per-Alkoxy-pillar[5]arenes as Electron Donors: Electrochemical Properties of Dimethoxy-Pillar[5]arene and Its Corresponding Rotaxane



Abstract
1. Introduction
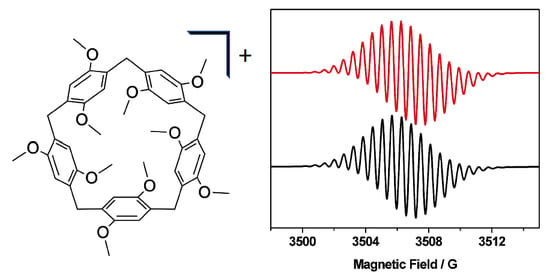
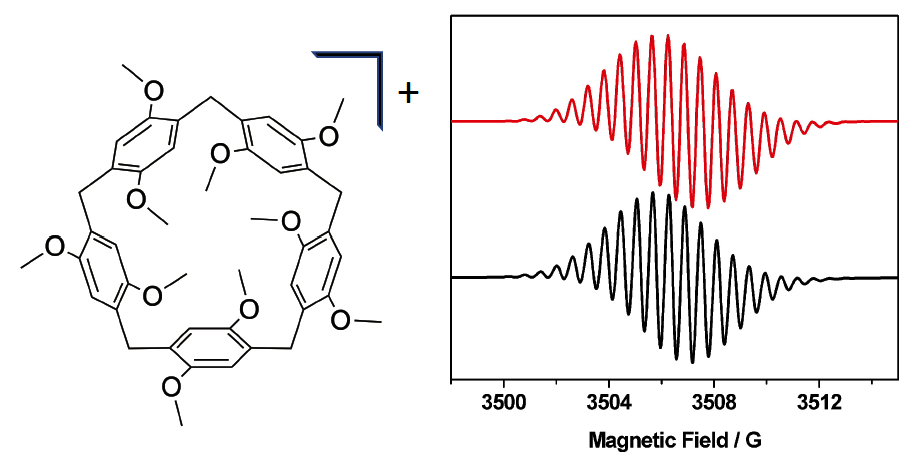
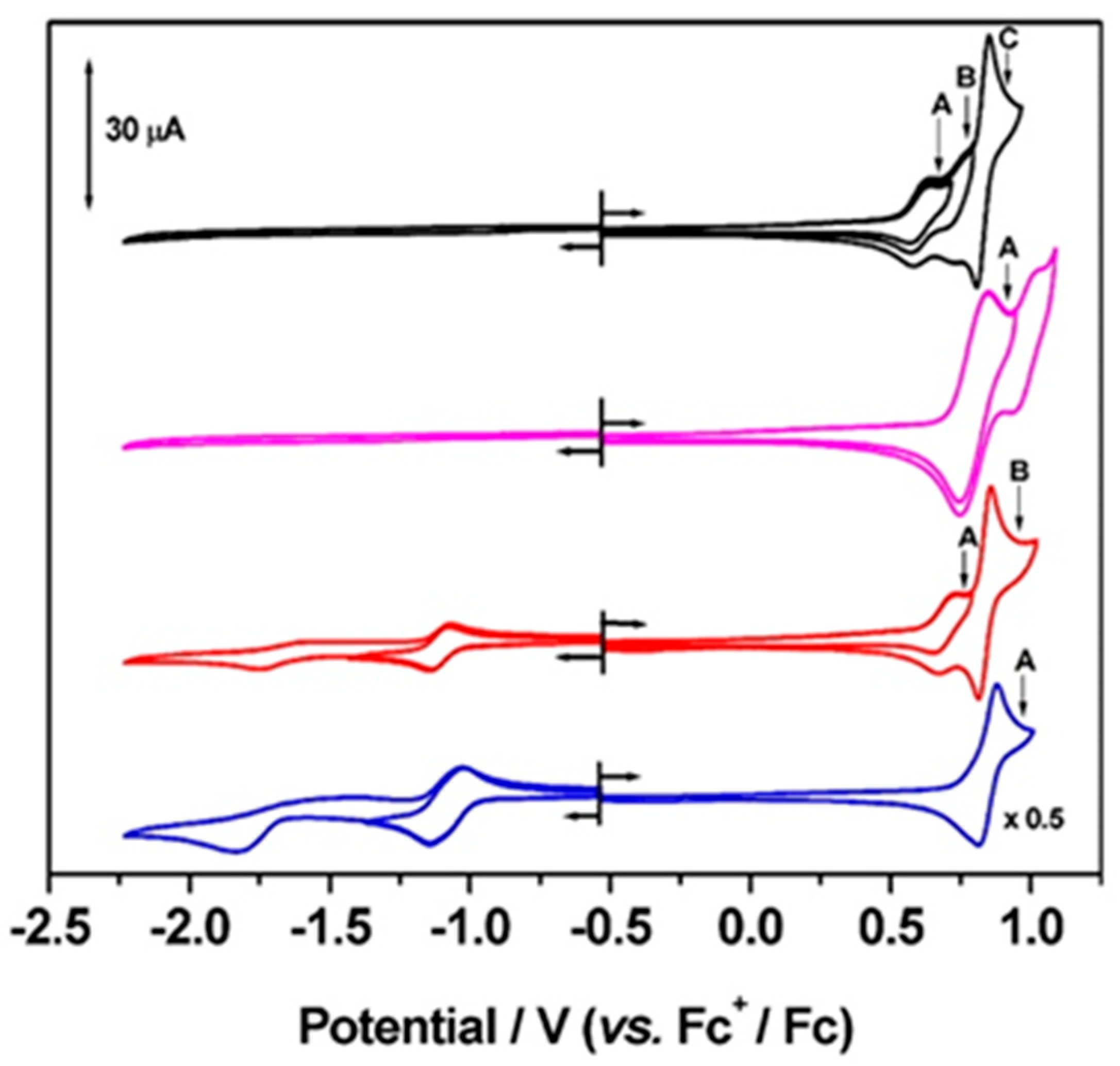
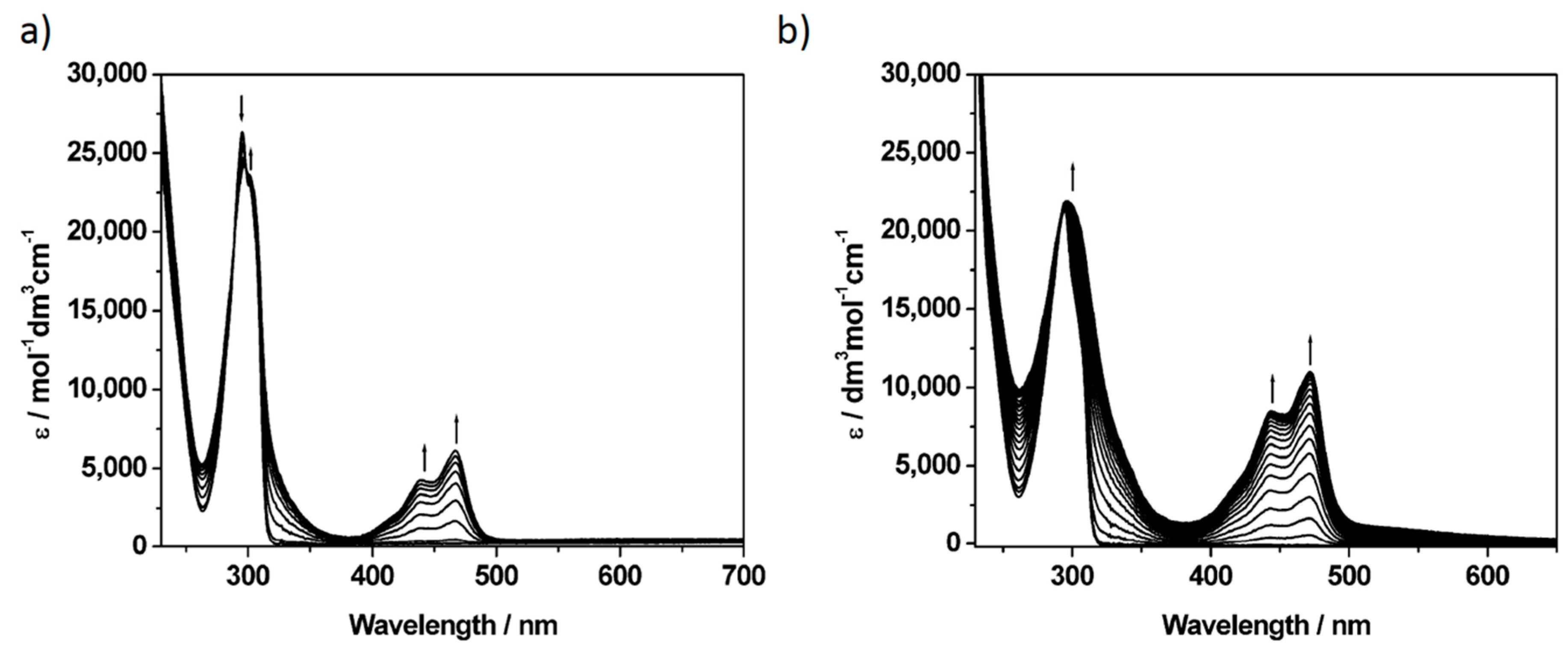
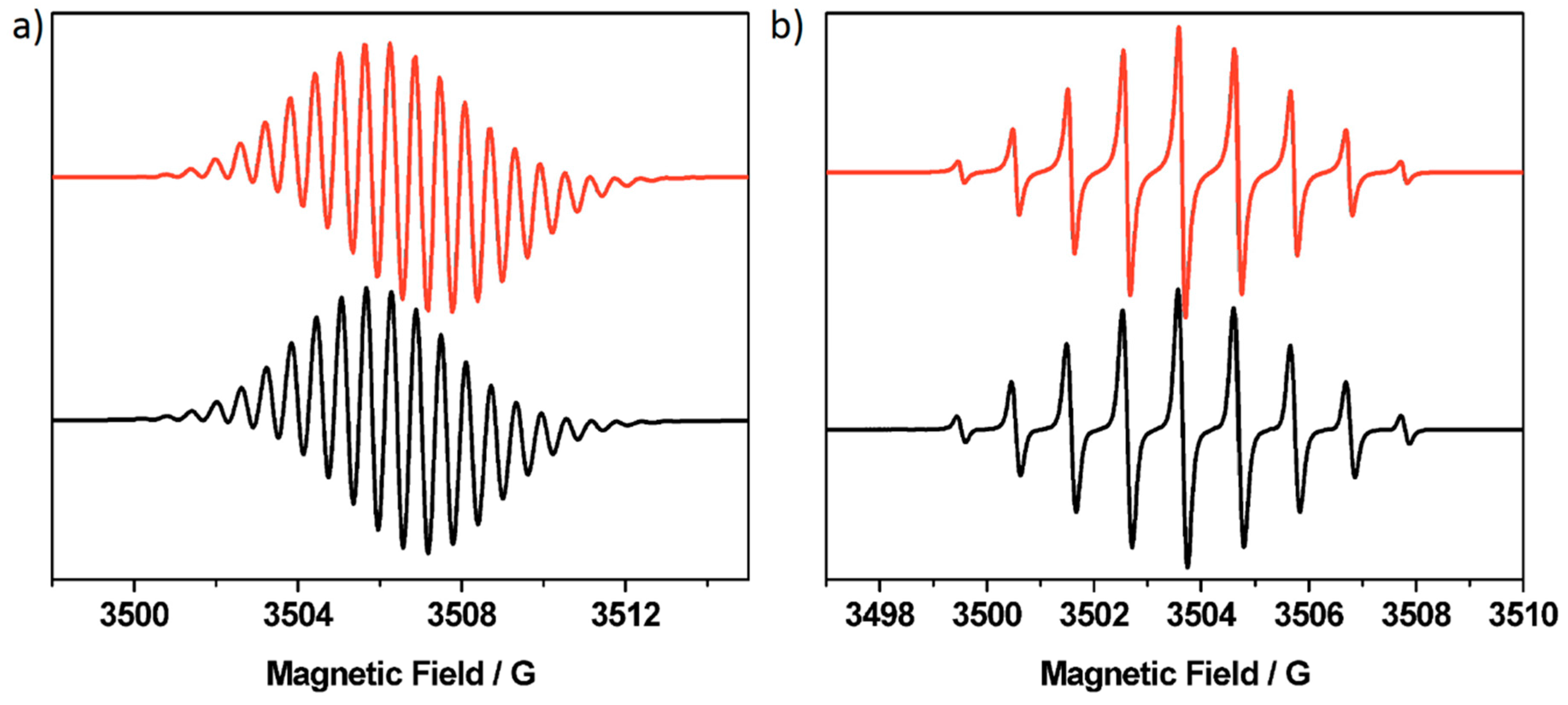
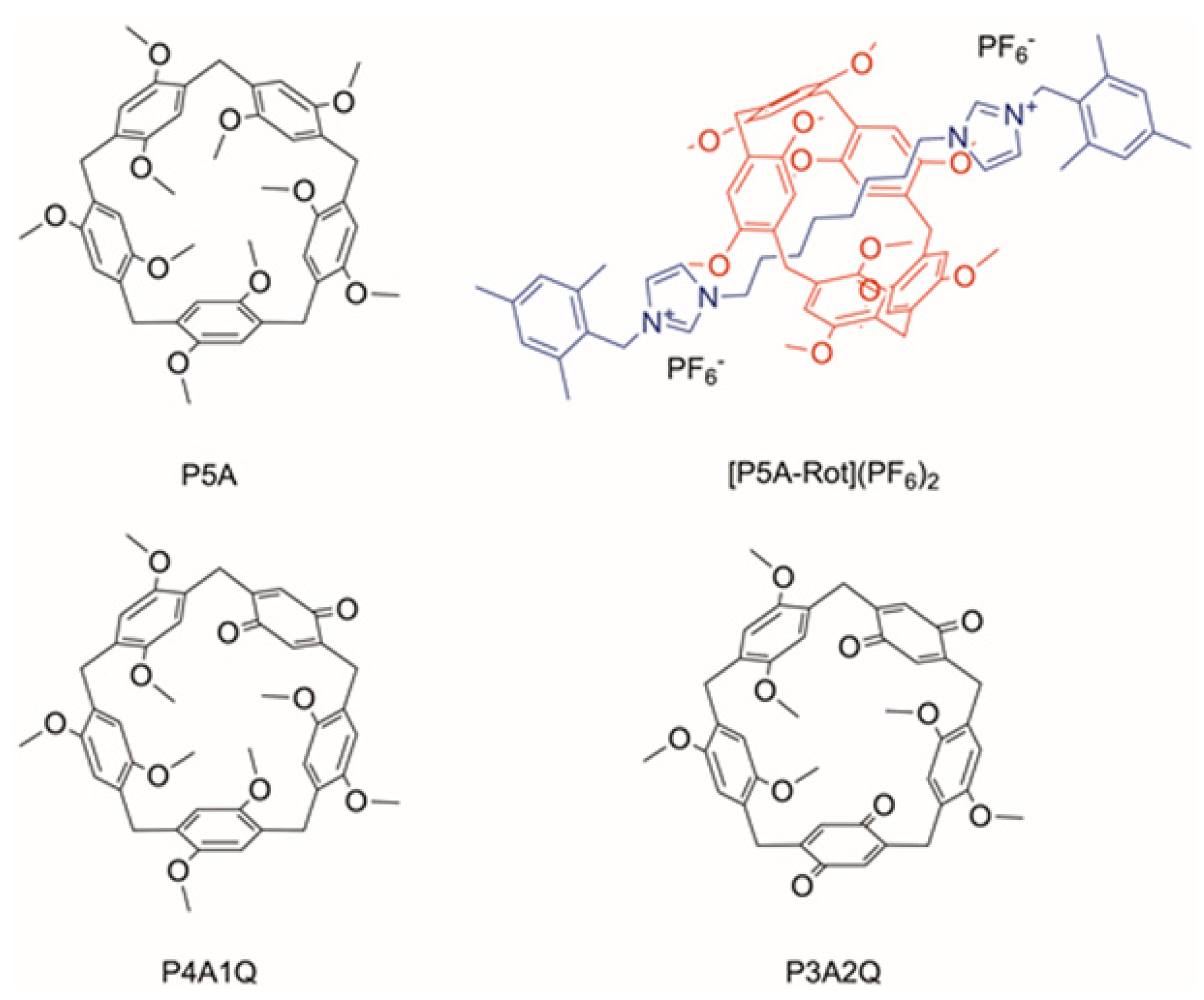
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ogoshi, T.; Kanai, S.; Fujinami, S.; Yamagashi, T.; Nakamoto, Y. para-Bridged symmetrical pillar[5]arenes: Their Lewis acid catalyzed synthesis and host-guest property. J. Am. Chem. Soc. 2008, 130, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Strutt, N.L.; Forgan, R.S.; Spruell, J.M.; Botros, Y.Y.; Stoddart, J.F. Monofunctionalized Pillar[5]arene as a Host for Alkanediamines. J. Am. Chem. Soc. 2011, 133, 5668–5671. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, S.; Li, J.; Han, K.; Xu, M.; Hu, B.; Yu, Y.; Jia, X. Novel neutral guest recognition and interpenetrated complex formation from pillar[5]arenes. Chem. Commun. 2011, 47, 11294–11296. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Chen, S.; Li, J.; Chen, Z.; Weng, L.; Jia, X.; Li, C. Highly effective binding of neutral dinitriles by simple pillar[5]arenes. Chem. Commun. 2012, 48, 2967–2969. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xu, Q.; Li, J.; Yao, F.; Jia, X. Complex interactions of pillar[5]arene with paraquats and bis(pyridinium) derivatives. Org. Biomol. Chem. 2010, 8, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Tanaka, S.; Yamagishi, T.; Nakamoto, Y. Ionic Liquid Molecules (ILs) as Novel Guests for Pillar[5]arene: 1:2 Host–Guest Complexes between Pillar[5]arene and ILs in Organic Media. Chem. Lett. 2011, 40, 96–98. [Google Scholar] [CrossRef]
- Hu, W.-B.; Hu, W.-J.; Zhao, X.-L.; Liu, Y.A.; Li, J.-S.; Jiang, B.; Wen, K. A1/A2-Diamino-Substituted Pillar[5]arene-Based Acid–Base-Responsive Host–Guest System. J. Org. Chem. 2016, 81, 3877–3881. [Google Scholar] [CrossRef]
- Yang, K.; Chao, S.; Zhang, F.; Pei, Y.; Pei, Z. Recent advances in the development of rotaxanes and pseudorotaxanes based on pillar[n]arenes: From construction to application. Chem. Commun. 2019, 55, 13198–13210. [Google Scholar] [CrossRef]
- Xue, M.; Yang, Y.; Chi, X.; Yan, X.; Huang, F. Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications. Chem. Rev. 2015, 115, 7398–7501. [Google Scholar] [CrossRef]
- Langer, P.; Yang, L.; Pfeiffer, C.R.; Lewis, W.; Champness, N.R. Restricting shuttling in bis(imidazolium)…pillar[5]arene rotaxanes using metal coordination. Dalton Trans. 2019, 48, 58–64. [Google Scholar] [CrossRef]
- Ogoshi, T.; Nishida, Y.; Yamagishi, T.; Nakamoto, Y. High Yield Synthesis of Polyrotaxane Constructed from Pillar[5]arene and Viologen Polymer and Stabilization of Its Radical Cation. Macromolecules 2010, 43, 7068–7072. [Google Scholar] [CrossRef]
