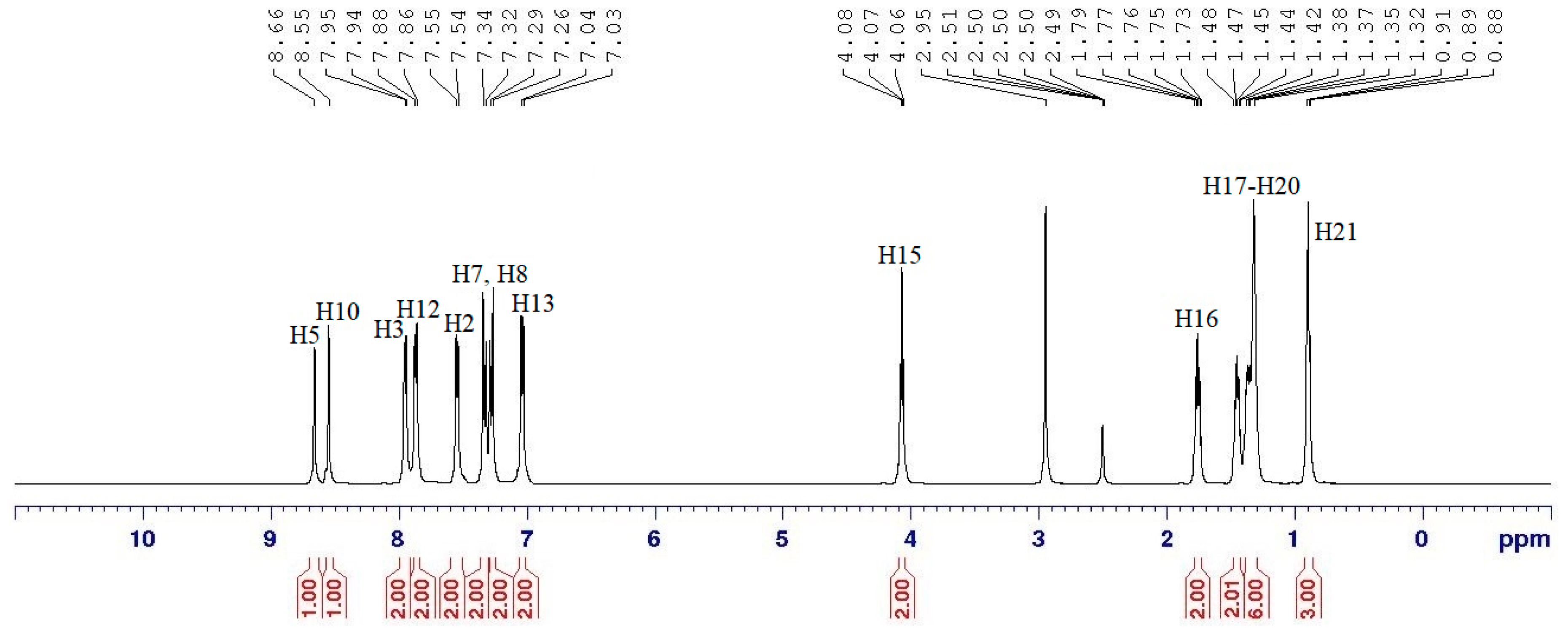
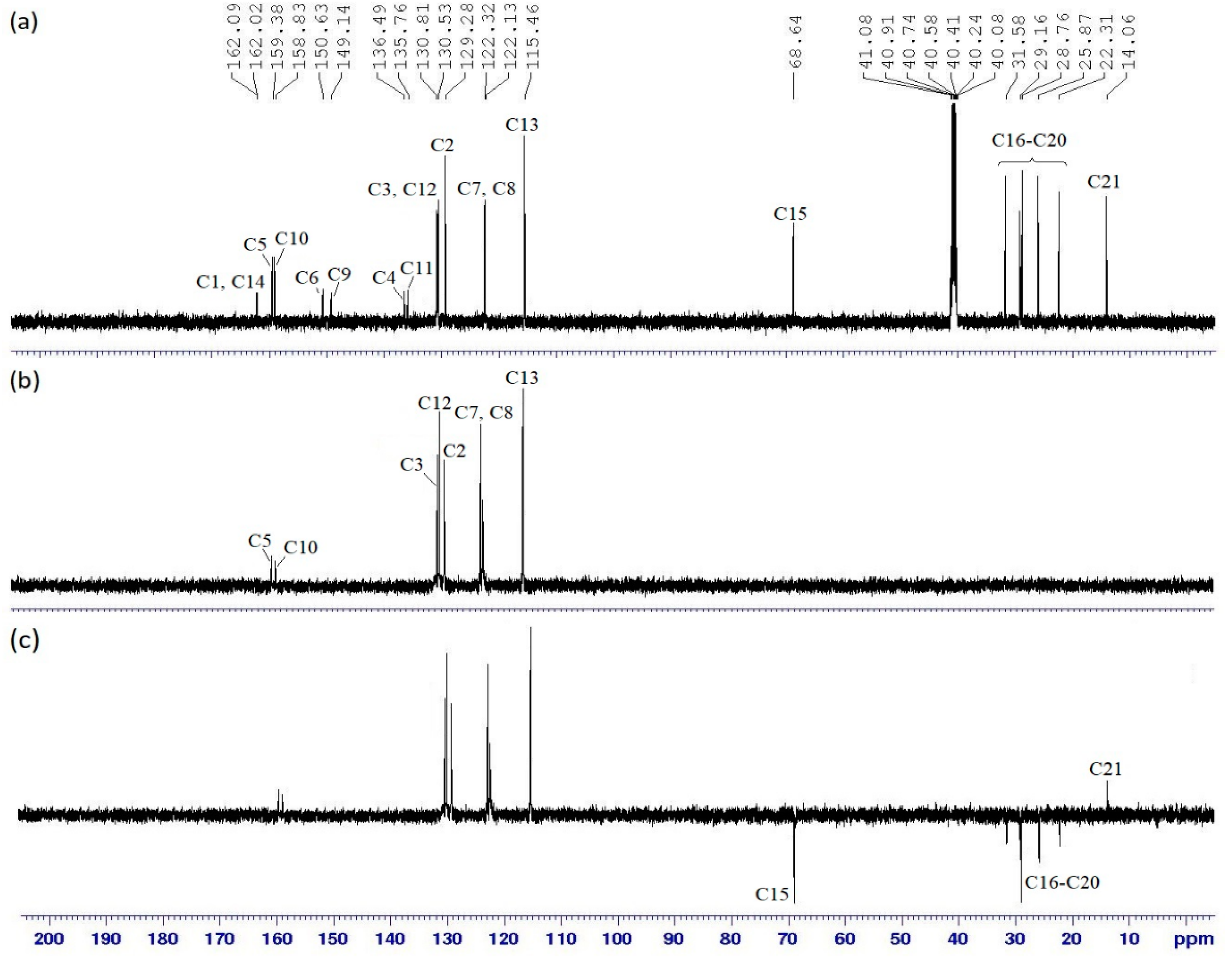
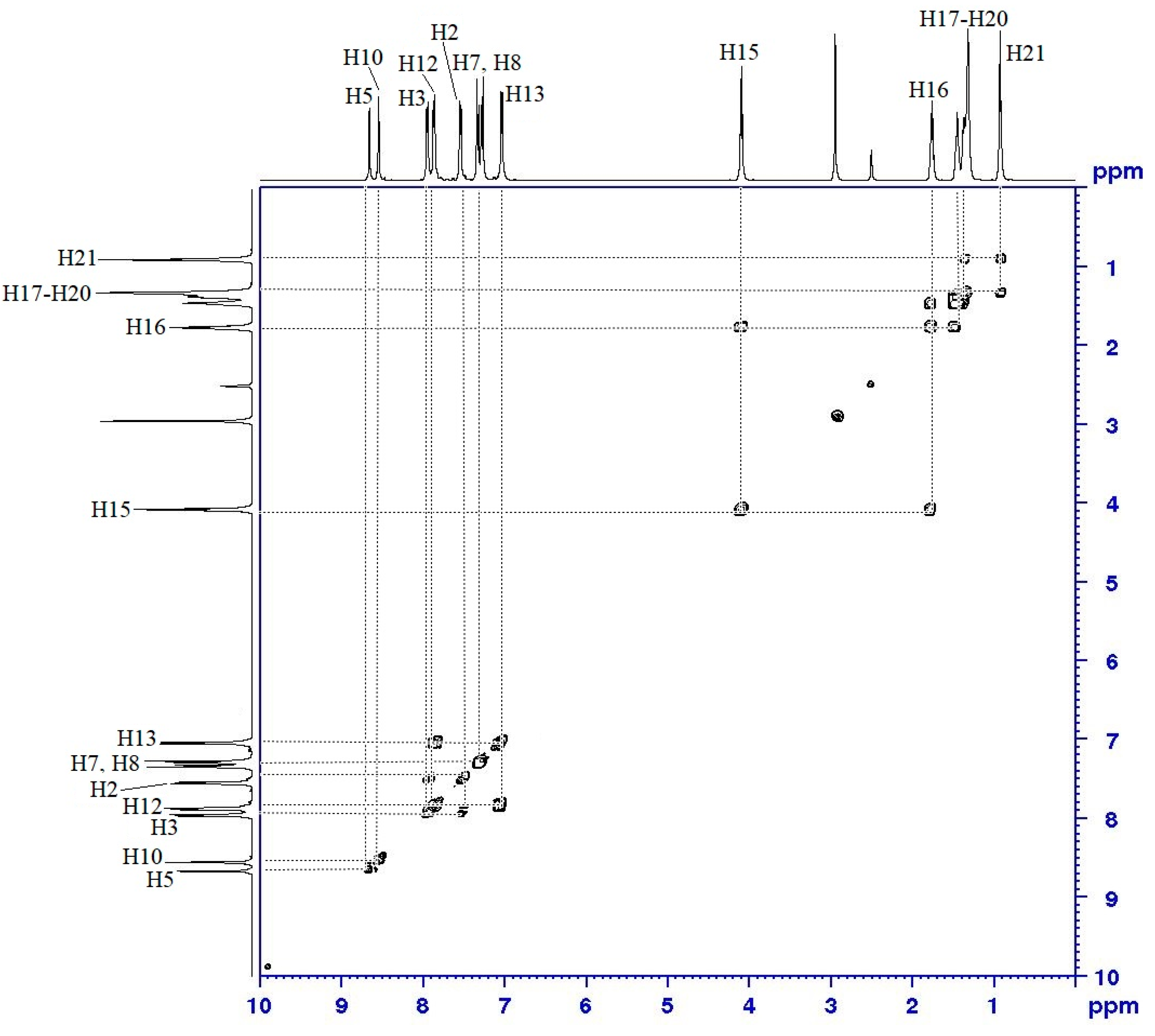
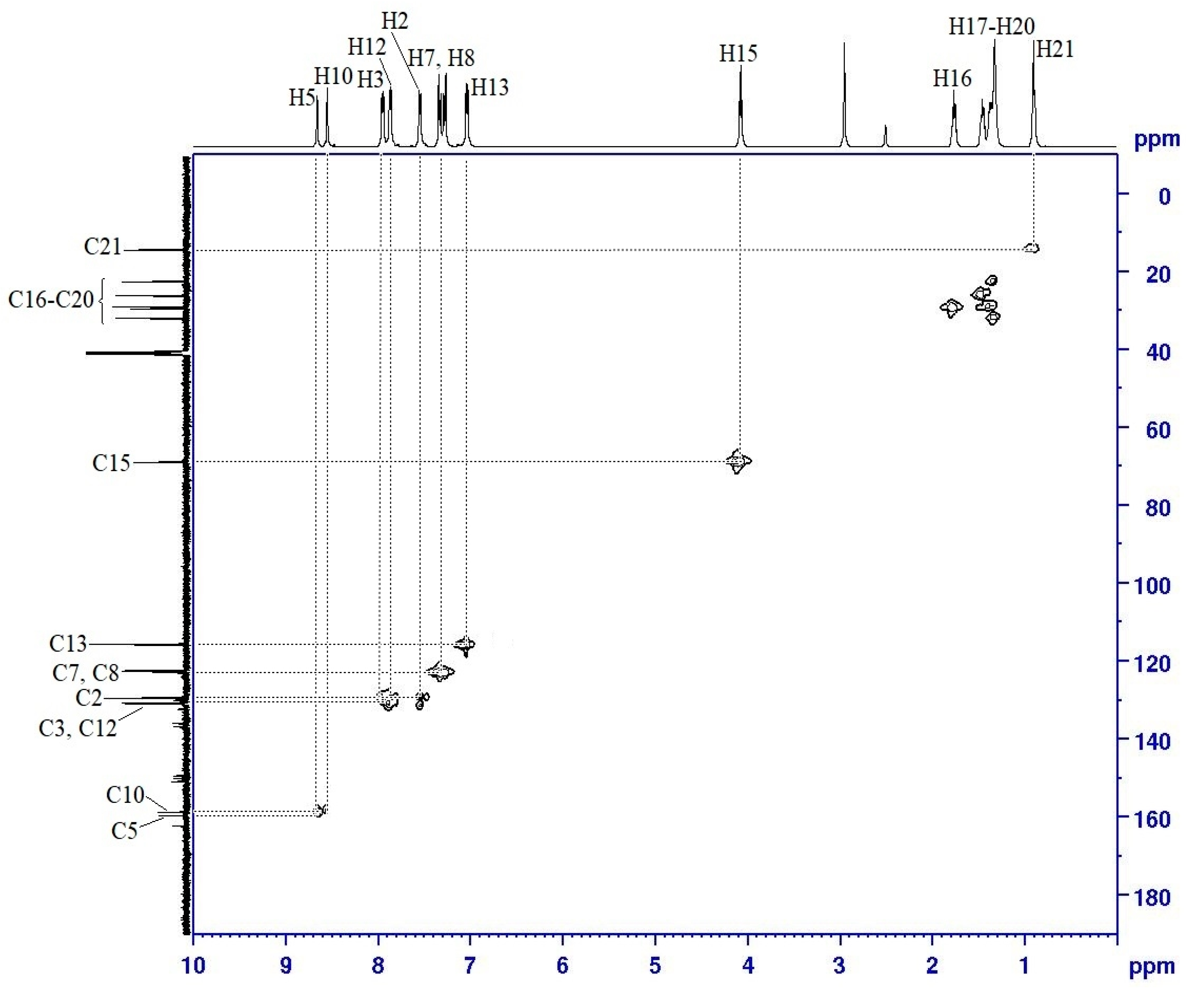
2.3. NMR Spectral Data of Final Compounds
2.4. Determination of Liquid-Crystal Properties Using POM
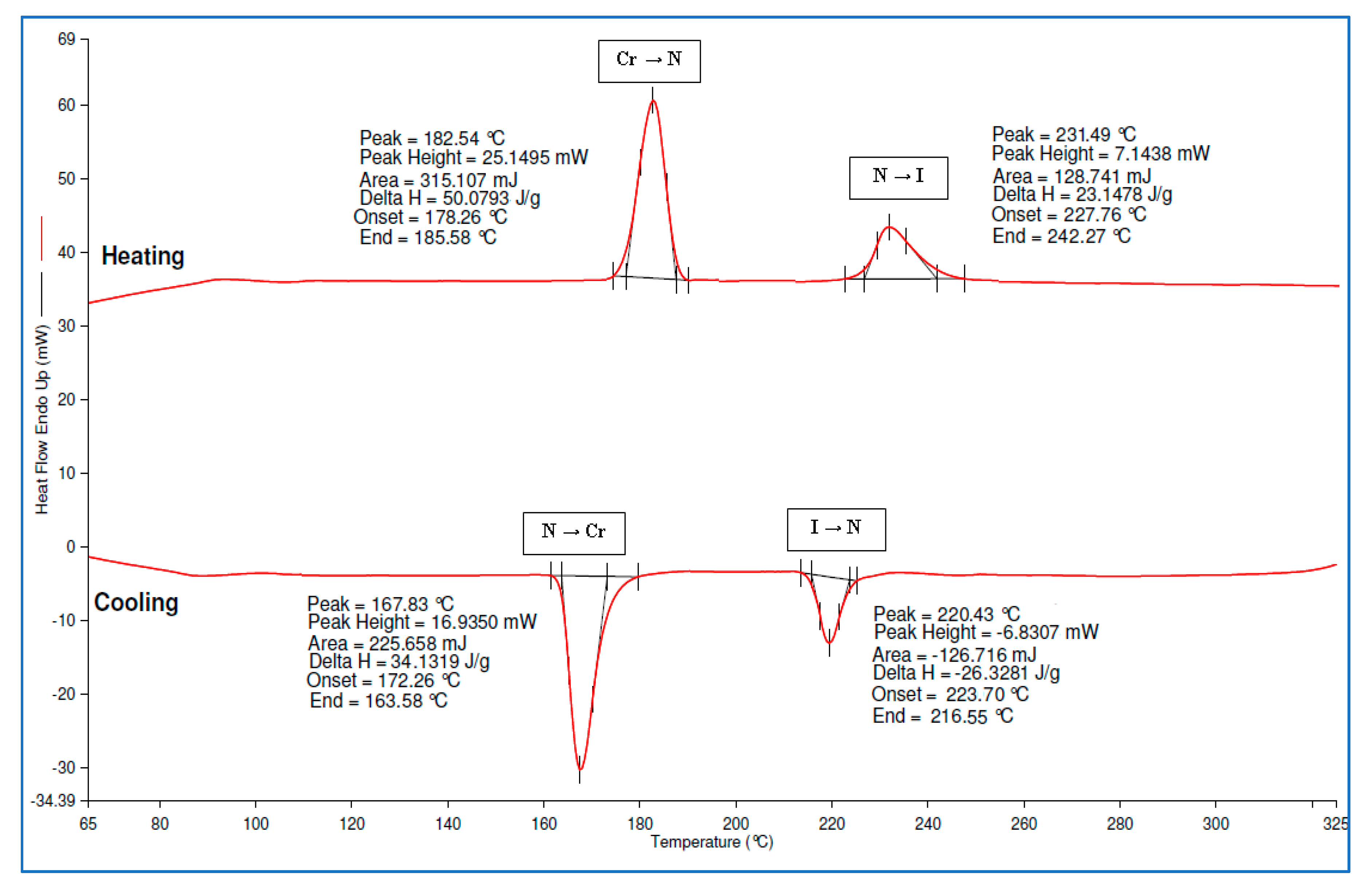
2.5. Determination of Thermal Transitions Using DSC
2.6. Structure–Properties Relationship
2.7. Determination of Fire-Retardant Properties Using LOI Testing
3. Materials and Methods
3.1. Chemicals
3.2. Instruments
3.3. Syntheses
3.3.1. Synthesis of 4-Alkoxybenzaldehyde, 1a–e
3.3.2. Synthesis of N-(4-Substitutedbenxylidene)benzene-1,4-diamine, 2a–i
3.3.3. Synthesis of Hexakis(4-Formlyphenoxy)cyclotriphosphazene, 3
3.3.4. Synthesis of Hexakis{4-((E)-((4-(((E)-4-substituted-benzylidene)amino)phenyl)imino)methyl) phenoxy}triazaphosphazene, 4a–i
3.3.5. Synthesis of Hexakis{4-((E)-((4-(((E)-4-amino-benzylidene)amino)phenyl)imino)methyl)phenoxy} triazaphosphazene, 4j
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Allcock, H.R. Recent advances in phosphazene (phosphonitrilic) chemistry. Chem. Rev. 1972, 72, 315–356. [Google Scholar] [CrossRef]
- Joaquín, B.; Manuel, B.; Josefina, J.; Antonio, L.; Pilar, M.M.; Luis, O.; José, L.S.; Irene, Z. Columnar mesomorphic organisations in cyclotriphosphazene. J. Am. Chem. Soc. 2005, 127, 8994–9002. [Google Scholar]
- He, Q.; Dai, H.; Tan, X.; Cheng, X.; Liu, F.; Tschierske, C. Synthesis and characterisation of room temperature columnar mesogens of cyclotriphosphazene with Schiff base unit. J. Mater. Chem. C 2013, 43, 7148–7154. [Google Scholar] [CrossRef]
- Chandrasekhar, S. Liquid Crystals, 2nd ed.; Cambridge University Press: Cambridge, UK, 1992; ISBN 0-521-41747-3. [Google Scholar]
- Allcock, H.R. Phosphorus-Nitrogen Compounds; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- Moriya, K.; Suzuki, T.; Kawanishi, Y.; Masuda, T.; Mizusaki, H.; Nakagawa, S.; Ikematsu, H.; Mizuno, K.; Yano, S.; Kajiwara, M. Liquid-crystalline phase transition in organophosphazene. Appl. Organomet. Chem. 1998, 12, 771–779. [Google Scholar] [CrossRef]
