
Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions


Abstract

1. Introduction
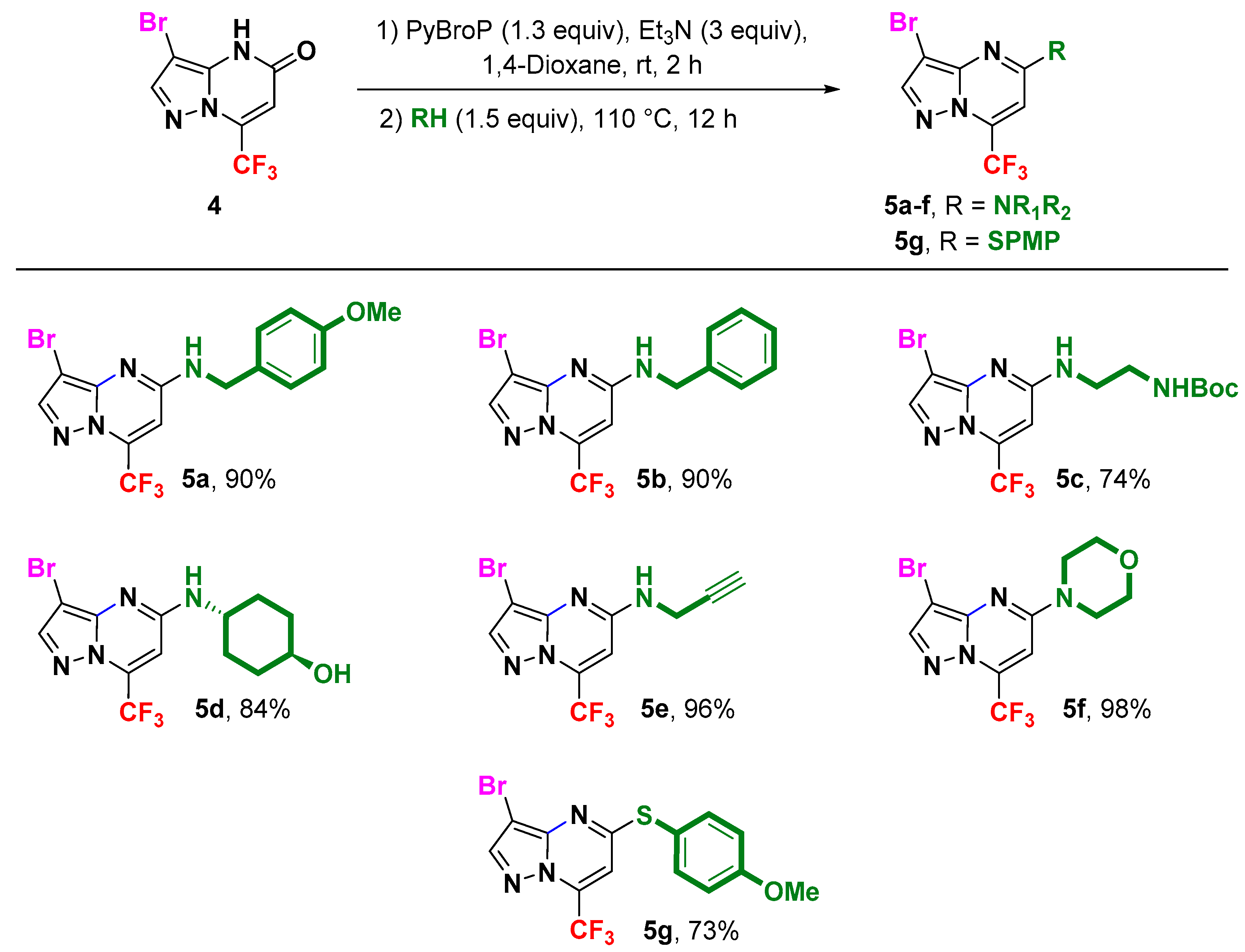
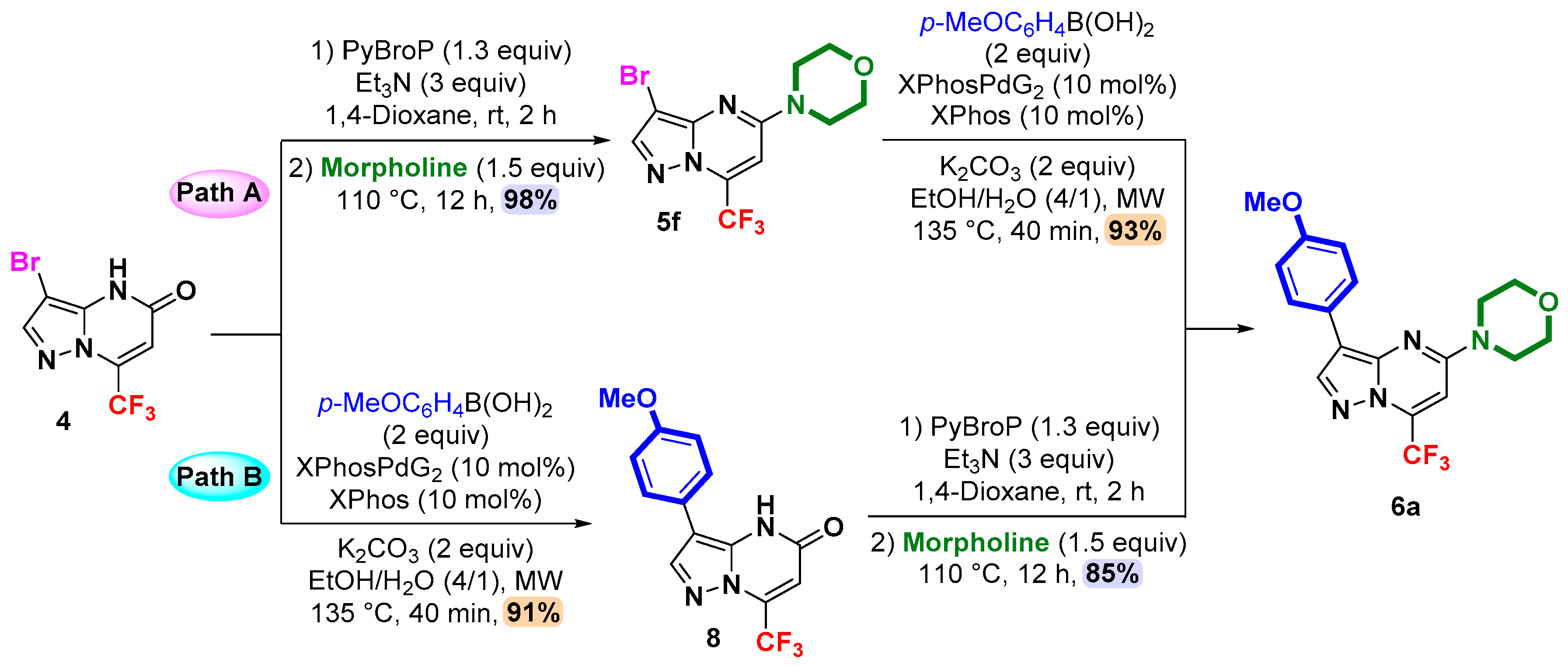
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
Microwave Assisted Reactions
3.2. Synthesis of 3-Bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one 4: Bromination of 3-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimdin-5-one 3
3.3. General Procedure for the Synthesis of 5-Amino (or Mercapto) 3-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidines 5a–g by the Direct Amination or Thiolation via C-OH Bond Activation with PyBroP
3.4. General Procedure for C-3 Suzuki–Miyaura Cross-Coupling: Synthesis of 3,5-Disubstituted 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimdines 6a–m
3.5. Deprotection of N-Boc Protected Amine by TFA: Synthesis of 5-[2-Amino-N-ethylamino]-3-(naphthalen-2-yl)-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidine (9)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Şener, N.; Erişkin, S.; Yavuz, S.; Şener, İ. Synthesis, characterization, solvatochromic properties, and antimicrobial-radical scavenging activities of new diazo dyes derived from pyrazolo[1,5-a]pyrimidine. J. Heterocyclic. Chem. 2017, 54, 3538–3548. [Google Scholar] [CrossRef]
- Bondock, S.; Fadaly, W.; Metwally, M.A. Synthesis and antimicrobial activity of some new thiazole, thiophene and pyrazole derivatives containing benzothiazole moiety. Eur. J. Med. Chem. 2010, 45, 3692–3701. [Google Scholar] [CrossRef]
- Hassan, A.S.; Mady, M.F.; Awad, H.M.; Hafez, T.S. Synthesis and antitumor activity of some new pyrazolo[1,5-a]pyrimidines. Chin. Chem. Lett. 2017, 28, 388–393. [Google Scholar] [CrossRef]
- Metwally, N.H.; Abdelrazek, F.M.; Eldaly, S.M. Synthesis and anticancer activity of some new heterocyclic compounds based on 1-cyanoacetyl-3,5-dimethylpyrazole. Res. Chem. Intermed. 2016, 42, 1071–1089. [Google Scholar] [CrossRef]
- Kamal, A.; Tamboli, J.R.; Nayak, V.L.; Adil, S.F.; Vishnuvardhan, M.V.P.S.; Ramakrishna, S. Synthesis of pyrazolo[1,5-a]pyrimidine linked aminobenzothiazole conjugates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2013, 23, 3208–3215. [Google Scholar] [CrossRef]