- Zhang, Z.; Han, C.; Yu, G.; Huang, F. A solvent-driven molecular spring. Chem. Sci. 2012, 3, 3026–3031. [Google Scholar] [CrossRef]
- Dong, S.; Han, C.; Zheng, B.; Zhang, M.; Huang, F. Preparation of two new [2]rotaxanes based on the pillar[5]arene/alkane recognition motif. Tetrahedron Lett. 2012, 53, 3668–3671. [Google Scholar] [CrossRef]
- Ogoshi, T.; Aoki, T.; Shiga, R.; Iizuka, R.; Ueda, S.; Demachi, K.; Kayama, H.; Yamagishi, T. Cyclic Host Liquids for Facile and High-Yield Synthesis of [2]Rotaxanes. J. Am. Chem. Soc. 2012, 134, 20322–20325. [Google Scholar] [CrossRef]
- Ke, C.; Strutt, N.L.; Li, H.; Hou, X.; Hartlieb, K.J.; McGonigal, P.R.; Ma, Z.; Iehl, J.; Stern, C.L.; Cheng, C.; et al. Pillar[5]arene as a Co-Factor in Templating Rotaxane Formation. J. Am. Chem. Soc. 2013, 135, 17019–17030. [Google Scholar] [CrossRef]
- Li, S.-H.; Zhang, H.-Y.; Xu, X.; Liu, Y. Mechanically selflocked chiral gemini-catenanes. Nat. Commun. 2015, 6, 7590. [Google Scholar] [CrossRef]
- Han, Y.; Huo, G.-F.; Sun, J.; Xie, J.; Yan, C.-G.; Zhao, Y.; Wu, X.; Lin, C.; Wang, L. Formation of a series of stable pillar[5]arene-based pseudo[1]-rotaxanes and their [1]rotaxanes in the crystal state. Sci. Rep. 2016, 6, 28748. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, Y.; Chen, J.; Dong, S.; Yu, Y.; Ma, Z.; Huang, F. Formation of Linear Supramolecular Polymers That Is Driven by C-H⋅⋅⋅π Interactions in Solution and in the Solid State. Angew. Chem. Int. Ed. 2011, 50, 1397–1401. [Google Scholar] [CrossRef]
