
Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases


Abstract
1. Introduction
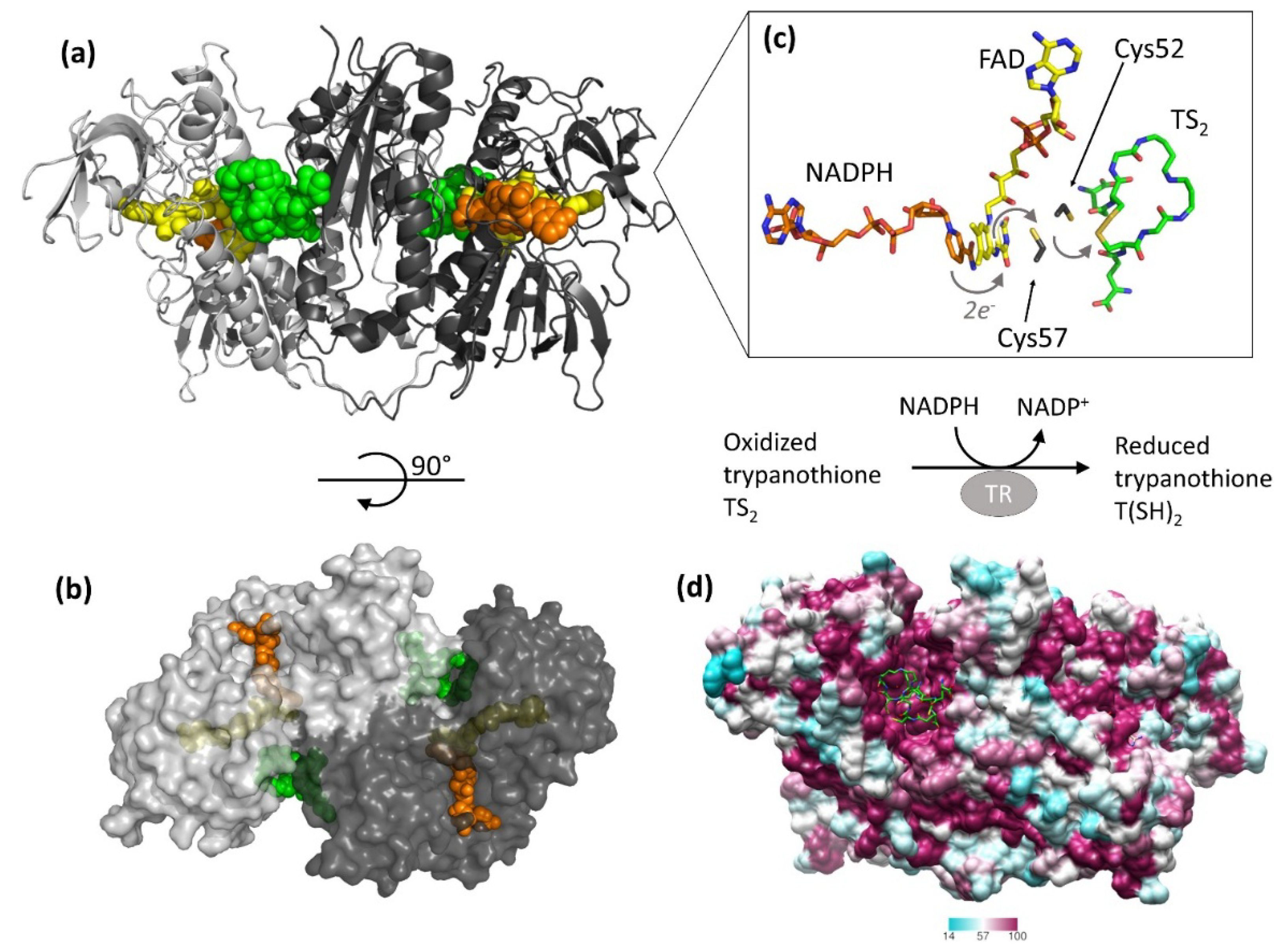
2. Relevant Structural Features of TR
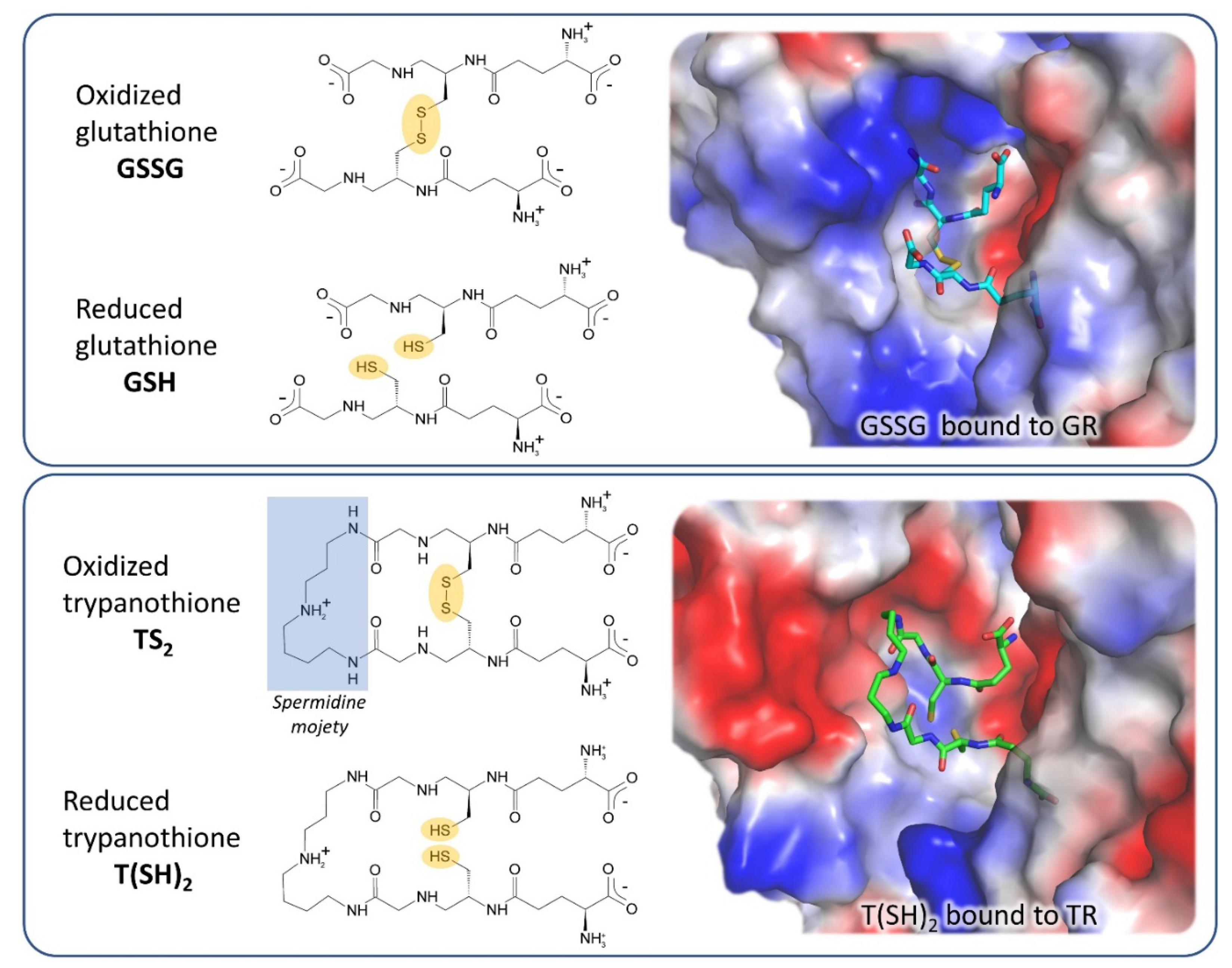