- Williamson, D.S.; Parratt, M.J.; Bower, J.F.; Moore, J.D.; Richardson, C.M.; Dokurno, P.; Jackson, P.S. Structure-guided design of pyrazolo[1,5-a]pyrimidines as inhibitors of human cyclin-dependent kinase 2. Bioorg. Med. Chem. Lett. 2005, 15, 863–867. [Google Scholar] [CrossRef]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Pyrazolo[1,5-a]pyrimidines as orally available inhibitors of cyclin-dependent kinase 2. Bioorg. Med. Chem. Lett. 2007, 17, 6220–6223. [Google Scholar] [CrossRef]
- Ali, S.; Heathcote, D.A.; Kroll, S.H.; Jogalekar, A.S.; Scheiper, B.; Patel, H.; Brackow, J.; Siwicka, A.; Fuchter, M.J.; Periyasamy, M.; et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009, 69, 6208–6215. [Google Scholar] [CrossRef]
- Selleri, S.; Bruni, F.; Costagli, C.; Costanzo, A.; Guerrini, G.; Ciciani, G.; Gratteri, P.; Besnard, F.; Costa, B.; Montali, M.; et al. A novel selective GABAA α1 receptor agonist displaying sedative and anxiolytic-like properties in rodents. J. Med. Chem. 2005, 48, 6756–6760. [Google Scholar] [CrossRef]
- Chen, Y.L. Substituted 6,5-Hetero-Bicyclic Derivatives. WO 98/08847, 5 March 1998. [Google Scholar]
- Boes, M.; Stadler, H.; Riemer, C. Pyrazolopyrimidine and Pyrazolines and Process for Preparation Thereof. U.S. Patent 6194410, 27 February 2001. [Google Scholar]
- Ramsey, S.J.; Attkins, N.J.; Fish, R.; van der Graaf, P.H. Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. Br. J. Pharmacol. 2011, 164, 992–1007. [Google Scholar] [CrossRef]
- Tellew, J.E.; Lanier, M.; Moorjani, M.; Lin, E.; Luo, Z.; Slee, D.H.; Zhang, X.; Hoare, S.R.; Grigoriadis, D.E.; St Denis, Y.; et al. Discovery of NBI-77860/GSK561679, a potent corticotropin-releasing factor (CRF1) receptor antagonist with improved pharmacokinetic properties. Bioorg. Med. Chem. Lett. 2010, 20, 7259–7264. [Google Scholar] [CrossRef] [PubMed]
- Dusza, J.P.; Tomcufcik, A.S.; Albright, J.D. Aryl and Heteroaryl[[7-(3-disubstituted amino)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]methanones. U.S. Patent 4654347, 31 March 1987. [Google Scholar]
- Ivashchenko, A.V.; Golovina, E.S.; Kadieva, M.G.; Kysil, V.M.; Mitkin, O.D.; Okun, I.M. Antagonists of serotonin 5-HT 6 receptors. III. Pyridine-substituted 3-(phenylsulfonyl)pyrazolo[1,5-a]pyrimidines: Synthesis and structure–activity relationship. Pharm. Chem. J. 2012, 46, 406–410. [Google Scholar] [CrossRef]