- Yang, L.; Langer, P.; Davies, E.S.; Baldoni, M.; Wickham, K.; Besley, N.A.; Besley, E.; Champness, N.R. Synthesis and characterisation of rylene diimide dimers using molecular handcuffs. Chem. Sci. 2019, 10, 3723–3732. [Google Scholar] [CrossRef]
- Kitajima, K.; Ogoshi, T.; Yamagishi, T. Diastereoselective synthesis of a [2]catenane from a pillar[5]arene and a pyridinium derivative. Chem. Commun. 2014, 50, 2925–2927. [Google Scholar] [CrossRef]
- Liu, L.; Cao, D.; Jin, Y.; Tao, H.; Kou, Y.; Meier, H. Efficient synthesis of copillar[5]arenes and their host–guest properties with dibromoalkanes. Org. Biomol. Chem. 2011, 9, 7007–7010. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Hu, X.-Y.; Jiang, J.; Wang, L. The self-complexation of mono-urea-functionalized pillar[5]arenes with abnormal urea behaviors. Chem. Commun. 2014, 50, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Demachi, K.; Kitajima, K.; Yamagishi, T. Monofunctionalized pillar[5]arenes: Synthesis and supramolecular structure. Chem. Commun. 2011, 47, 7164–7166. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Kitajima, K.; Fujinami, S.; Yamagishi, T. Synthesis and X-ray crystal structure of a difunctionalized pillar[5]arene at A1/B2 positions by in situcyclization and deprotection. Chem. Commun. 2011, 47, 10106–10108. [Google Scholar] [CrossRef]
- Deng, H.; Shu, X.; Hu, X.; Li, J.; Jia, X.; Li, C. Synthesis of a fully functionalized pillar[5]arene by ‘click chemistry’ and its effective binding toward neutral alkanediamines. Tetrahedron Lett. 2012, 53, 4609–4612. [Google Scholar] [CrossRef]