- Zhu, L.; Zhu, Y.; Pan, Y.; Huang, Y.W.; Huang, X.B.; Tang, X.Z. Fully cross-linked poly[-cyclotriphosphazene-co-(4, 40-sulfonyldiphenol)] microspheres via precipitation polymerisation and their superior thermal properties. Macromol. React. Eng. 2007, 1, 45–52. [Google Scholar] [CrossRef]
- Lejeune, N.; Dez, I.; Jaffres, P.A.; Lohier, J.F.; Madec, P.J.; Santos, J.S.O. Synthesis, crystal structure and thermal properties of phosphorylated cyclotriphosphazenes. Eur. J. Inorg. Chem. 2008, 1, 138–143. [Google Scholar] [CrossRef]
- Allcock, H.R. The crucial role of inorganic ring chemistry in the development of new polymers. Phosphorus Sulfur Silicon Relat. Elem. 2004, 179, 661–671. [Google Scholar] [CrossRef]
- Barberá, J.; Bardají, M.; Jiménez, J.; Laguna, A.; Martínez, M.P.; Oriol, L.; Serrano, J.L.; Zaragozano, I. Columnar mesomorphic organizations in cyclotriphosphazenes. J. Am. Chem. Soc. 2005, 127, 8994–9002. [Google Scholar] [CrossRef]
- Heal, H.G. The Inorganic Heterocyclic Chemistry of Sulphur, Nitrogen and Phosphorus; Academic Press: New York, NY, USA, 1980; p. 214. [Google Scholar]
- Allen, C.W. The Chemistry of Inorganic Homo- and Heterocycles; Haiduc, I., Sowerby, D.B., Eds.; Academic Press: New York, NY, USA, 1987; p. 2. [Google Scholar]
- Allen, C.W. Organophosphorus Chemistry; Hutchinson, D.W., Walker, B.J., Eds.; Royal Society of Chemistry: London, UK, 1990; Volume 21, p. 368. [Google Scholar]
- Allcock, H.R. Chemistry and Applications of Polyphosphazenes; Wiley Interscience: New York, NY, USA, 2003. [Google Scholar]
- Chen-Yang, Y.W.; Lee, H.F.; Yuan, C.Y. A Flame-retardant phosphate and cyclotriphosphazene containing epoxy resin: Synthesis and properties. J. Polym. Sci. A Polym. Chem. 2000, 38, 972–981. [Google Scholar] [CrossRef]
