Synthesis and Structure Insights of Two Novel Broad-Spectrum Antibacterial Candidates Based on (E)-N′-[(Heteroaryl)methylene]adamantane-1-carbohydrazides

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis
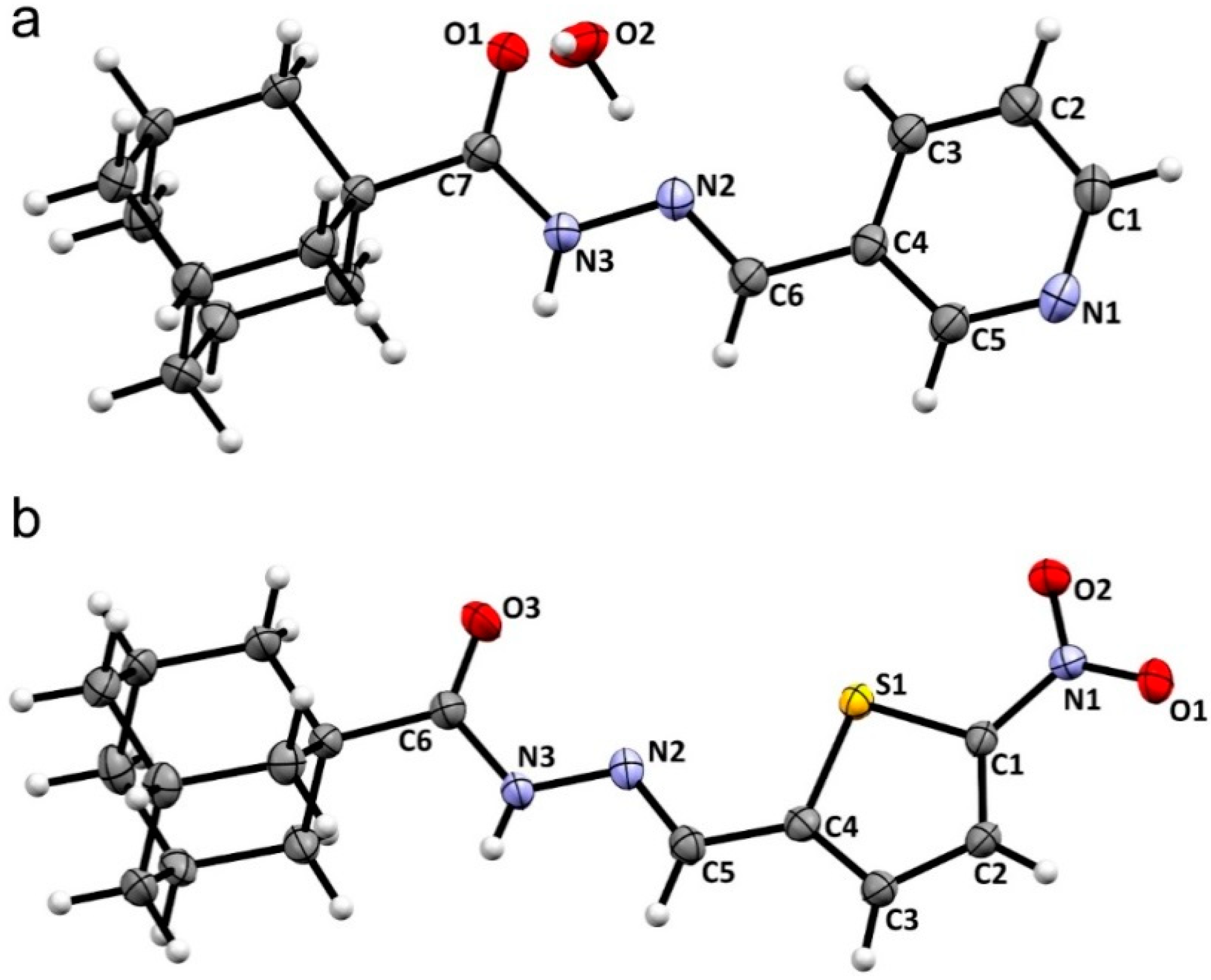
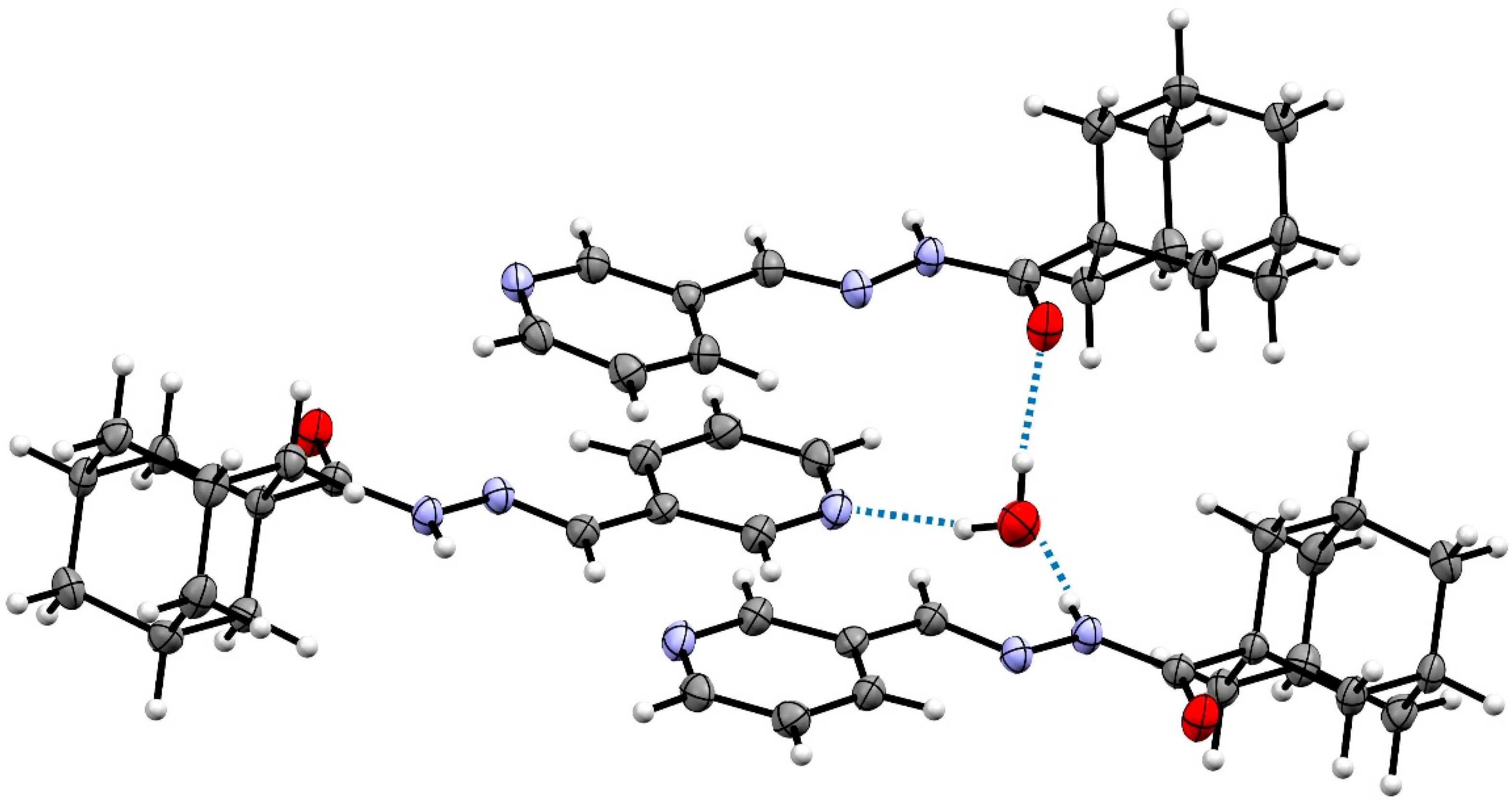
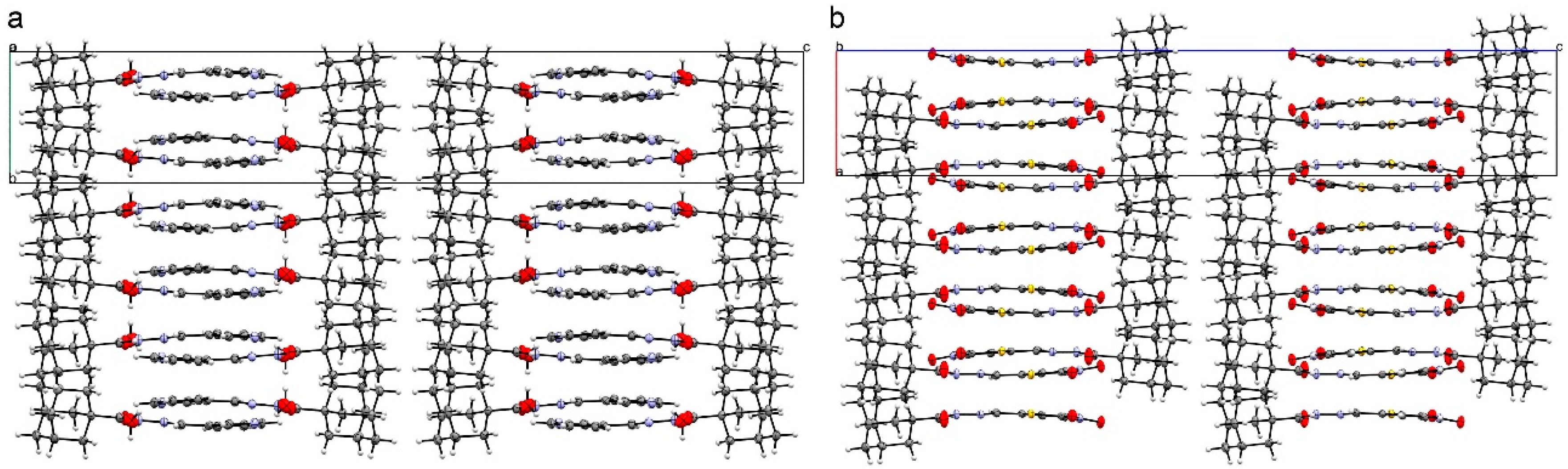
2.2. Crystal Structures
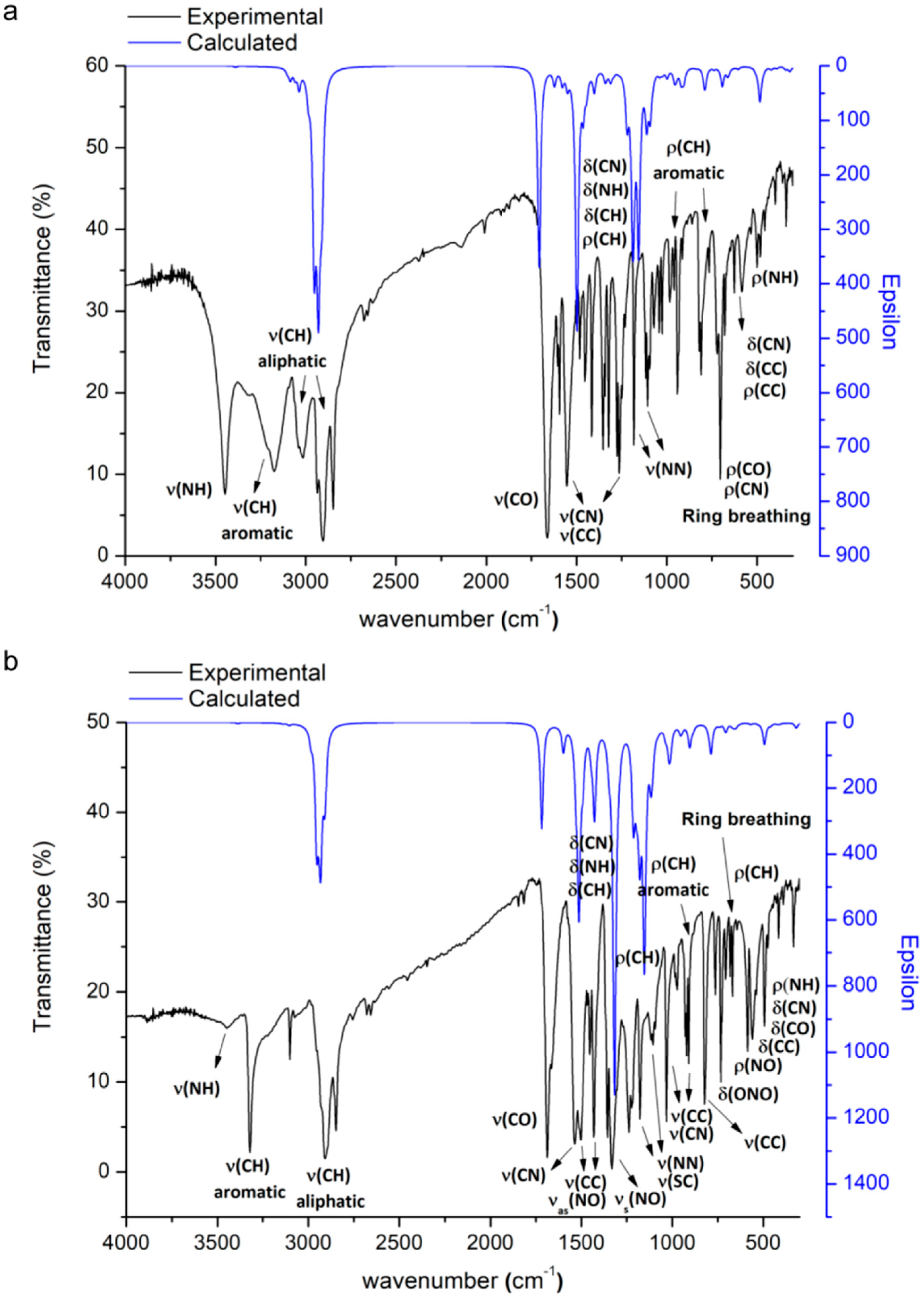
2.3. Infrared Spectra
2.3.1. Vibration Modes of the Adamantyl Component
2.3.2. Vibration Modes of the of the N′-Methyleneacetohydrazide Moiety
2.3.3. Vibration Modes of the of the Pyridine or 2-Nitrothiophene Groups
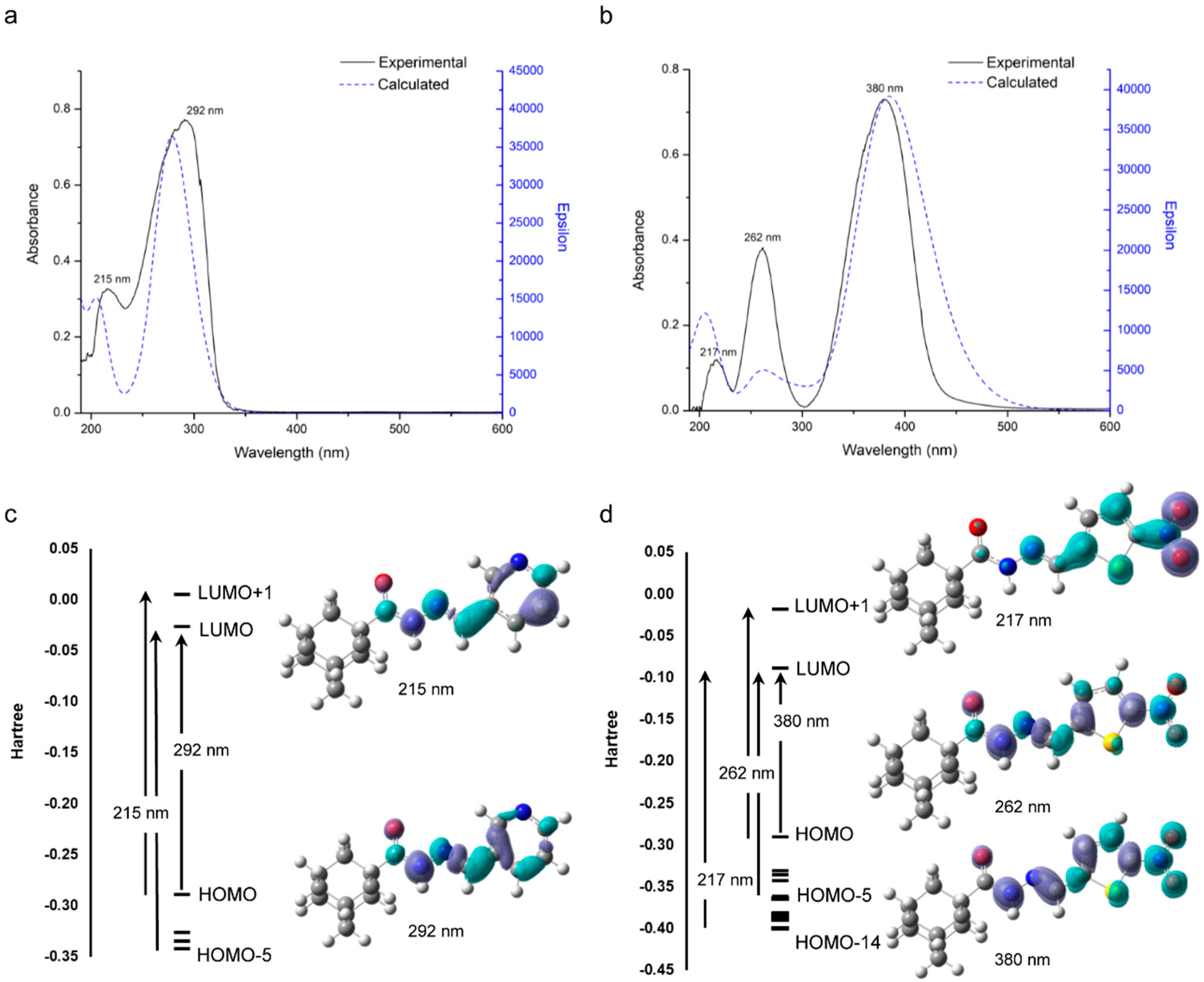
2.4. Electronic Spectra
2.5. In Vitro Antimicrobial and Antifungal Activity
3. Materials and Methods
3.1. Instrumentation
3.2. Synthesis, Crystallization, and Single Crystal X Ray Determination
3.2.1. E-N′-[(Pyridine-3-yl)methylidene]adamantane-1-carbohydrazide 1
3.2.2. E-N′-[(5-Nitrothiophen-2-yl)methylidene]adamantane-1-carbohydrazide 2
3.3. Computational Details
3.3.1. Hirshfeld Surface Analysis
3.3.2. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fox, H.H. Synthetic tuberculstats. I. Pyridine carboxylic acid derivatives. J. Org. Chem. 1952, 17, 542–546. [Google Scholar] [CrossRef]
- Fox, H.H.; Gibas, J.T. Synthetic tuberculstats. IX. Dialkyl derivatives of isonicotinylhydrazine. J. Org. Chem. 1955, 20, 60–69. [Google Scholar] [CrossRef]