- Guo, M.; Wang, W.; Zhan, C.; Demay-Drouhard, P.; Li, W.; Du, K.; Olson, M.A.; Zuilhof, H.; Sue, A.C.-H. Rim-Differentiated C5-Symmetric Tiara-Pillar[5]arenes. J. Am. Chem. Soc. 2018, 140, 74–77. [Google Scholar] [CrossRef]
- Demay-Drouhard, P.; Du, K.; Samanta, K.; Wan, X.; Yang, W.; Srinivasan, R.; Sue, A.C.-H.; Zuilhof, H. Functionalization at Will of Rim-Differentiated Pillar[5]arenes. Org. Lett. 2019, 21, 3976–3980. [Google Scholar] [CrossRef]
- Yang, W.; Samanta, K.; Wan, X.; Thikekar, T.U.; Chao, Y.; Li, S.; Du, K.; Xu, J.; Gao, Y.; Zuilhof, H.; et al. Tiara[5]arenes: Synthesis, Solid-State Conformational Studies, Host–Guest Properties, and Application as Nonporous Adaptive Crystals. Angew. Chem. Int. Ed. 2020, 59, 3994–3999. [Google Scholar] [CrossRef]
- Ogoshi, T.; Yamafuji, D.; Kotera, D.; Aoki, T.; Fujinami, S.; Yamagishi, T. Clickable Di- and Tetrafunctionalized Pillar[n]arenes (n = 5, 6) by Oxidation–Reduction of Pillar[n]arene Units. J. Org. Chem. 2012, 77, 11146–11152. [Google Scholar] [CrossRef]
- Han, C.; Zhang, Z.; Yu, G.; Huang, F. Syntheses of a pillar[4]arene[1]quinone and a difunctionalized pillar[5]arene by partial oxidation. Chem. Commun. 2012, 48, 9876–9878. [Google Scholar] [CrossRef]
- Xie, C.; Hu, W.; Hu, W.; Liu, Y.A.; Huo, J.; Li, J.; Jiang, B.; Wen, K. Synthesis of Pillar[n]arene[5−n]quinines via Partial Oxidation of Pillar[5]arene. Chin. J. Chem. 2015, 33, 379–383. [Google Scholar] [CrossRef]