- Shin, Y.J.; Ham, Y.R.; Kim, S.H.; Lee, D.H.; Kim, S.B.; Park, C.S.; Yoo, Y.M.; Kim, J.G.; Kwon, S.H.; Shin, J.S. Application of cyclotriphosphazene derivatives as flame retardants for ABS. J. Ind. Eng. Chem. 2010, 16, 364–367. [Google Scholar] [CrossRef]
- Salmeia, K.A.; Gaan, S.; Malucelli, G. Recent advances for flame retardancy of textiles based on phosphorus chemistry. Polymers 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Wendels, S.; Chavez, T.; Bonnet, M.; Salmeia, K.A.; Gaan, S. Recent development in organophosphorus flame retardants containing P-C bond and their applications. Materials 2017, 10, 784. [Google Scholar] [CrossRef] [PubMed]
- Senthil, S.; Kannan, P. Ferrocene-based organophosphorus liquid-crystalline polymers: Synthesis and characterization. J. Polym. Sci. A Polym. Chem. 2001, 39, 2396–2403. [Google Scholar] [CrossRef]
- Kim, C.; Allcock, H.R. A liquid crystalline poly (organophosphazene). Macromolecules 1987, 20, 1726–1727. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kim, C. Liquid crystalline phosphazenes. High polymeric and cyclic trimeric systems with aromatic azo side groups. Macromolecules 1989, 22, 2596–2602. [Google Scholar] [CrossRef]
- Singh, S. Phase transitions in liquid crystals. Phys. Rep. 2000, 324, 107–269. [Google Scholar] [CrossRef]
- Iqbal, D.; Samiullah, M.S. Photo-responsive shape-memory and shape-changing liquid-crystal polymer networks. Materials 2013, 6, 116–142. [Google Scholar] [CrossRef]
- Balakrishnan, V.; Phan, H.P.; Dinh, T.; Dao, D.V.; Nguyen, N.T. Thermal flow sensors for harsh environments. Sensors 2017, 17, 2061. [Google Scholar] [CrossRef]
- Selvarasu, C.; Kannan, O. Effect of azo and ester linkages on rod shapd Schiff base liquid crystals and their photophysical investigations. J. Mol. Struct. 2016, 1125, 234–240. [Google Scholar] [CrossRef]
- El-Wahab, H.S. Synthesis and characterisation of the flame retardant properties and corrosion resistance of Schiff’s base compounds incorporated into organic coating. Pigment. Resin Technol. 2015, 44, 101–108. [Google Scholar] [CrossRef]