3. Off-Target Evaluation: Comparison with Glutathione Reductase (GR)
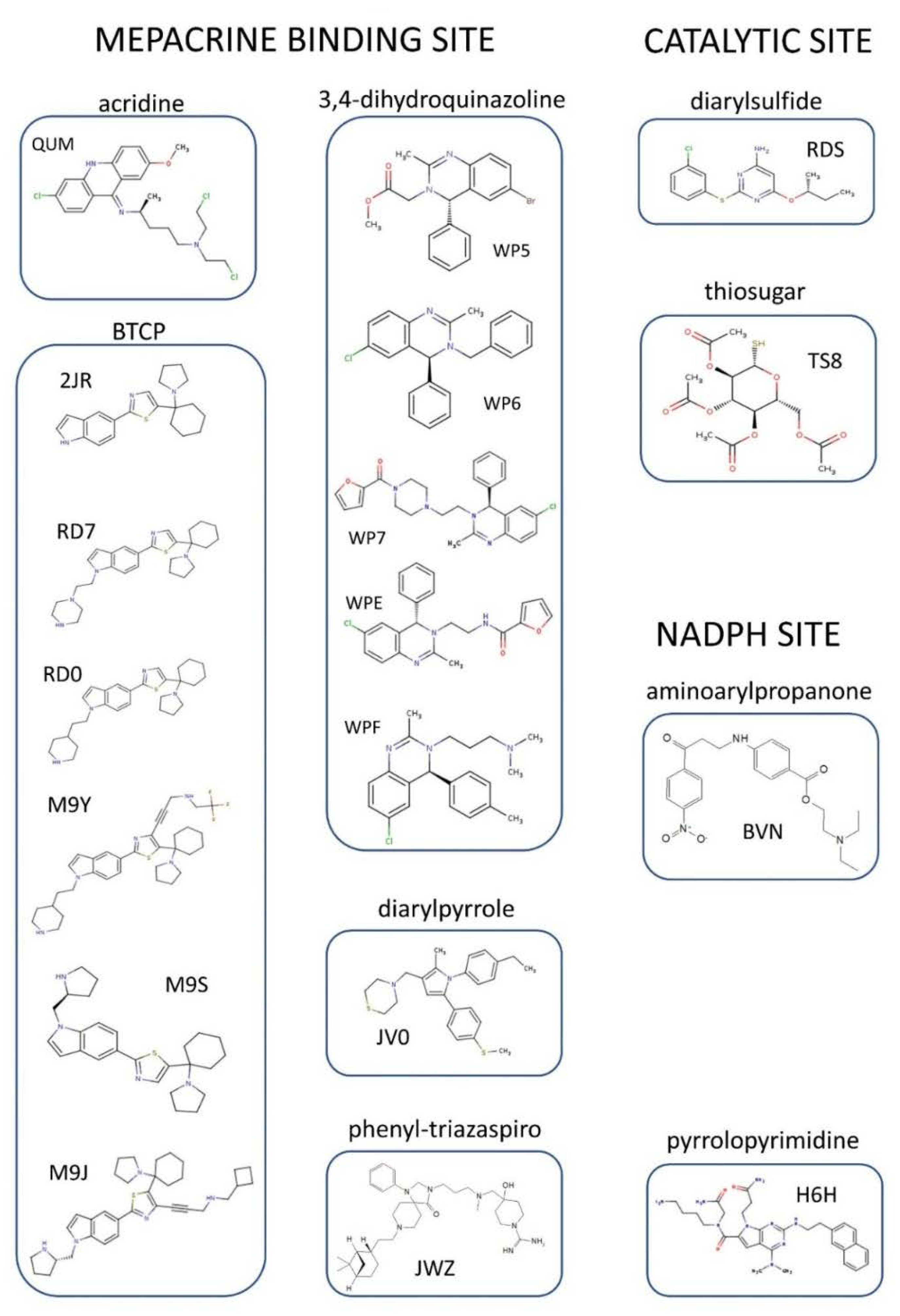
4. Structural Characterization of TR Inhibitors
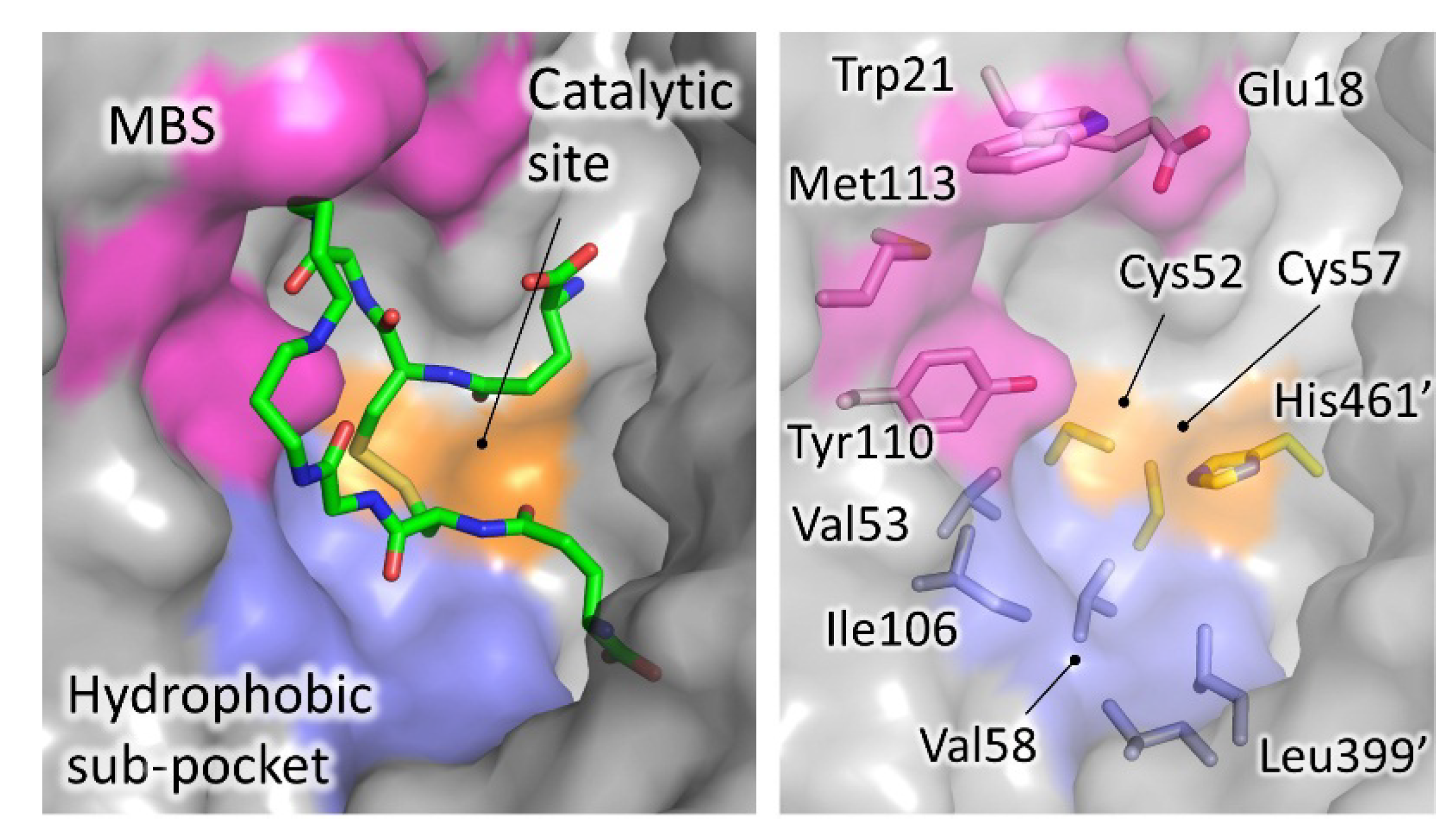