- Manvar, A.; Bavishi, A.; Radadiya, A.; Patel, J.; Vora, V.; Dodia, N.; Rawal, K.; Shah, A. Diversity oriented design of various hydrazides and their in vitro evaluation against Mycobacterium tuberculosis H37Rv strains. Bioorg. Med. Chem. Lett. 2011, 21, 4728–4731. [Google Scholar] [CrossRef] [PubMed]
- Manvar, A.; Malde, A.; Verma, J.; Virsodia, V.; Mishra, A.; Upadhyay, K.; Acharya, H.; Coutinho, E.; Shah, A. Synthesis, anti-tubercular activity and 3D-QSAR study of coumarin-4-acetic acid benzylidene hydrazides. Eur. J. Med. Chem. 2008, 43, 2395–2403. [Google Scholar] [CrossRef]
- Maccari, R.; Ottanà, R.; Vigorita, M.G. In vitro advanced antimycobacterial screening of isoniazid-related hydrazones, hydrazides and cyanoboranes: Part 14. Bioorg. Med. Chem. Lett. 2005, 15, 2509–2513. [Google Scholar] [CrossRef]
- Kumar, P.; Narasimhan, B.; Yogeeswari, P.; Sriram, D. Synthesis and antitubercular activities of substituted benzoic acid N′-(substituted benzylidene/furan-2-ylmethylene)-N-(pyridine-3-carbonyl)- hydrazides. Eur. J. Med. Chem. 2010, 45, 6085–6089. [Google Scholar] [CrossRef]
- Martins, F.; Santos, S.; Ventura, C.; Elvas-Leitão, R.; Santos, L.; Vitorino, S.; Reis, M.; Miranda, V.; Correia, H.F.; Aires-de-Sousa, J.; et al. Design, synthesis and biological evaluation of novel isoniazid derivatives with potent antitubercular activity. Eur. J. Med. Chem. 2014, 81, 119–138. [Google Scholar] [CrossRef]
- Özdemir, A.; Turan-Zitouni, G.; Kaplancikli, Z.A.; Tunali, Y. Synthesis and biological activities of new hydrazide derivatives. J. Enzyme Inhib. Med. Chem. 2009, 24, 825–831. [Google Scholar] [CrossRef]