- Gouri, M.E.; Bachiri, A.E.; Hegazi, S.E.; Ziraoui, R.; Rafik, M.; Harfi, A.E. Thermal degradation of a reactive flame retardant based on cyclotriphosphazene and its blend with DGEBA epoxy resin. Polym. Degrad. Stab. 2009, 94, 2101–2106. [Google Scholar] [CrossRef]
- Habib, T.; Morteza, J.; Azam, N. Mechanical and thermo-gravimetric properties of unsaturated polyester resin blended with FGD gypsum. Constr. Build. Mater. 2018, 163, 438–445. [Google Scholar]
- Boyce, R.S.; Cherian, J.; Calanasan, C.; Sihombing, M.S.A. Lipopeptide Compounds and Their Use. PCT Int. Appl. Patent No. WO2010062264A1, 2010. [Google Scholar]
- Raghuwanshi, P.B.; Malalle, P.V. Synthesis and substituted Schiff’s bases and their antimicrobial activity. Pharma Chem. 2014, 6, 262–266. [Google Scholar]
- Rong, Y.; Wentian, H.; Liang, X.; Yan, S.; Jinchun, L. Synthesis, mechanical and fire behaviours of rigid polyurethane foam with a reactive flame retardant containing phosphazene and phosphate. Polym. Degrad. Stab. 2015, 122, 102–109. [Google Scholar]
- Jamain, Z.; Khairuddean, M.; Saidin, S.A. Synthesis and characterisation of 1, 4-phenylenediamine derivatives containing hydroxyl and cyclotriphosphazene as terminal group. J. Mol. Struct. 2019, 1186, 293–302. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, Y.; Zhang, Y.; Cao, Z. Phosphorus-Containing Schiff Base Derivative Expanded Flame Retardant and Its Preparation. Patent No. CN102732041A, 2012. [Google Scholar]
- Deyan, Z.; Yangyang, W.; Jiong, J.; Wenzhu, Y.; Baofeng, Q.; Xia, L.; Xuan, S. H-Bonding and charging mediated affregation and emission for fluorescene turn-on detection of hydrazine hydrate. Chem. Comm. 2015, 51, 10656–10659. [Google Scholar]
- Thaker, B.T.; Patel, P.H.; Vansadiya, A.D.; Kanojiya, J.B. Substitution effects on the liquid crystalline properties of thermotropic liquid crystals containing Schiff base chalcone linkages. Mol. Cryst. Liq. Cryst. 2009, 515, 135–147. [Google Scholar] [CrossRef]