- Saba, H.; An, J.; Yang, Y.; Xue, M.; Liu, Y. Voltammetric Behavior of 1,4-Dimethoxypillar[m]arene[n]quinones. Chin. J. Chem. 2016, 34, 861–865. [Google Scholar] [CrossRef]
- Cheng, B.; Kaifer, A.E. Cathodic Voltammetric Behavior of Pillar[5]quinone in Nonaqueous Media. Symmetry Effects on the Electron Uptake Sequence. J. Am. Chem. Soc. 2015, 137, 9788–9791. [Google Scholar] [CrossRef] [PubMed]
- Smolko, V.A.; Shurpik, D.N.; Shamagsumova, R.V.; Porfireva, A.V.; Evtugyn, V.G.; Yakimova, L.S.; Stoikov, I.I.; Evtugyn, G.A. Electrochemical Behavior Of Pillar[5]Arene On Glassy Carbon Electrode And Its Interaction With Cu2+ And Ag+ Ions. Electrochim. Acta 2014, 147, 726–734. [Google Scholar] [CrossRef]
- Zweig, A.; Hodgson, W.G.; Jura, W.H. The Oxidation of Methoxybenzenes. J. Am. Chem. Soc. 1964, 86, 4124–4129. [Google Scholar] [CrossRef]
- Buck, R.P.; Wagoner, D.E. Selective anodic oxidation of p-alkylaryl ethers—Pathways and products. J. Electroanal. Chem. 1980, 115, 89–113. [Google Scholar] [CrossRef]
- Foos, J.S.; Erker, S.M.; Rembetsy, L.M. Synthesis and Characterization of Semiconductive Poly-1,4-Dimethoxybenzene and Its Derived Polyquinone. J. Electrochem. Soc. 1986, 133, 836–841. [Google Scholar] [CrossRef]
- Le Berre, V.; Angely, L.; Simonet, J. Electrochemical polymerization of paradialkoxybenzenes: Part I. Anodic oxidation of paradimethoxybenzene in dry acetonitrile. J. Electroanal. Chem. 1987, 218, 173–185. [Google Scholar] [CrossRef]