4.1. Inhibitors Targeting the TS2 Binding Cavity
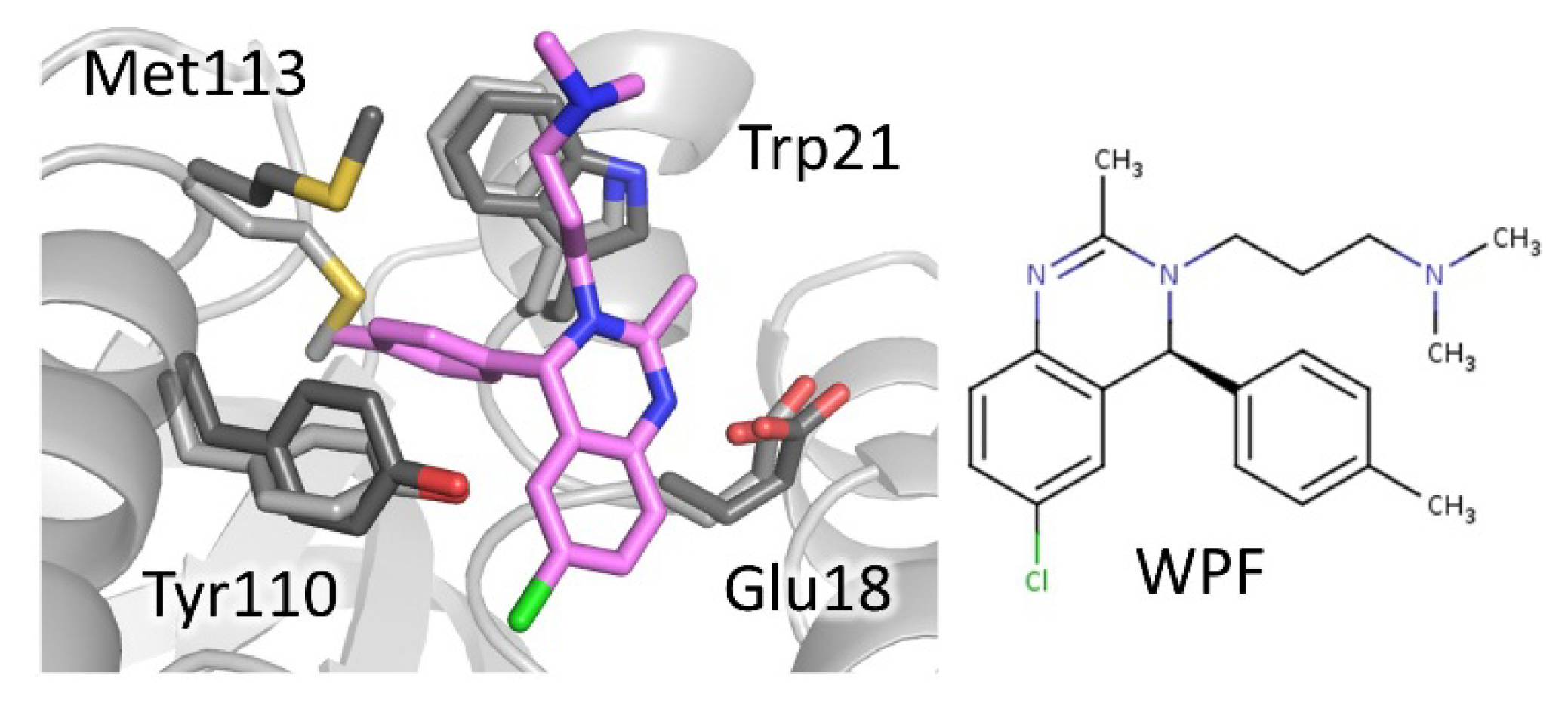
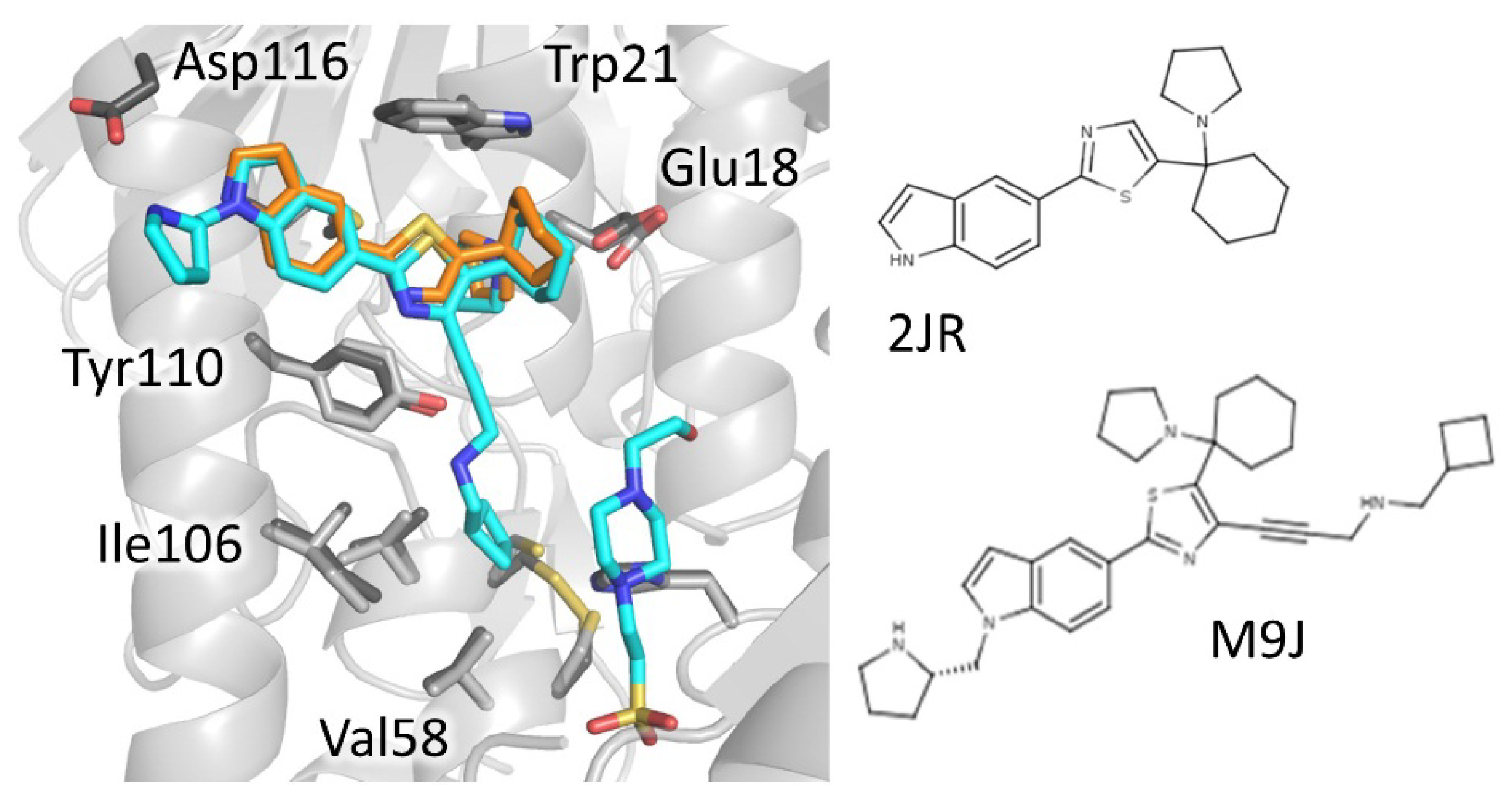
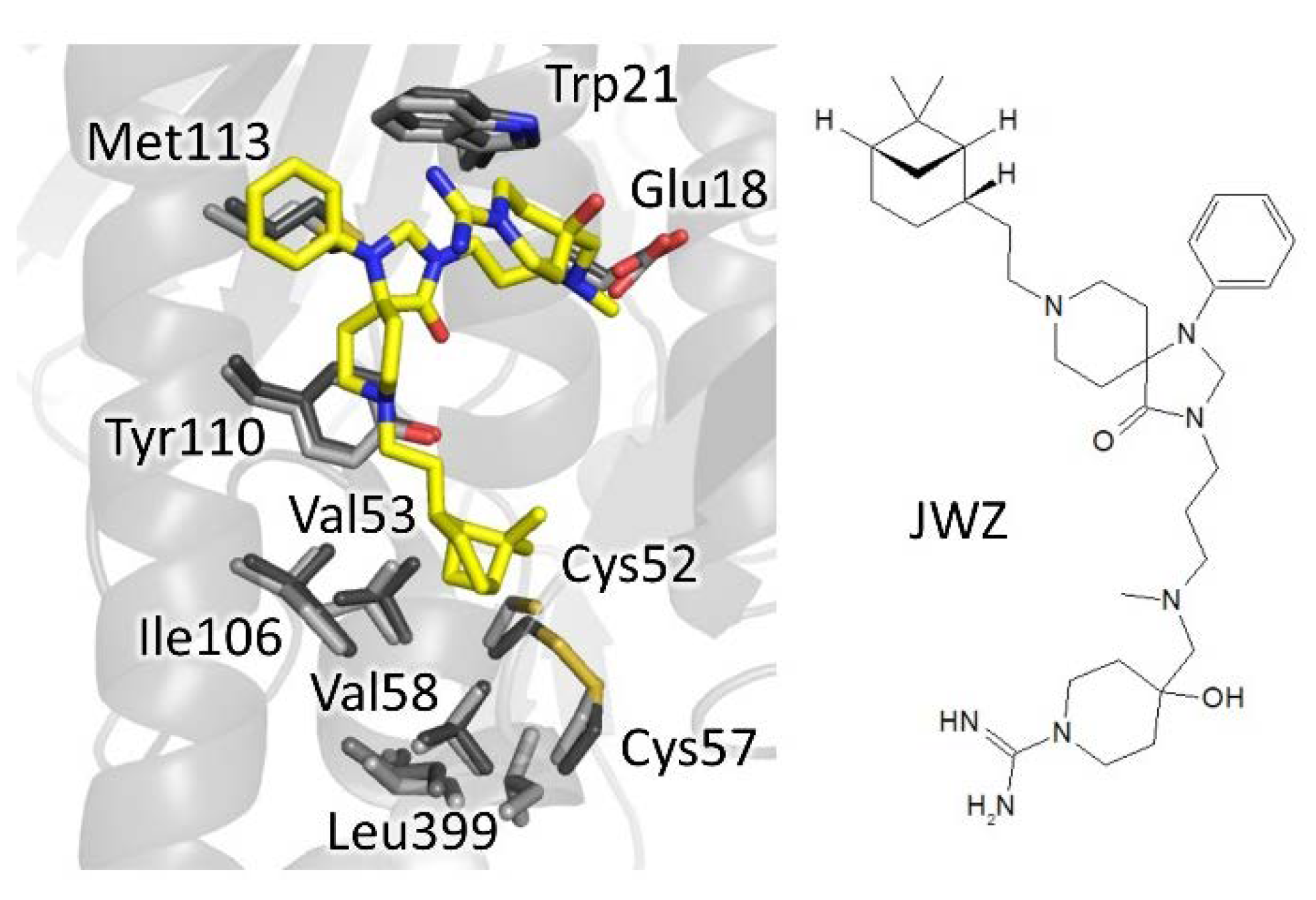