- Backes, G.L.; Neumann, D.M.; Jursic, B.S. Synthesis and antifungal activity of substituted salicylaldehyde hydrazones, hydrazides and sulfohydrazides. Bioorg. Med. Chem. 2014, 22, 4629–4636. [Google Scholar] [CrossRef]
- Vicini, P.; Zani, F.; Cozzini, P.; Doytchinova, I. Hydrazones of 1,2-benzisothiazole hydrazides: Synthesis, antimicrobial activity and QSAR investigations. Eur. J. Med. Chem. 2001, 37, 553–564. [Google Scholar] [CrossRef]
- Metwally, K.A.; Abdel-Aziz, L.M.; Lashine, E.S.; Husseiny, M.I.; Badawy, R.H. Hydrazones of 2-aryl-quinoline-4-carboxylic acid hydrazides: Synthesis and preliminary evaluation as antimicrobial agents. Bioorg. Med. Chem. 2006, 14, 8675–8682. [Google Scholar] [CrossRef] [PubMed]
- Turan-Zitouni, G.; Altıntop, M.D.; Özdemir, A.; Demirci, F.; Abu Mohsen, U.; Kaplancıklı, Z.A. Synthesis and antifungal activity of new hydrazide derivatives. J. Enzyme Inhib. Med. Chem. 2013, 28, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Judge, V.; Narang, R.; Sangwan, S.; De Clercq, E.; Balzarini, J.; Narasimhan, B. Benzylidene/2-chlorobenzylidene hydrazides: Synthesis, antimicrobial activity, QSAR studies and antiviral evaluation. Eur. J. Med. Chem. 2010, 45, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Drewe, J.; Tseng, B.; Kasibhatla, S.; Cai, S.X. Discovery and SAR of indole-2-carboxylic acid benzylidene-hydrazides as a new series of potent apoptosis inducers using a cell-based HTS assay. Bioorg. Med. Chem. 2004, 12, 3649–3655. [Google Scholar] [CrossRef]