- Khoo, I.C. Liquid Crystals, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 60–65. [Google Scholar]
- Andrienko, D. Introduction to liquid crystals. J. Mol. Liq. 2018, 267, 520–541. [Google Scholar] [CrossRef]
- Kelker, H.; Hatz, R. Handbook of Liquid Crystals; Verlag Chemie: Weinheim, Germany; Deerfield Beach, FL, USA, 1980. [Google Scholar]
- Sudhakar, S.; Narasimhaswamy, T.; Srinivasan, K.S.V. Towards the molecular-statistical modelling of the optically isotropic mesophase in neat systems: From the thermodynamic point of view. Liq. Cryst. 2000, 27, 1525–1559. [Google Scholar] [CrossRef]
- Sakagami, S.; Nakamizo, M. Liquid crystalline properties of N-(alkoxybenzylidene)-4-halogenoanilines. Bull. Chem. Soc. Jpn. 1980, 53, 265–266. [Google Scholar] [CrossRef]
- Singh, S.; Dunmur, D.A. Liquid Crystals: Fundamentals; World Scientific Publishing Co. Pte. Ltd.: London, UK, 2002. [Google Scholar]
- Sharma, V.S.; Patel, R.B. Effect of alkyl chain in the terminal ester group on mesomorphic properties of new rod like homologous series: Synthesis and characterization. Mol. Cryst. Liq. Cryst. 2017, 643, 62–75. [Google Scholar] [CrossRef]
- Galewski, Z.; Coles, H.J. Liquid crystalline properties and phase situations in 4-chlorobenzylidene-4′-alkylanilines. J. Mol. Liq. 1999, 79, 77–87. [Google Scholar] [CrossRef]
- Frenkel, D.; Mulder, B.M. The hard ellipsoid-of-revolution fluid: I. Monte Carlo stimulations. Mol. Phys. 1985, 55, 1171–1192. [Google Scholar] [CrossRef]
- Demus, D.; Goodby, J.; Gray, G.W.; Spiess, H.-W.; Vill, V. Handbook of Liquid Crystals, Low Molecular Weight Liquid Crystals I; Wiley-VCH: New York, NY, USA, 1998; Volume 2A. [Google Scholar]
- Jamain, Z.; Khairuddean, M.; Zulbaharen, N.N.; Chung, T.K. Synthesis, characterisation and determination of mesophase transition of azo-azomethine derivatives with different terminal chain lengths. Malays. J. Chem. 2019, 22, 73–85. [Google Scholar]