4.1.1. Mepacrine Binding Site (MBS)
4.1.2. Catalytic Site
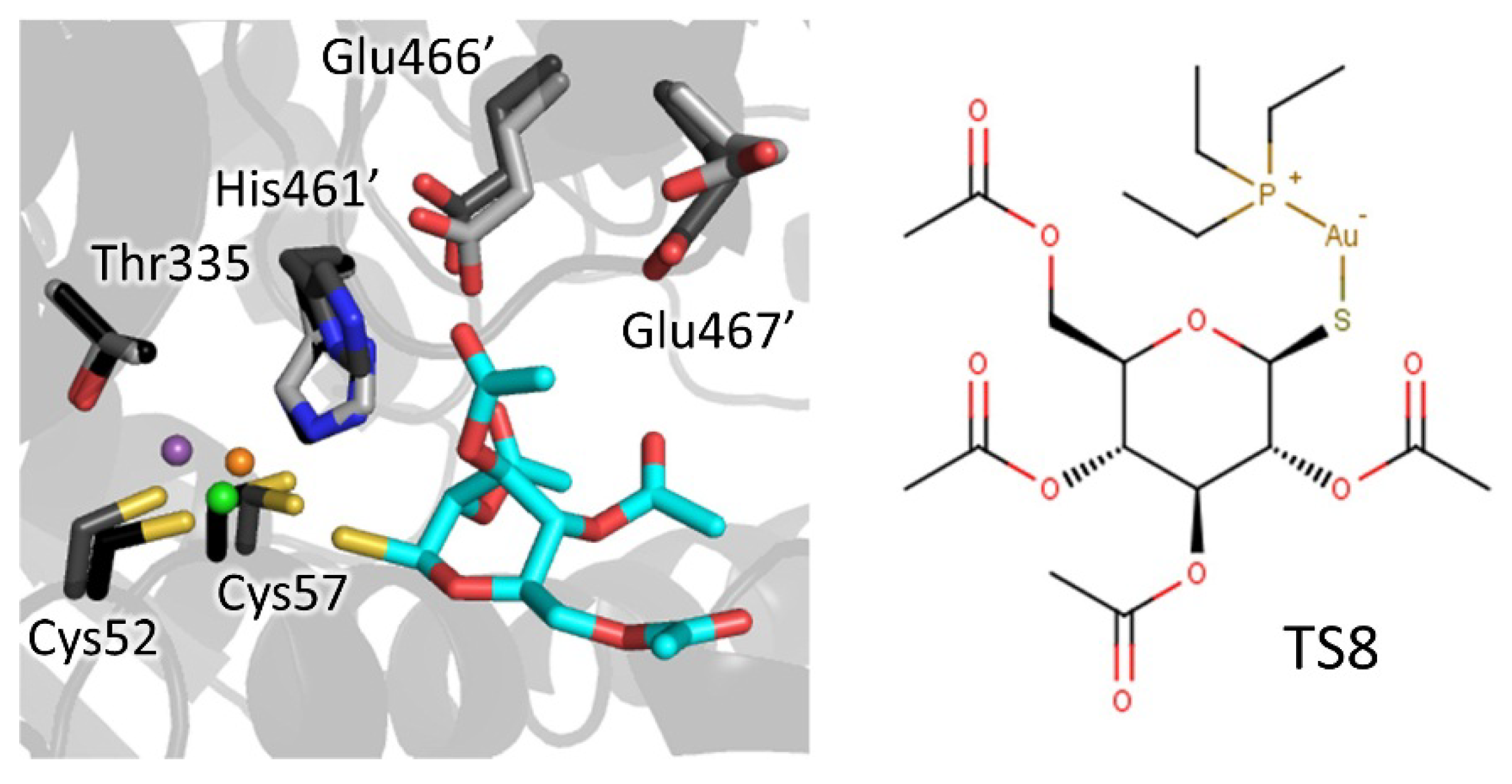
4.1.3. Metal Inhibitors
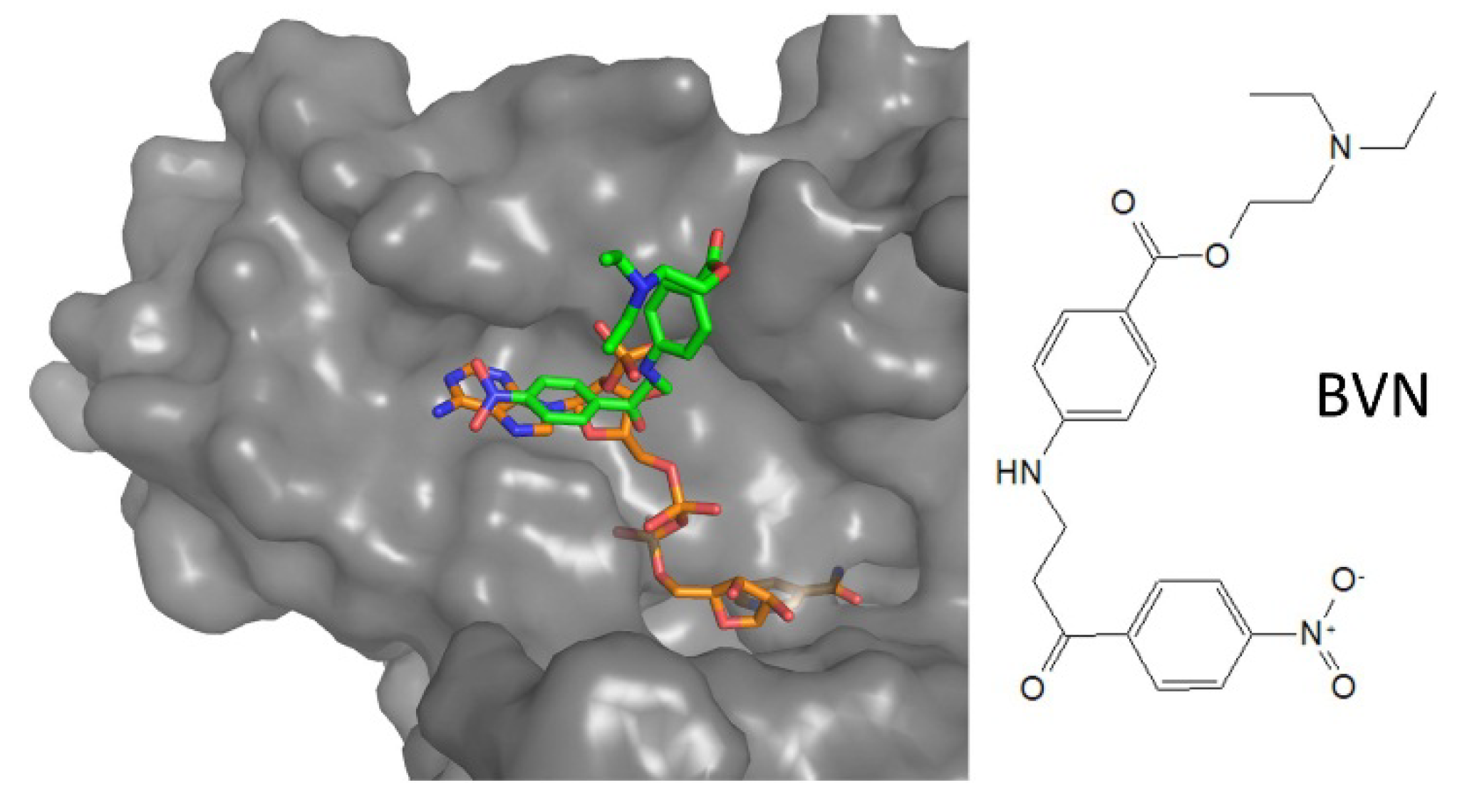
4.2. Inhibitors Targeting NADPH Binding Cavity
4.3. Nonpeptidic Dimerization Inhibition
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Publishes On-line Key Information about Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 30 March 2020).
