Protein Extraction, Enrichment and MALDI MS and MS/MS Analysis from Bitter Orange Leaves (Citrus aurantium)

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Protein Identification
2.2. Bioinformatic Analysis
2.2.1. Prediction of Biological Processes and Protein Class
2.2.2. Subcellular Localization Prediction
2.2.3. Pathways Enrichment Analysis
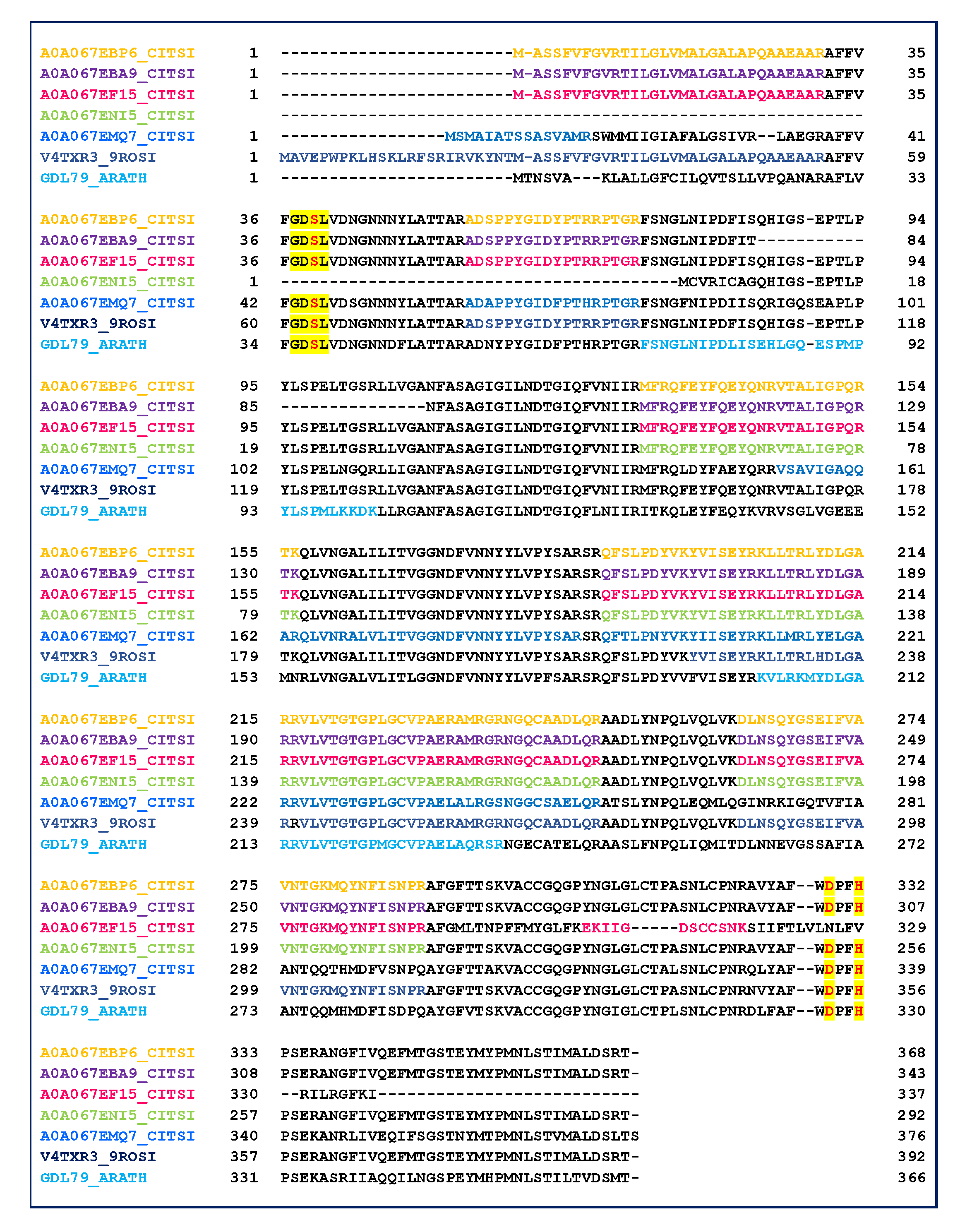
2.3. GDSL Esterase-Lipase Characterization
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Plant Materials
4.3. Protein Extraction
4.4. Solid Phase Extraction (SPE) Procedures
4.5. SDS PAGE
4.6. In-Solution Digestion
4.7. Mass Spectrometry Analysis
4.8. Database Proteomics, Targeting Predictions and Functional Classification
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Castle, W.S. A Career Perspective on Citrus Rootstocks, Their Development, and Commercialization. HortScience 2010, 45, 11–15. [Google Scholar] [CrossRef]
- Nimbolkar, P.K.; Awachare, C.; Reddy, Y.T.N.; Chander, S.; Hussain, F. Role of Rootstocks in Fruit Production—A Review. J. Agric. Eng. Food Technol. 2016, 3, 183–188. [Google Scholar]
- Vensel, W.H.; Tanaka, C.K.; Cai, N.; Wong, J.H.; Buchanan, B.B.; Hurkman, W.J. Developmental changes in the metabolic protein profiles of wheat endosperm. Proteomics 2005, 5, 1594–1611. [Google Scholar] [CrossRef]
- Balmer, Y.; Vensel, W.H.; Dupont, F.M.; Buchanan, B.B.; Hurk-Man, W.J. Proteome of amyloplasts isolated from developing wheat endosperm presents evidence of broad metabolic capability. J. Exp. Bot. 2006, 57, 1591–1602. [Google Scholar] [CrossRef]
- Song, X.; Ni, Z.; Yao, Y.; Xie, C.; Li, Z.; Wu, H.; Zhang, Y.; Professo, Q.S. Wheat (Triticumaestivum L.) root proteome and differentially expressed root proteins between hybridand parents. Proteomics 2007, 7, 3538–3557. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, B.E.; Madden, R.D.; Ayoubi, P.; Porter, R.; Dillwith, J.W. The wheat (Triticum aestivum L.) leaf proteome. Proteomics 2005, 5, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.M.; Echevarria-Zomeno, S.; Jean-Baptiste, S.; Hernandez, M.; Jorrin-Novo, J.V. Evaluation of three different protocols of protein extraction for Arabidopsis thaliana leaf proteome analysis by two-dimensional electrophoresis. J. Proteom. 2008, 71, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.P.; Ventura, J.A.; Zingali, R.B.; Fernandes, R.M.B. Evaluation of sample preparation methods for the analysis of papaya leaf proteins through 2wo-dimensional gel electrophoresis. Phytochem. Anal. 2009, 20, 456–464. [Google Scholar] [CrossRef]
- Da Silva, M.A.O.; Garcia, J.S.; de Souza, G.; Eberlin, M.N.; Gozzo, F.C.; Arruda, M.A.Z. Evaluation of sample preparation protocols for proteomic analysis of sunflower leaves. Talanta 2010, 80, 1545–1551. [Google Scholar] [CrossRef]
- Xie, C.J.; Wang, D.; Yang, X.Y. Protein extraction methods compatible with proteomic analysis for the cotton seedling. Crop Sci. 2009, 49, 395–402. [Google Scholar] [CrossRef]