- Machakanur, S.S.; Patil, B.R.; Badiger, D.S.; Bakale, R.P.; Gudasi, K.B.; Bligh, S.W.A. Synthesis, characterization and anticancer evaluation of novel tri-arm star shaped 1,3,5-triazine hydrazones. J. Mol. Struct. 2012, 1011, 121–127. [Google Scholar] [CrossRef]
- Liao, W.; Xu, C.; Ji, X.; Hu, G.; Ren, L.; Liu, Y.; Li, R.; Gong, P.; Sun, T. Design and optimization of novel 4-(2-fluorophenoxy)quinoline derivatives bearing a hydrazine moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2014, 87, 508–518. [Google Scholar] [CrossRef]
- Garkani-Nejad, Z.; Ahmadi-Roudi, B. Modeling the antileishmanial activity screening of 5-nitro-2-heterocyclic benzylidene hydrazides using different. chemometrics methods. Eur. J. Med. Chem. 2010, 45, 719–726. [Google Scholar] [CrossRef]
- Khan, K.M.; Rasheed, M.; Ullah, Z.; Hayat, S.; Kaukab, F.; Choudhary, M.I.; ur-Rahman, A.; Perveen, S. Synthesis and in vitro leishmanicidal activity of some hydrazides and their analogues. Bioorg. Med. Chem. 2003, 11, 1381–1387. [Google Scholar] [CrossRef]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef]
- Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011, 46, 1949–1963. [Google Scholar] [CrossRef]
- Lamoureux, G.; Artavia, G. Use of the adamantane structure in medicinal chemistry. Curr. Med. Chem. 2010, 17, 2967–2978. [Google Scholar] [CrossRef]
- Ficarra, R.; Ficarra, P.; Tommasini, A.; Fenech, G.; Pizzimenti, F.C.; Bisignano, G. 1-Adamantanecarboxylic acid hydrazides with presumed antimicrobial activity. Boll. Chim. Farm. 1984, 123, 317–321. [Google Scholar] [PubMed]