- Thaker, B.T.; Kanojiya, J.B.; Tandel, R.S. Effects of different terminal substituent on the mesomorphic behavior of some azo-Schiff base and azo-ester-based liquid crystals. Mol. Cryst. Liq. Cryst. 2010, 528, 120–137. [Google Scholar] [CrossRef]
- Prajapati, A.K.; Pandya, H.M. Effect of a lateral chloro group on azomesogens containing the naphthalene moiety. Mol. Cryst. Liq. Cryst. 2003, 393, 31–39. [Google Scholar] [CrossRef]
- Rong, Y.; Bo, W.; Xiaofeng, H.; Binbin, M.; Jinchun, L. Synthesis and characterisation of flame retardant rigid polyurethane foam based on a reactive flame retardant containing phosphazene and cyclotriphosphazene. Polym. Degrad. Stab. 2017, 144, 62–69. [Google Scholar]
- Liu, Y.L.; Chiu, Y.C.; Chen, T.Y. Phosphorus-containing polyaryloxydiphenylsilanes with high flame retardance arising from a phosphorus-silicon synergic effect. Polym. Int. 2003, 52, 1256–1261. [Google Scholar] [CrossRef]
- Faghihi, K.; Hagibeygi, M. New aromatic polyamide with azo and phosphine oxide groups in the main chain. Turk. J. Chem. 2007, 31, 65–73. [Google Scholar]
- Zahra, S.; Nasrin, J.; Shahla, S. Flame retardant cotton fibres produced using novel synthesised halogen-free phosphoramide nanoparticles. Carbohydr. Polym. 2015, 118, 183–198. [Google Scholar]
- Bian, X.C.; Chen, L.; Wang, J.S.; Wang, Y.Z. A novel thermotropic liquid crystalline copolyester containing phosphorus and aromatic ether moiety toward high flame retardancy and low mesophase temperature. J. Polym. Sci. A Polym. Chem. 2010, 48, 1182–1189. [Google Scholar] [CrossRef]
- Mequanint, K.; Sanderson, R.; Pasch, H. Thermogravimetric study of phosphated polyurethane ionomers. Polym. Degrad. Stab. 2002, 77, 121–128. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
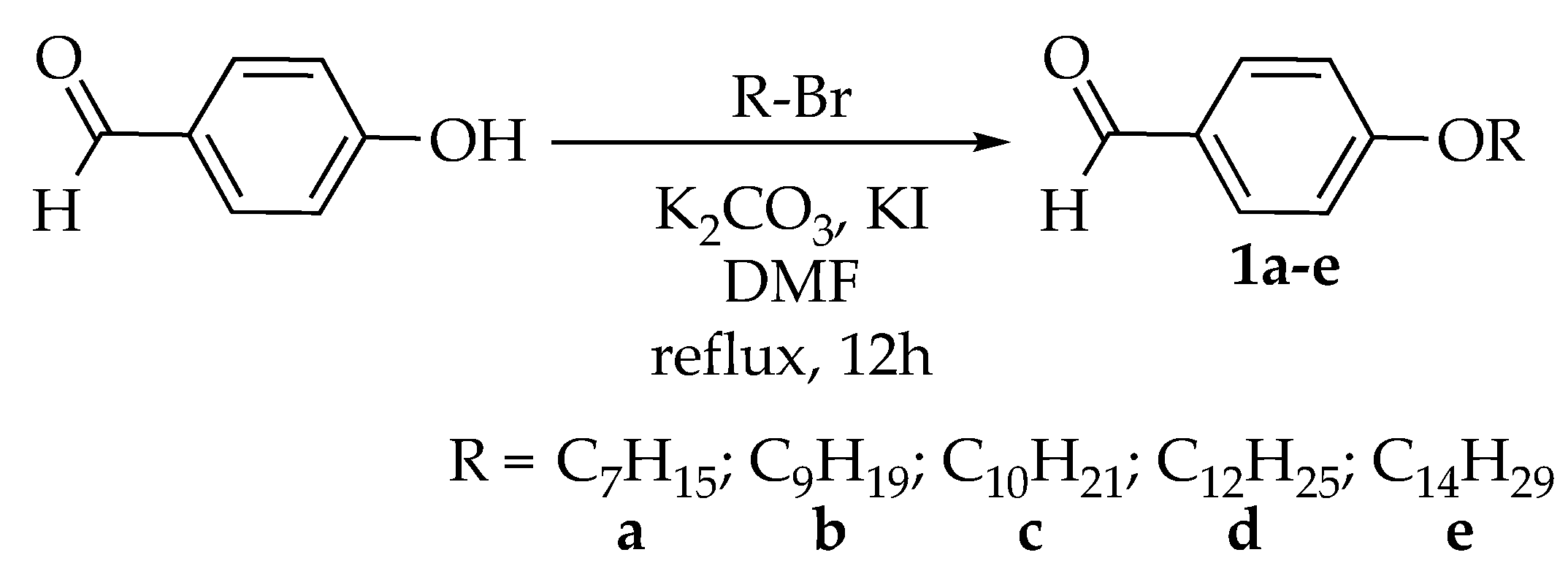{kind=link}
{kind=link}
{kind=link}
| Proton | 1H [δ (ppm), Multiplicity, Coupling Constant (Hz)] | COSY (1H–1H) Correlation | HSQC (1H–13C) Correlation (ppm) |
|---|---|---|---|
| H2 | 7.55 (d, J = 5.0 Hz) | H3 | C2 (129.28) |
| H3 | 7.95 (d, J = 5.0 Hz) | H2 | C3 (136.49) |