- World Health Organization. WHO Publishes On-line Key Information about Chagas Disease. Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (accessed on 30 March 2020).
- World Health Organization. WHO Publishes On-line Key Information about HAT. Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 30 March 2020).
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Sorci, G.; Faivre, B. Inflammation and oxidative stress in vertebrate host-parasite systems. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kraeva, N.; Horáková, E.; Kostygov, A.Y.; Kořený, L.; Butenko, A.; Yurchenko, V.; Lukeš, J. Catalase in Leishmaniinae: With me or against me? Infect. Genet. Evol. 2017, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Tran, A.N.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; Blandin, G.; et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 2005, 309, 409–415. [Google Scholar] [CrossRef]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef]
- Ilari, A.; Fiorillo, A.; Genovese, I.; Colotti, G. Polyamine-trypanothione pathway: An update. Future Med. Chem. 2017, 9, 61–77. [Google Scholar] [CrossRef]
- Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Target assessment for antiparasitic drug discovery. Trends Parasitol. 2007, 23, 589–595. [Google Scholar] [CrossRef]
- Tovar, J.; Cunningham, M.L.; Smith, A.C.; Croft, S.L.; Fairlamb, A.H. Down-regulation of Leishmania donovani trypanothione reductase by heterologous expression of a trans-dominant mutant homologue: Effect on parasite intracellular survival. Proc. Natl. Acad. Sci. USA 1998, 95, 5311–5316. [Google Scholar] [CrossRef]
- Krieger, S.; Schwarz, W.; Ariyanayagam, M.R.; Fairlamb, A.H.; Krauth-Siegel, R.L.; Clayton, C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000, 35, 542–552. [Google Scholar] [CrossRef]
- Cunningham, M.L.; Fairlamb, A.H. Trypanothione reductase from Leishmania donovani. Purification, characterisation and inhibition by trivalent antimonials. Eur. J. Biochem. 1995, 230, 460–468. [Google Scholar] [CrossRef]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Genovese, I.; Fiorillo, F.; Battista, T.; De Ionna, I.; Fiorillo, A.; Colotti, G. Toward a Drug Against All Kinetoplastids: From LeishBox to Specific and Potent Trypanothione Reductase Inhibitors. Mol. Pharm. 2018, 15, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Beig, M.; Oellien, F.; Garoff, L.; Noack, S.; Krauth-Siegel, R.L.; Selzer, P.M. Trypanothione reductase: A target protein for a combined in vitro and in silico screening approach. PLoS Negl. Trop. Dis. 2015, 9, e0003773. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.A.; Charman, W.N.; Fairlamb, A.H.; Brun, R.; Kaiser, M.; Kostewicz, E.; Novello, P.M.; Parisot, J.P.; Richardson, J.; Street, I.P.; et al. Trypanothione reductase high-throughput screening campaign identifies novel classes of inhibitors with antiparasitic activity. Antimicrob. Agents Chemother. 2009, 53, 2824–2833. [Google Scholar] [CrossRef]
- Maccari, G.; Jaeger, T.; Moraca, F.; Biava, M.; Flohé, L.; Botta, M. A fast virtual screening approach to identify structurally diverse inhibitors of trypanothione reductase. Bioorg. Med. Chem. Lett. 2011, 21, 5255–5258. [Google Scholar] [CrossRef]
- Martyn, D.C.; Jones, D.C.; Fairlamb, A.H.; Clardy, J. High-throughput screening affords novel and selective trypanothione reductase inhibitors with anti-trypanosomal activity. Bioorg. Med. Chem. Lett. 2007, 17, 1280–1283. [Google Scholar] [CrossRef]
- Perez-Pineiro, R.; Burgos, A.; Jones, D.C.; Andrew, L.C.; Rodriguez, H.; Suarez, M.; Fairlamb, A.H.; Wishart, D.S. Development of a novel virtual screening cascade protocol to identify potential trypanothione reductase inhibitors. J. Med. Chem. 2009, 52, 1670–1680. [Google Scholar] [CrossRef]
- Richardson, J.L.; Nett, I.R.; Jones, D.C.; Abdille, M.H.; Gilbert, I.H.; Fairlamb, A.H. Improved tricyclic inhibitors of trypanothione reductase by screening and chemical synthesis. ChemMedChem 2009, 4, 1333–1340. [Google Scholar] [CrossRef]
- Salmon-Chemin, L.; Buisine, E.; Yardley, V.; Kohler, S.; Debreu, M.A.; Landry, V.; Sergheraert, C.; Croft, S.L.; Krauth-Siegel, R.L.; Davioud-Charvet, E. 2- and 3-substituted 1,4-naphthoquinone derivatives as subversive substrates of trypanothione reductase and lipoamide dehydrogenase from Trypanosoma cruzi: Synthesis and correlation between redox cycling activities and in vitro cytotoxicity. J. Med. Chem. 2001, 44, 548–565. [Google Scholar] [CrossRef]
- Turcano, L.; Torrente, E.; Missineo, A.; Andreini, M.; Gramiccia, M.; Di Muccio, T.; Genovese, I.; Fiorillo, A.; Harper, S.; Bresciani, A.; et al. Identification and binding mode of a novel Leishmania Trypanothione reductase inhibitor from high throughput screening. PLoS Negl. Trop. Dis. 2018, 12, e0006969. [Google Scholar] [CrossRef]
- Chacón-Vargas, K.F.; Nogueda-Torres, B.; Sánchez-Torres, L.E.; Suarez-Contreras, E.; Villalobos-Rocha, J.C.; Torres-Martinez, Y.; Lara-Ramirez, E.E.; Fiorani, G.; Krauth-Siegel, R.L.; Bolognesi, M.L.; et al. Trypanocidal Activity of Quinoxaline 1,4 Di-N-oxide Derivatives as Trypanothione Reductase Inhibitors. Molecules 2017, 22, 220. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Saccoliti, F.; Gramiccia, M.; Di Muccio, T.; Prakash, J.; Yadav, S.; Dubey, V.K.; Vistoli, G.; Battista, T.; Mocci, S.; et al. Structure-guided approach to identify a novel class of anti-leishmaniasis diaryl sulfide compounds targeting the trypanothione metabolism. Amino Acids 2019, 52, 247–259. [Google Scholar] [CrossRef]