- Fang, Z.; Wang, T.Q.; Li, H.; Zhang, G.; Wu, X.A.; Yang, L.; Peng, Y.L.; Zou, J.; Li, L.L.; Xiang, R.; et al. Discovery of pyrazolo[1,5-a]pyrimidine-3-carbonitrile derivatives as a new class of histone lysine demethylase 4D (KDM4D) inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3201–3204. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Brenning, B.G.; Kultgen, S.G.; Foulks, J.M.; Clifford, A.; Lai, S.; Chan, A.; Merx, S.; McCullar, M.V.; Kanner, S.B.; et al. Synthesis and biological evaluation of pyrazolo[1,5-a]pyrimidine compounds as potent and selective Pim-1 inhibitors. ACS Med. Chem. Lett. 2015, 6, 63–67. [Google Scholar] [CrossRef]
- Wang, X.; Magnuson, S.; Pastor, R.; Fan, E.; Hu, H.; Tsui, V.; Deng, W.; Murray, J.; Steffek, M.; Wallweber, H.; et al. Discovery of novel pyrazolo[1,5-a]pyrimidines as potent pan-Pim inhibitors by structure-and property-based drug design. Bioorg. Med. Chem. Lett. 2013, 23, 3149–3153. [Google Scholar] [CrossRef]
- Liu, Y.; Laufer, R.; Patel, N.K.; Ng, G.; Sampson, P.B.; Li, S.W.; Lang, Y.; Feher, M.; Brokx, R.; Beletskaya, I.; et al. Discovery of pyrazolo[1, 5-a]pyrimidine TTK inhibitors: CFI-402257 is a potent, selective, bioavailable anticancer agent. ACS Med. Chem. Lett. 2016, 7, 671–675. [Google Scholar] [CrossRef]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef]
- Mackman, R.L.; Sangi, M.; Sperandio, D.; Parrish, J.P.; Eisenberg, E.; Perron, M.; Hui, H.; Zhang, L.; Siegel, D.; Yang, H.; et al. Discovery of an oral respiratory syncytial virus (RSV) fusion inhibitor (GS-5806) and clinical proof of concept in a human RSV challenge study. J. Med. Chem. 2015, 58, 1630–1643. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Windisch, M.P.; Jo, S.; Kim, K.; Kong, S.; Kim, H.C.; Kim, S.; Kim, H.; Lee, M.E.; Kim, Y.; et al. Discovery and characterization of a novel 7-aminopyrazolo[1,5-a]pyrimidine analog as a potent hepatitis C virus inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 7297–7301. [Google Scholar] [CrossRef]
- Mikami, S.; Kawasaki, M.; Ikeda, S.; Negoro, N.; Nakamura, S.; Nomura, I.; Ashizawa, T.; Kokubo, H.; Hoffman, I.D.; Zou, H.; et al. Discovery of a novel series of pyrazolo[1,5-a]pyrimidine-based phosphodiesterase 2A inhibitors structurally different from N-((1S)-1-(3-fluoro-4-(trifluoromethoxy)phenyl)-2-methoxyethyl)-7-methoxy-2-oxo-2,3-dihydropyrido[2,3-b]pyrazine-4(1H)-carboxamide (TAK-915), for the treatment of cognitive disorders. Chem. Pharm. Bull. 2017, 65, 1058–1077. [Google Scholar]