| H5 | 8.66 (s, 1H) | - | C5 (159.38) |
| H7 | 7.33 (d, J = 10.0 Hz) | H8 | C7 (122.32) |
| H8 | 7.28 (d, J = 20.0 Hz) | H7 | C8 (122.13) |
| H10 | 8.55 (s, 1H) | - | C10 (158.83) |
| H12 | 7.87 (d, J = 10.0 Hz) | H13 | C12 (135.76) |
| H13 | 7.03 (d, J = 5.0 Hz) | H12 | C13 (115.46) |
| H15 | 4.07 (t, J = 5.0 Hz) | H16 | C15 (68.64) |
| H16–H20 | 1.32–1.79 (m) | H15–H21 | C16–C20 (22.31–31.58) |
| H21 | 0.89 (t, J = 7.5 Hz) | H20 | C21 (14.06) |
| Cpd | Mode | Transition Temperature (°C) Enthalpy, ΔH (kJ/mol) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4a | H | Cr | SmC | SmA | N | I | ||||
| • | 156.33 48.30 | • | 210.97 32.42 | • | 235.89 15.74 | • | ||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 232.89 −12.16 | • | 202.65 −28.05 | • | 144.82 −27.92 | • | ||||
| 4b | H | Cr | SmC | SmA | N | I | ||||
| • | 152.47 9.25 | • | 206.81 7.68 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 200.53 −7.57 | • | 144.76 −7.16 | • | ||||||
| 4c | H | Cr | SmC | SmA | N | I | ||||
| • | 151.60 36.70 | • | 201.38 19.81 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 192.87 −10.25 | • | 140.08 −28.35 | • | ||||||
| 4d | H | Cr | SmC | SmA | N | I | ||||
| • | 149.69 19.02 | • | 198.44 12.79 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 193.87 −9.04 | • | 136.91 −26.59 | • | ||||||
| 4e | H | Cr | SmC | SmA | N | I | ||||
| • | 147.75 34.59 | • | 174.33 31.48 | • | 197.81 22.18 | • | ||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 195.64 −14.34 | • | 148.24 −38.11 | • | 132.86 −34.60 | • | ||||
| 4f | H | Cr | SmC | SmA | N | I | ||||
| • | 182.54 97.20 | • | 231.49 44.93 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 220.43 −51.10 | • | 167.83 −66.25 | • | ||||||
| 4g | H | Cr | SmC | SmA | N | I | ||||
| • | 172.62 15.15 | • | 243.79 10.22 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 240.96 −33.81 | • | 167.81 −18.90 | • | ||||||
| 4h | H | Cr | SmC | SmA | N | I | ||||