- Da Paixão, V.G.; Pita, S.S.D.R. In silico identification and evaluation of new Trypanosoma cruzi trypanothione reductase (TcTR) inhibitors obtained from natural products database of the Bahia semi-arid region (NatProDB). Comput. Biol. Chem. 2019, 79, 36–47. [Google Scholar] [CrossRef] [PubMed]
- De Gasparo, R.; Halgas, O.; Harangozo, D.; Kaiser, M.; Pai, E.F.; Krauth-Siegel, R.L.; Diederich, F. Targeting a Large Active Site: Structure-Based Design of Nanomolar Inhibitors of Trypanosoma brucei Trypanothione Reductase. Chemistry 2019, 25, 11416–11421. [Google Scholar] [CrossRef] [PubMed]
- Jacomini, A.P.; Silva, M.J.V.; Silva, R.G.M.; Gonçalves, D.S.; Volpato, H.; Basso, E.A.; Paula, F.R.; Nakamura, C.V.; Sarragiotto, M.H.; Rosa, F.A. Synthesis and evaluation against Leishmania amazonensis of novel pyrazolo[3,4-d]pyridazinone-N-acylhydrazone-(bi)thiophene hybrids. Eur. J. Med. Chem. 2016, 124, 340–349. [Google Scholar] [CrossRef]
- Jagu, E.; Pomel, S.; Diez-Martinez, A.; Rascol, E.; Pethe, S.; Loiseau, P.M.; Labruère, R. Synthesis and antikinetoplastid evaluation of bis(benzyl)spermidine derivatives. Eur. J. Med. Chem. 2018, 150, 655–666. [Google Scholar] [CrossRef]
- Ortalli, M.; Ilari, A.; Colotti, G.; De Ionna, I.; Battista, T.; Bisi, A.; Gobbi, S.; Rampa, A.; Di Martino, R.M.C.; Gentilomi, G.A.; et al. Identification of chalcone-based antileishmanial agents targeting trypanothione reductase. Eur. J. Med. Chem. 2018, 152, 527–541. [Google Scholar] [CrossRef]
- Pandey, R.K.; Kumbhar, B.V.; Srivastava, S.; Malik, R.; Sundar, S.; Kunwar, A.; Prajapati, V.K. Febrifugine analogues as Leishmania donovani trypanothione reductase inhibitors: Binding energy analysis assisted by molecular docking, ADMET and molecular dynamics simulation. J. Biomol. Struct. Dyn. 2017, 35, 141–158. [Google Scholar] [CrossRef]
- Saccoliti, F.; Angiulli, G.; Pupo, G.; Pescatori, L.; Madia, V.N.; Messore, A.; Colotti, G.; Fiorillo, A.; Scipione, L.; Gramiccia, M.; et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzyme. Inhib. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef]
- Zimmermann, L.A.; de Moraes, M.H.; da Rosa, R.; de Melo, E.B.; Paula, F.R.; Schenkel, E.P.; Steindel, M.; Bernardes, L.S.C. Synthesis and SAR of new isoxazole-triazole bis-heterocyclic compounds as analogues of natural lignans with antiparasitic activity. Bioorg. Med. Chem. 2018, 26, 4850–4862. [Google Scholar] [CrossRef]
- Bailey, S.; Smith, K.; Fairlamb, A.H.; Hunter, W.N. Substrate interactions between trypanothione reductase and N1-glutathionylspermidine disulphide at 0.28-nm resolution. Eur. J. Biochem. 1993, 213, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Poce, G.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Colotti, G.; Biava, M.; Moraca, F.; Botta, M.; Yardley, V.; et al. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: A comparative analysis with its physiological substrate by X-ray crystallography. ChemMedChem 2013, 8, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Ariza, A.; Chow, W.H.; Oza, S.L.; Fairlamb, A.H. Comparative structural, kinetic and inhibitor studies of Trypanosoma brucei trypanothione reductase with T. cruzi. Mol. Biochem. Parasitol. 2010, 169, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Alphey, M.S.; Jones, D.C.; Shanks, E.J.; Street, I.P.; Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: Discovery, synthesis, and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011, 54, 6514–6530. [Google Scholar] [CrossRef] [PubMed]
- Fairlamb, A.H.; Cerami, A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef]
- Stoll, V.S.; Simpson, S.J.; Krauth-Siegel, R.L.; Walsh, C.T.; Pai, E.F. Glutathione reductase turned into trypanothione reductase: Structural analysis of an engineered change in substrate specificity. Biochemistry 1997, 36, 6437–6447. [Google Scholar] [CrossRef]
- Persch, E.; Bryson, S.; Todoroff, N.K.; Eberle, C.; Thelemann, J.; Dirdjaja, N.; Kaiser, M.; Weber, M.; Derbani, H.; Brun, R.; et al. Binding to large enzyme pockets: Small-molecule inhibitors of trypanothione reductase. ChemMedChem 2014, 9, 1880–1891. [Google Scholar] [CrossRef]
- Revuelto, A.; Ruiz-Santaquiteria, M.; de Lucio, H.; Gamo, A.; Carriles, A.A.; Gutiérrez, K.J.; Sánchez-Murcia, P.A.; Hermoso, J.A.; Gago, F.; Camarasa, M.J.; et al. Pyrrolopyrimidine vs. Imidazole-Phenyl-Thiazole Scaffolds in Nonpeptidic Dimerization Inhibitors of Leishmania infantum Trypanothione Reductase. ACS Infect. Dis. 2019, 5, 873–891. [Google Scholar] [CrossRef]
- Jacoby, E.M.; Schlichting, I.; Lantwin, C.B.; Kabsch, W.; Krauth-Siegel, R.L. Crystal structure of the Trypanosoma cruzi trypanothione reductase.mepacrine complex. Proteins 1996, 24, 73–80. [Google Scholar] [CrossRef]
- Saravanamuthu, A.; Vickers, T.J.; Bond, C.S.; Peterson, M.R.; Hunter, W.N.; Fairlamb, A.H. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: A template for drug design. J. Biol. Chem. 2004, 279, 29493–29500. [Google Scholar] [CrossRef]
- Patterson, S.; Jones, D.C.; Shanks, E.J.; Frearson, J.A.; Gilbert, I.H.; Wyatt, P.G.; Fairlamb, A.H. Synthesis and evaluation of 1-(1-(Benzo[b]thiophen-2-yl)cyclohexyl) piperidine (BTCP) analogues as inhibitors of trypanothione reductase. ChemMedChem 2009, 4, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Eberle, C.; Lauber, B.S.; Fankhauser, D.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Improved inhibitors of trypanothione reductase by combination of motifs: Synthesis, inhibitory potency, binding mode, and antiprotozoal activities. ChemMedChem 2011, 6, 292–301. [Google Scholar] [CrossRef] [PubMed]