- Koizumi, Y.; Tanaka, Y.; Matsumura, T.; Kadoh, Y.; Miyoshi, H.; Hongu, M.; Takedomi, K.; Kotera, J.; Sasaki, T.; Taniguchi, H.; et al. Discovery of a pyrazolo[1,5-a]pyrimidine derivative (MT-3014) as a highly selective PDE10A inhibitor via core structure transformation from the stilbene moiety. Bioorg. Med. Chem. 2019, 27, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Yamagami, T.; Kobayashi, R.; Moriyama, N.; Horiuchi, H.; Toyofuku, E.; Kadoh, Y.; Kawanishi, E.; Izumoto, S.; Hiramatsu, H.; Nanjo, T.; et al. Scalable process design for a PDE10A inhibitor consisting of pyrazolopyrimidine and quinoxaline as key units. Org. Process Res. Dev. 2019, 23, 578–587. [Google Scholar] [CrossRef]
- Raheem, I.T.; Schreier, J.D.; Fuerst, J.; Gantert, L.; Hostetler, E.D.; Huszar, S.; Joshi, A.; Kandebo, M.; Kim, S.H.; Li, J.; et al. Discovery of pyrazolopyrimidine phosphodiesterase 10A inhibitors for the treatment of schizophrenia. Bioorg. Med. Chem. Lett. 2016, 26, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: New structural trends and therapeutic areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Jeffries, B.; Wang, Z.; Graton, J.; Holland, S.D.; Brind, T.; Greenwood, R.D.R.; Le Questel, J.Y.; Scott, J.S.; Chiarparin, E.; Linclau, B. Reducing the Lipophilicity of Perfluoroalkyl Groups by CF2–F/CF2–Me or CF3/CH3 Exchange. J. Med. Chem. 2018, 61, 10602–10618. [Google Scholar] [CrossRef]
- Aggarwal, R.; Masan, E.; Kaushik, P.; Kaushik, D.; Sharma, C.; Aneja, K.R. Synthesis and biological evaluation of 7-trifluoromethylpyrazolo[1,5-a]pyrimidines as anti-inflammatory and antimicrobial agents. J. Fluor. Chem. 2014, 168, 16–24. [Google Scholar] [CrossRef]
- Yoshida, M.; Mori, A.; Inaba, A.; Oka, M.; Makino, H.; Yamaguchi, M.; Fujita, H.; Kawamoto, T.; Goto, M.; Kimura, H.; et al. Synthesis and structure–activity relationship of tetrahydropyrazolopyrimidine derivatives—A novel structural class of potent calcium-sensing receptor antagonists. Bioorg. Med. Chem. 2010, 18, 8501–8511. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Selivanov, S.I.; Nelyubina, Y.V.; Antipin, M.Y. Highly regioselective synthesis of trifluoromethyl derivatives of pyrazolo[1,5-a]pyrimidines bearing fused cycloalkane rings using (2-ethoxycycloalkenyl)-2,2,2-trifluoroethanones. J. Fluor. Chem. 2009, 130, 861–869. [Google Scholar] [CrossRef]