- Pham, V.H.; Phan, T.P.D.; Phan, D.C.; Vu, B.D. Synthesis and bioactivity of hydrazide-hydrazones with the 1-adamantyl-carbonyl moiety. Molecules 2019, 24, 4000. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; El-Emam, A.A.; Gad, L.M.; Barghash, A.E.M. Synthesis, antimicrobial and antiviral testing of some new 1-adamantyl analogues. Saudi Pharm. J. 2010, 18, 123–128. [Google Scholar] [CrossRef]
- El-Emam, A.A.; Alrashood, K.A.; Al-Omar, M.A.; Al-Tamimi, A.-M.S. Synthesis and antimicrobial activity of N’-heteroarylidene-1-adamantylcarbohydrazides and (±)-2-(1-adamantyl)-4-acetyl-5-[5-(4-substituted phenyl-3-isoxazolyl)]-1,3,4-oxadiazolines. Molecules 2012, 17, 3475–3483. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Hassan, H.M.; Abo-Kamar, A.M.; Ghabbour, H.A.; El-Emam, A.A. Adamantane-isothiourea hybrid derivatives: Synthesis, characterization, in vitro antimicrobial, and in vivo hypoglycemic activities. Molecules 2017, 22, 710. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Sujay, S.; Muthu, G.G.; El-Emam, A.A.; Venkataramanan, N.S.; Al-Omary, F.A.M.; Ghabbour, H.A.; Percino, J.; Thamotharan, S. Theoretical investigations of two adamantane derivatives: A combined X-ray, DFT, QTAIM analysis and molecular docking. J. Mol. Struct. 2018, 1159, 233–245. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Alsfouk, A.; El-Emam, A.A.; Blacque, O. Crystal structures and Hirshfeld surface analysis of 2-(adamantan-1-yl)-5-(4-fluorophenyl)-1,3,4-oxadiazole and 2-(adamantan-1-yl)-5-(4- chlorophenyl)-1,3,4-oxadiazole. Acta Crystallog. Cryst. Commun. 