- De Gasparo, R.; Brodbeck-Persch, E.; Bryson, S.; Hentzen, N.B.; Kaiser, M.; Pai, E.F.; Krauth-Siegel, R.L.; Diederich, F. Biological Evaluation and X-ray Co-crystal Structures of Cyclohexylpyrrolidine Ligands for Trypanothione Reductase, an Enzyme from the Redox Metabolism of Trypanosoma. ChemMedChem 2018, 13, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Turcano, L.B.T.; Torrente De Haro, E.; Missineo, A.; Alli, C.; Paonessa, G.; Colotti, G.; Harper, S.F.A.; Ilari, A.; Bresciani, A. Spiro-containing derivateives show antiparasitic activity against Trypanosoma brucei through inhibition of the trypanothione reductase enzyme. PLoS Negl. Trop. Dis. 2020. accepted. [Google Scholar]
- Chivers, J.; Jenner, P.; Marsden, C.D. Pharmacological characterization of binding sites identified in rat brain following in vivo administration of [3H]-spiperone. Br. J. Pharmacol. 1987, 90, 467–478. [Google Scholar] [CrossRef]
- Janssen, P.A.; Niemegeers, C.J.; Schellekens, K.H.; Lenaerts, F.M.; Verbruggen, F.J.; van Nueten, J.M.; Marsboom, R.H.; Hérin, V.V.; Schaper, W.K. The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug. Arzneimittelforschung 1970, 20, 1689–1698. [Google Scholar]
- Stump, B.; Eberle, C.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Diaryl sulfide-based inhibitors of trypanothione reductase: Inhibition potency, revised binding mode and antiprotozoal activities. Org. Biomol. Chem. 2008, 6, 3935–3947. [Google Scholar] [CrossRef]
- Eberle, C.; Burkhard, J.A.; Stump, B.; Kaiser, M.; Brun, R.; Krauth-Siegel, R.L.; Diederich, F. Synthesis, inhibition potency, binding mode, and antiprotozoal activities of fluorescent inhibitors of trypanothione reductase based on mepacrine-conjugated diaryl sulfide scaffolds. ChemMedChem 2009, 4, 2034–2044. [Google Scholar] [CrossRef]
- Colotti, G.; Fiorillo, A.; Ilari, A. Metal- and metalloid-containing drugs for the treatment of trypanosomatid diseases. Front. Biosci. 2018, 23, 954–966. [Google Scholar]
- Baiocco, P.; Ilari, A.; Ceci, P.; Orsini, S.; Gramiccia, M.; Di Muccio, T.; Colotti, G. Inhibitory Effect of Silver Nanoparticles on Trypanothione Reductase Activity and Leishmania infantum Proliferation. ACS Med. Chem. Lett. 2011, 2, 230–233. [Google Scholar] [CrossRef]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; Di Muccio, T.; Colotti, G. A gold-containing drug against parasitic polyamine metabolism: The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids 2012, 42, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Colotti, G.; Ilari, A.; Fiorillo, A.; Baiocco, P.; Cinellu, M.A.; Maiore, L.; Scaletti, F.; Gabbiani, C.; Messori, L. Metal-based compounds as prospective antileishmanial agents: Inhibition of trypanothione reductase by selected gold complexes. ChemMedChem 2013, 8, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Auranofin. Available online: https://www.drugbank.ca/drugs/DB00995 (accessed on 14 April 2020).
- Toro, M.A.; Sánchez-Murcia, P.A.; Moreno, D.; Ruiz-Santaquiteria, M.; Alzate, J.F.; Negri, A.; Camarasa, M.J.; Gago, F.; Velázquez, S.; Jiménez-Ruiz, A. Probing the dimerization interface of Leishmania infantum trypanothione reductase with site-directed mutagenesis and short peptides. Chembiochem 2013, 14, 1212–1217. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Scaffold | PDB Code | Source | Inhibitor PDB ID (Paper ID a) | Potency b | Reference |
|---|---|---|---|---|---|---|
| MBS | Acridine | Not available | Tc | (Quinacrine or mepacrine) | Ki: 25 μM | Jacoby, 1996 |
| 1gxf | Tb | QUM (Quin. mustard) | Irreversible inhibition | Saravanamuthu, 2004 | ||
| 3,4-dihydro quinazoline  | 2wp5 | Tb | WP5 (1a) | IC50: 6.8 μM | Patterson, 2011 | |
| 2wp6 | Tb | WP6 (6a) | IC50: 0.93 μM | |||
| 2wpc | Tb | WP7 (13e) | IC50: 0.42 μM | |||
| 2wpe | Tb | WPE (11e) | IC50: 0.86 μM | |||
| 2wpf | Tb | WPF (29a) | IC50: 0.23 μM | |||
BTCP | 4nev | Tb | 2JR (10a) | Ki: 12 μM Inh. [%] b: 43 | Persch, 2014 | |
| 4new | Tc | 2JR (10a) | Ki: 4 μM Inh. [%] b: 79 | |||
| 6btl | Tb | RD7 (18) | Ki: 3.8 μM Inh. [%] c: 80 | De Gasparo, 2018 | ||
| 6bu7 | Tb | RD0 (19) | Ki: 6.4 μM Inh. [%] c: 78 | |||
| 6oez | Tb | M9J ((+)-2) | Ki: 73 nM | De Gasparo, 2019 | ||
| 6oey | Tb | M9S ((+)-4)) | Ki: 2.1 μM | |||
| 6oex | Tb | M9Y (5) | Ki: 1.5 μM | |||
| diarylpyrrole | 4apn (B) | Li | JV0 (1) | Ki: 4.6 μM IC50: 13.8 μM | Baiocco, 2013 | |
| phenyl- triazaspiro | 6br5 | Tb | JWZ (1) | IC50: 5.7 µM | Turcano, 2020 | |
| Pyrrolopyrimi- dine | 6i7n (B) | Li | H6H (2f) | IC50: 52.2 µM | Revuelto, 2019 | |
| Catalytic site | diaryl sulfide | 5ebk | Li | RDS (RDS 777) | Ki: 0.25 µM | Saccoliti, 2017 |
| Catalytic site/cysteines | Metal/thiosugar | 2yau | Li | AU-TS8 (auranofin) | Ki: 0.15 µM | Ilari, 2012 |
| Catalytic cysteines | Metal | 2w0h | Li | SB | Ki: 1.5 µM | Baiocco, 2009 |
| 2x50 | Li | AG | Ki (Ag1): 500 nM Ki (Ag0): 50 nM | Baiocco, 2010 | ||
| NADPH-cavity | 3-amino-1-arylpropan-1-one | 6er5 | Li | BVN (3) | IC50: 12.4 µM | Turcano, 2018 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924. https://doi.org/10.3390/molecules25081924
Battista T, Colotti G, Ilari A, Fiorillo A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules. 2020; 25(8):1924. https://doi.org/10.3390/molecules25081924
Chicago/Turabian StyleBattista, Theo, Gianni Colotti, Andrea Ilari, and Annarita Fiorillo. 2020. "Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases" Molecules 25, no. 8: 1924. https://doi.org/10.3390/molecules25081924
APA StyleBattista, T., Colotti, G., Ilari, A., & Fiorillo, A. (2020). Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules, 25(8), 1924. https://doi.org/10.3390/molecules25081924