- Shaaban, M.R. Microwave-assisted synthesis of fused heterocycles incorporating trifluoromethyl moiety. J. Fluor. Chem. 2008, 129, 1156–1161. [Google Scholar] [CrossRef]
- Petrov, A.A.; Emelina, E.E.; Selivanov, S.I. α-aminoazoles in synthesis of heterocycles: IV. Regiodirection of 3(5)-amino-5(3)-methylpyrazole reaction with hexafluoroacetylacetone. Russ. J. Org. Chem. 2008, 44, 263–269. [Google Scholar] [CrossRef]
- Martins, M.A.P.; Cunico, W.; Scapin, E.; Emmerich, D.J.; Fiss, G.F.; Rosa, F.A.; Bonacorso, H.G.; Zanatta, N.; Flores, A.F.C. Microwave-assisted regiospecific synthesis of 2-trifluoromethyl-7-trihalomethylated pyrazolo[1,5-a]pyrimidines. Lett. Org. Chem. 2006, 3, 358–362. [Google Scholar] [CrossRef]
- Krasovsky, A.L.; Hartulyari, A.S.; Nenajdenko, V.G.; Balenkova, E.S. Efficient syntheses of new CF3-containing diazolopyrimidines. Synthesis 2002, 2002, 133–137. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Firsov, A.V. Aminoazoles in Heterocycles Synthesis: II. Trifluoromethyl-containing Diketones in the Synthesis of Pyrazolo[1,5-a]pyrimidines. Russ. J. Org. Chem. 2001, 37, 852–858. [Google Scholar] [CrossRef]
- Jismy, B.; Guillaumet, G.; Allouchi, H.; Akssira, M.; Abarbri, A. Concise and efficient eccess to 5,7-disubstituted pyrazolo[1, 5-a]pyrimidines by Pd-Catalyzed sequential arylation, alkynylation and SNAr Reaction. Eur. J. Org. Chem. 2017, 6168–6178. [Google Scholar] [CrossRef]
- Petrignet, J.; Thiery, E.; Silpa, L.; Abarbri, M. Mild and Direct Access to 7-Substituted-4-trifluoromethylpyrimido[1,2-b]pyridazin-2-one Systems. Synthesis 2014, 46, 947–954. [Google Scholar] [CrossRef]
- Silpa, L.; Petrignet, J.; Abarbri, M. Direct access to fluorinated thiadiazolo[3,2-a]pyrimidin-7-one systems. Synlett 2014, 25, 1827–1830. [Google Scholar]
- Silpa, L.; Niepceron, A.; Laurent, F.; Brossier, F.; Pénichon, M.; Enguehart-Gueiffier, C.; Abarbri, M.; Silvestre, A.; Petrignet, J. Synthesis and evaluation of the anticoccidial activity of trifluoropyrido[1,2-a]pyrimidin-2-one derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 114–120. [Google Scholar] [CrossRef]