2019, E75, 611–615. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Joubert, J.; Blacque, O.; Al-Shaalan, N.H.; El-Emam, A.A. Crystal structure, Hirshfeld surface analysis and DFT studies of 5-(adamantan-1-yl)-3-[(4-chlorobenzyl)sulfanyl]-4-methyl-4H- 1,2,4-triazole, a potential 11β-HSD1 inhibitor. Sci. Rep. 2019, 9, 19745. [Google Scholar] [CrossRef]
- El-Emam, A.A.; Ibrahim, T.M. Synthesis and anti-inflammatory and analgesic activity of some 3-(1-adamantyl)-4-substituted-5-mercapto-1,2,4-triazoles. Arzneim. Forsch. Drug Res. 1991, 41, 1260–1264. [Google Scholar]
- Babjaková, E.; Dastychová, L.; Hanulí, B.; Kuřitka, I.; Nečas, M.; Vašková, H.; Vícha, R. Synthesis, molecular structure and vibrational spectra of 1,3-bis(1-adamantyl)-2-phenylpropan-1,3-diones. J. Mol. Struct. 2015, 1085, 207–214. [Google Scholar] [CrossRef]
- El-Emam, A.A.; Al-Tamimi, A.-M.S.; Al-Rashood, K.A.; Misra, H.N.; Narayan, V.; Prasad, O.; Sinha, L. Structural and spectroscopic characterization of a novel potential chemotherapeutic agent 3-(1-adamantyl)-1-{[4-(2-methoxyphenyl)piperazin-1-yl]methyl}-4-methyl-1H-1,2,4-triazole-5(4H)-thione by first principle calculations. J. Mol. Struct. 2012, 1022, 49–60. [Google Scholar] [CrossRef]
- Johnson, R.D. Computational Chemistry Comparison and Benchmark DataBase. Available online: https://cccbdb.nist.gov/vibscalejust.asp (accessed on 17 April 2020).