| • | 166.47 19.71 | • | 226.30 15.71 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 223.55 −8.00 | • | 149.33 −18.74 | • | ||||||
| 4i | H | Cr | SmC | SmA | N | I | ||||
| • | 157.47 7.25 | • | 219.11 4.98 | • | ||||||
| C | I | N | SmA | SmC | Cr | |||||
| • | 211.47 −5.94 | • | 157.26 −5.67 | • | ||||||
| Material | POM Observation | Material | POM Observation |
|---|---|---|---|
| Compound 4a | Smectic A and Nematic | Compound 4f | Nematic |
| Compound 4b | Smectic A | Compound 4g | Nematic |
| Compound 4c | Smectic A | Compound 4h | Nematic |
| Compound 4d | Smectic A | Compound 4i | Nematic |
| Compound 4e | Smectic A and C | Compound 4j | Non-mesogenic |
| Material | LOI Value (%) | Material | LOI Value (%) |
|---|---|---|---|
| Pure polyester resin | 22.53 (± 0.00) | Polyester resin + 1 wt.% of HCCP | 24.71 (± 0.00) |
| Compound 4a | 26.90 (± 0.00) | Compound 4f | 27.54 (± 0.01) |
| Compound 4b | 26.71 (± 0.00) | Compound 4g | 27.73 (± 0.03) |
| Compound 4c | 26.71 (± 0.00) | Compound 4h | 27.90 (± 0.00) |
| Compound 4d | 26.53 (± 0.00) | Compound 4i | 28.37 (± 0.00) |
| Compound 4e | 26.37 (± 0.00) | Compound 4j | 27.28 (± 0.00) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamain, Z.; Khairuddean, M.; Guan-Seng, T. Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units. Molecules 2020, 25, 2122. https://doi.org/10.3390/molecules25092122
Jamain Z, Khairuddean M, Guan-Seng T. Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units. Molecules. 2020; 25(9):2122. https://doi.org/10.3390/molecules25092122
Chicago/Turabian StyleJamain, Zuhair, Melati Khairuddean, and Tay Guan-Seng. 2020. "Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units" Molecules 25, no. 9: 2122. https://doi.org/10.3390/molecules25092122
APA StyleJamain, Z., Khairuddean, M., & Guan-Seng, T. (2020). Liquid-Crystal and Fire-Retardant Properties of New Hexasubstituted Cyclotriphosphazene Compounds with Two Schiff Base Linking Units. Molecules, 25(9), 2122. https://doi.org/10.3390/molecules25092122