- Jismy, B.; Allouchi, H.; Guillaumet, G.; Akssira, M.; Abarbri, A. An efficient synthesis of new 7-trifluoromethyl-2,5-disubstituted pyrazolo[1,5-a]pyrimidines. Synthesis 2018, 50, 1675–1686. [Google Scholar]
- Dwyer, M.P.; Keertikar, K.; Paruch, K.; Alvarez, C.; Labroli, M.; Poker, C.; Fischmann, T.O.; Mayer-Ezell, R.; Bond, R.; Wang, Y.; et al. Discovery of pyrazolo[1,5-a]pyrimidine-based Pim inhibitors: A template-based approach. Bioorg. Med. Chem. Lett. 2013, 23, 6178–6182. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. The Pim kinases control rapamycin-resistant T cell survival and activation. J. Exp. Med. 2005, 201, 259–266. [Google Scholar] [CrossRef]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Organic Solvent | Base | [Pd] | Ligand | T (°C) | Time | Ratio a | |
|---|---|---|---|---|---|---|---|---|
| 6a | 7 | |||||||
| 1 | Dioxane | Na2CO3 | PdCl2(PPh3)2 | - | 110 | 12 h | 10 | 90 |
| 2 | Dioxane | Na2CO3 | PdCl2dppf | - | 110 | 12 h | 20 | 80 |
| 3 | Dioxane | K2CO3 | PdCl2dppf | - | 110 | 12 h | 22 | 78 |
| 4 | Dioxane | K2CO3 | XPhosPdG2 | XPhos | 135 | 12 h | 30 | 70 |
| 5 b | Dioxane | K2CO3 | XPhosPdG2 | XPhos | 135 b | 40 min | 45 | 55 |
| 6 b | EtOH | K2CO3 | XPhosPdG2 | XPhos | 135 b | 40 min | 100 (93) c | 0 |
| 7 b | EtOH | Na2CO3 | XPhosPdG2 | XPhos | 135 b | 40 min | 84 | 16 |
| 8 b | EtOH | K2CO3 | XPhosPdG2 | - | 135 b | 40 min | 56 | 44 |
| 9 b,d | EtOH | K2CO3 | XPhosPdG2 | - | 135 | 40 min | 100 (85) c | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jismy, B.; Tikad, A.; Akssira, M.; Guillaumet, G.; Abarbri, M. Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions. Molecules 2020, 25, 2062. https://doi.org/10.3390/molecules25092062
Jismy B, Tikad A, Akssira M, Guillaumet G, Abarbri M. Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions. Molecules. 2020; 25(9):2062. https://doi.org/10.3390/molecules25092062
Chicago/Turabian StyleJismy, Badr, Abdellatif Tikad, Mohamed Akssira, Gérald Guillaumet, and Mohamed Abarbri. 2020. "Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions" Molecules 25, no. 9: 2062. https://doi.org/10.3390/molecules25092062
APA StyleJismy, B., Tikad, A., Akssira, M., Guillaumet, G., & Abarbri, M. (2020). Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions. Molecules, 25(9), 2062. https://doi.org/10.3390/molecules25092062