- Yamamoto, K.; Asada, T.; Nishide, H.; Tsuchida, E. Electro-oxidative polymerization of p-dialkoxybenzenes and its mechanism. Polym. Bull. 1988, 19, 533–538. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors by request. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1st Oxidation E1/2/V | 2nd Oxidation E1/2/V | Reduction | ΔE Fc+/Fc /V |
|---|---|---|---|---|
| P5A | +0.61 (0.08) | +0.83 (0.04) b,c | (0.07) | |
| [P5A-Rot](PF6)2 | +0.80 (0.10) | - | (0.07) | |
| P4A1Q | +0.69 (0.08) | +0.84 (0.04) c | −1.11 (0.07) d | (0.07) |
| P3A2Q | +0.85 (0.07) c | - | −1.08 (0.12) e,f | (0.07) |
| Compound | Neutral | 1st Oxidation | 2nd Oxidation | 3rd Oxidation | 1st Reduction |
|---|---|---|---|---|---|
| P5A | 295 (2.6) | 297 (2.4), 439 (0.4), 467 (0.6) | 298 (2.4), 440 (0.7), 467 (0.9) | 301 (2.4), 439 (1.0), 467 (1.2) | - |
| [P5A-Rot](PF6)2 | 294 (2.2) | 296 (2.2), 443 (0.8), 472 (1.1) | |||
| P4A1Q | 261 (1.3), 269 (1.4), 294 (1.7) | 258 (1.6), 266 (1.6), 296 (1.7), 440 (0.4), 467 (0.5) | 259 (1.8), 299 (1.7), 440 (0.5), 466 (0.7) | 295 (1.9), 325 (0.7), 436 (0.6) | |
| P3A2Q | 260 (2.2), 267 (2.1), 293 (1.3), 436 (0.1) | 254 (4.1), 456 (0.7) | 294 (1.5), 323 (1.4), 432 (1.0) |
| Compound | giso | aiso/x 10−4 cm−1 | Linewidth/G | Lineshape |
|---|---|---|---|---|
| [P5A]+ | 2.0038 | 1.720 (2H), 1.136 (4H), 0.570 (8H) | 0.25 | Gaussian |
| [P5A-Rot]3+ | 2.0037 | 1.780 (2H), 1.220 (4H), 0.580 (8H) | 0.37 | Lorentzian |
| [P4A1Q]- | 2.0050 | 1.947 (2H), 0.958 (4H) | 0.12 | Lorentzian |