- Wang, Y.; Saebø, S.; Pittman, C.U. The structure of aniline by ab initio studies. J. Mol. Struct. THEOCHEM 1993, 281, 91–98. [Google Scholar] [CrossRef]
- Altun, A.; Gölcük, K.; Kumru, M. Structure and vibrational spectra of p-methylaniline: Hartree-Fock, MP2 and density functional theory studies. J. Mol. Struct. THEOCHEM 2003, 637, 155–169. [Google Scholar] [CrossRef]
- Varsányi, G. Vibrational Spectra of Benzene Derivatives, 1st ed.; Elsevier Science: Burlington, NJ, USA, 2012. [Google Scholar]
- Socrates, G. Infrared and Raman characteristic group frequencies: Tables and charts, 3rd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Tamer, Ö.; Avcı, D.; Atalay, Y. Quantum chemical characterization of N-(2-hydroxybenzylidene)- acetohydrazide (HBAH): A detailed vibrational and NLO analysis. Spectrochim. Acta A 2014, 117, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Varsányi, G.; Láng, L. Assignments for vibrational spectra of seven hundred benzene derivatives; Wiley: New York, NY, USA, 1974. [Google Scholar]
- Perelygin, I.S.; Klimchuk, M.A. Infrared spectra of coordinated pyridine. J. Appl. Spectrosc. 1976, 24, 43–46. [Google Scholar] [CrossRef]
- Arcoria, A.; Maccarone, E.; Tomaselli, G.A. Electronic spectra and infrared characteristics of trans-2-styrylthiophene and some nitro derivatives. Spectrochim. Acta A 1973, 29, 1601–1609. [Google Scholar] [CrossRef]
- Feuer, H.; Savides, C.; Rao, C.N.R. The infrared spectra of the salts of nitro compounds. Characteristic frequencies of the carbonitronate group, C NO2−. Spectrochim. Acta 1963, 19, 431–434. [Google Scholar] [CrossRef]
- Woods, G.L.; Washington, J.A. Antibacterial susceptibility tests: Dilution and disk diffusion methods. In Manual of Clinical Microbiology; Murray, P.R., Baron, E.J., Pfaller, M.A., Tenover, F.C., Yolken, R.H., Eds.; American Society of Microbiology: Washington, DC, USA, 1995. [Google Scholar]
- National Committee for Clinical Laboratory Standards. Approved Standard Document M-7A; NCCS: Villanova, PA, USA, 1985. [Google Scholar]
- Clark, R.C.; Reid, J.S. The analytical calculation of absorption in multifaceted crystals. Acta Cryst. A 1995, 51, 887–897. [Google Scholar] [CrossRef]
- CrysAlisPro (version 1.171.40.39a); Rigaku Oxford Diffraction: Oxford, UK, 2018.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. [Google Scholar] [CrossRef]
- Lewars, E.G. Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics, 2nd ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 101–191. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Al-Wahaibi, L.H.; Sert, Y.; Ucun, F.; Al-Shaalan, N.H.; Alsfouk, A.; El-Emam, A.A.; Karakaya, M. Theoretical and experimental spectroscopic studies, XPS analysis, dimer interaction energies and molecular docking study of 5-(adamantan-1-yl)-N-methyl 1,3,4-thiadiazol-2-amine. J. Phys. Chem. Solids 2019, 135, 109091. [Google Scholar] [CrossRef]
- Jamróz, M. VEDA 4 program for Vibrational Energy Distribution Analysis; Drug Institute: Warsaw, Poland, 2004. [Google Scholar]
- Labute, P. Computer-alded Molecular Design; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014. [Google Scholar]
- Labute, P. LowModeMD--implicit low-mode velocity filtering applied to conformational search of macrocycles and protein loops. J. Chem. Inf. Model. 2010, 50, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Accelrys Software, Material Studio Package, version 4; Accelrys Inc.: San Diego, CA, USA, 2009.