| [P4A1Q]+ | 2.0036 | |||
| [P3A2Q]- | 2.0051 | 1.947 (2H), 0.958 (4H) | 0.22 | Lorentzian |
| Compound | 1st Oxidation /V | 2nd Oxidation /V | 3rd Oxidation /V | 1st Reduction /V | 2nd Reduction /V |
|---|---|---|---|---|---|
| P5A | +0.61 | +0.75 | +0.83 | - | - |
| [P5A-Rot](PF6)2 | +0.80 | +0.98 | - | - | - |
| P4A1Q | +0.69 | +0.83 | - | −1.10 | −1.66 |
| P3A2Q | +0.84b | - | - | −1.05c | −1.64 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearce, N.; Davies, E.S.; Champness, N.R. per-Alkoxy-pillar[5]arenes as Electron Donors: Electrochemical Properties of Dimethoxy-Pillar[5]arene and Its Corresponding Rotaxane. Molecules 2020, 25, 1627. https://doi.org/10.3390/molecules25071627
Pearce N, Davies ES, Champness NR. per-Alkoxy-pillar[5]arenes as Electron Donors: Electrochemical Properties of Dimethoxy-Pillar[5]arene and Its Corresponding Rotaxane. Molecules. 2020; 25(7):1627. https://doi.org/10.3390/molecules25071627
Chicago/Turabian StylePearce, Nicholas, E. Stephen Davies, and Neil R. Champness. 2020. "per-Alkoxy-pillar[5]arenes as Electron Donors: Electrochemical Properties of Dimethoxy-Pillar[5]arene and Its Corresponding Rotaxane" Molecules 25, no. 7: 1627. https://doi.org/10.3390/molecules25071627
APA StylePearce, N., Davies, E. S., & Champness, N. R. (2020). per-Alkoxy-pillar[5]arenes as Electron Donors: Electrochemical Properties of Dimethoxy-Pillar[5]arene and Its Corresponding Rotaxane. Molecules, 25(7), 1627. https://doi.org/10.3390/molecules25071627