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5.0.8; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
Sample Availability: Samples of compounds 1 and 2 are available from the correspondent author. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Parameters | Compound 1 | Compound 2 |
|---|---|---|
| Empirical formula | C17H23N3O2 | C16H19N3O3S |
| Formula weight | 301.38 | 333.40 |
| Temperature | 160(1) K | 160 (1) K |
| Wavelength | 1.54184 Å | 1.54184 Å |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | I 1 2/a 1 | Pbca |
| Unit cell dimensions | a = 11.34690(10) Å b = 6.66810(10) Å c = 40.4828(5) Å β = 94.9240(10)° | a = 6.78143(15) Å b = 11.4079(3) Å c = 39.1760(8) Å β = 90° |
| Volume (Å3) | 3051.72(7) | 3030.74(12) |
| Z | 8 | 8 |
| Calculated density (mg/m3) | 1.312 | 1.461 |
| Absorption coefficient (mm−1) | 0.700 | 2.073 |
| F(000) | 1296 | 1408 |
| Crystal size (mm3) | 0.13 × 0.04 × 0.02 | 0.12 × 0.065 × 0.015 |
| Theta range for data collection | 4.385 to 74.475° | 4.515 to 74.488° |
| Index ranges | −14 ≤ h ≤ 14, −8 ≤ k ≤ 7, −50 ≤ l ≤ 49 | −8 ≤ h ≤ 7, −14 ≤ k ≤ 14, −47 ≤ l ≤ 48 |
| Reflections collected | 25245 | 13432 |
| Independent reflections | 3122 [R(int) = 0.0342] | 3102 [R(int) = 0.0384] |
| Completeness | 100.0% | 100.0% |
| Max. and min. transmission | 0.985 and 0.929 | 0.973 and 0.846 |
| Data/restraints/parameters | 3122/0/211 | 3102/0/212 |
| Goodness-of-fit on F2 | 1.045 | 1.071 |
| Final R indices [I > 2σ(I)] | R1 = 0.0365, wR2 = 0.0957 | R1 = 0.0395, wR2 = 0.1001 |
| R indices (all data) | R1 = 0.0424, wR2 = 0.0993 | R1 = 0.0493, wR2 = 0.1046 |
| Largest diff. peak and hole (e.Å−3) | 0.252 and −0.216 | 0.505 and −0.273 |
| CCDC number | 1992005 | 1992009 |
| D-H···A | d(D-H) | d(H···A) | d(D···A) | <(DH···A) |
|---|---|---|---|---|
| Compound 1 | ||||
| N(3)-H(3A) ···O(2)#1a | 0.881(17) | 1.959(17) | 2.8345(14) | 172.8(15) |
| O(2)-H(2A) ···N(1)#2a | 0.90(2) | 1.95(2) | 2.8368(14) | 165(2) |
| O(2)-H(2B) ···O(1) | 0.89(3) | 1.93(3) | 2.8118(16) | 172(2) |
| Compound 2 | ||||
| N(3)-H(3) ···O(2)#1b | 0.82(3) | 2.19(3) | 2.881(2) | 142(3) |
| Diameter of Growth Inhibition Zone (mm) | ||||||
|---|---|---|---|---|---|---|
| SA | BS | ML | EC | PA | CA | |
| Compound 1 | 28 | 26 | 23 | 12 | 14 | 15 |
| Compound 2 | 31 | 32 | 22 | 22 | 19 | 11 |
| Gentamicin sulfate | 27 | 26 | 20 | 22 | 21 | NT |
| Ampicillin trihydrate | 22 | 23 | 20 | 16 | 16 | NT |
| Clotrimazole | NT | NT | NT | NT | NT | 21 |
| MIC, µg/mL (µM) | ||||||
|---|---|---|---|---|---|---|
| SA | BS | ML | EC | PA | CA | |
| Compound 1 | 1 (3.53) | 1 (3.53) | 2 (7.06) | 64 (225.85) | >64 | 64 (225.85) |
| Compound 2 | 0.5 (1.50) | 0.5 (1.50) | 1 (3.0) | 2 (6.0) | 2 (6.0) | >64 |
| Gentamicin | 1 (2.09) 1 | 2 (4.19) 1 | 2 (4.19) 1 | 0.5 (1.05) 1 | 0.5 (1.05) 1 | NT |
| Ampicillin | 2 (5.72) 2 | 1 (2.86) 2 | 2 (5.72) 2 | 8 (22.90) 2 | 8 (22.90) 2 | NT |
| Clotrimazole | NT | NT | NT | NT | NT | 4 (11.60) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Alvarez, N.; Blacque, O.; Veiga, N.; Al-Mutairi, A.A.; El-Emam, A.A. Synthesis and Structure Insights of Two Novel Broad-Spectrum Antibacterial Candidates Based on (E)-N′-[(Heteroaryl)methylene]adamantane-1-carbohydrazides. Molecules 2020, 25, 1934. https://doi.org/10.3390/molecules25081934
Al-Wahaibi LH, Alvarez N, Blacque O, Veiga N, Al-Mutairi AA, El-Emam AA. Synthesis and Structure Insights of Two Novel Broad-Spectrum Antibacterial Candidates Based on (E)-N′-[(Heteroaryl)methylene]adamantane-1-carbohydrazides. Molecules. 2020; 25(8):1934. https://doi.org/10.3390/molecules25081934
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Natalia Alvarez, Olivier Blacque, Nicolás Veiga, Aamal A. Al-Mutairi, and Ali A. El-Emam. 2020. "Synthesis and Structure Insights of Two Novel Broad-Spectrum Antibacterial Candidates Based on (E)-N′-[(Heteroaryl)methylene]adamantane-1-carbohydrazides" Molecules 25, no. 8: 1934. https://doi.org/10.3390/molecules25081934
APA StyleAl-Wahaibi, L. H., Alvarez, N., Blacque, O., Veiga, N., Al-Mutairi, A. A., & El-Emam, A. A. (2020). Synthesis and Structure Insights of Two Novel Broad-Spectrum Antibacterial Candidates Based on (E)-N′-[(Heteroaryl)methylene]adamantane-1-carbohydrazides. Molecules, 25(8), 1934. https://doi.org/10.3390/molecules25081934