- Zheng, Q.; Song, J.; Doncaster, K.; Rowland, E.; Byers, D.M. Qualitative and quantitative evaluation of protein extraction protocols for apple and strawberry fruit suitable for two-dimensional electrophoresis and mass spectrometry analysis. J. Agric. Food Chem. 2007, 55, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Napoli, A.; Aiello, D.; Di Donna, D.; Moschidis, P.; Sindona, G. Vegetable proteomics: The detection of Ole e 1 isoallergens by peptide matching of MALDI MS/MS spectra of underivatized and dansylated glycopeptides. J. Prot. Res. 2008, 7, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Delaplace, P.; van der Wal, F.; Dierick, J.F.; Cordewener, J.H.G.; Fauconnier, M.L.; du Jardin, P.; America, A.H.P. Potato tuber proteomics: Comparison of two complementary extraction methods designed for 2-DE of acidic proteins. Proteomics 2006, 6, 6494–6497. [Google Scholar] [CrossRef] [PubMed]
- Jellouli, N.; Ben Salem, A.; Ghorbel, A.; Ben Jouira, H. Evaluation of protein extraction methods for Vitis vinifera leaf and root proteome analysis by two-dimensional electrophoresis. J. Integr. Plant Biol. 2010, 52, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.S.; Prinsi, B.; Scienza, A.; Morgutti, S.; Cocucci, M.; Espen, L. Analysis of grape berry cell wall proteome: A comparative evaluation of extraction methods. J. Plant Physiol. 2008, 165, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Shi, M.J.; Lu, X.L.; Ma, R.F.; Wu, C.G.; Guo, A.P.; Peng, M.; Tian, W. A method for protein extraction from different subcellular fractions of laticifer latex in Hevea brasiliensis compatible with 2-DE and MS. Proteome Sci. 2010, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yang, Y.W.; Liu, J.Y. An efficient protein preparation for proteomic analysis of developing cotton fibers by 2-DE. Electrophoresis 2006, 27, 4559–4569. [Google Scholar] [CrossRef]
- Carpentier, S.C.; Witters, E.; Laukens, K.; Deckers, P.; Swennen, R.; Panis, B. Preparation of protein extracts from recalcitrant plant tissues: An evaluation of different methods for two-dimensional gel electrophoresis analysis. Proteomics 2005, 5, 2497–2507. [Google Scholar] [CrossRef]
- Napoli, A.; Aiello, D.; Di Donna, L.; Sajjad, A.; Perri, E.; Sindona, G. Profiling of hydrophilic proteins from Olea europaea olive pollen by MALDI TOF mass spectrometry. Anal. Chem. 2006, 78, 3434–3443. [Google Scholar] [CrossRef]
- Fan, P.X.; Wang, X.C.; Kuang, T.Y.; Li, Y.X. An efficient method for the extraction of chloroplast proteins compatible for 2-DE and MS analysis. Electrophoresis 2009, 30, 3024–3033. [Google Scholar] [CrossRef]
- Witzel, K.; Shahzad, M.; Matros, A.; Mock, H.P.; Mühling, K.H. Comparative evaluation of extraction methods for apoplastic proteins from maize leaves. Plant Methods 2011, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Zukas, A.A.; Breksa, A.P., III. Extraction methods for analysis of Citrus leaf proteins by two-dimensional gel electrophoresis. J. Chromatogr. A 2005, 1078, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Maserti, B.E.; Della Croce, C.M.; Luro, F.; Morillon, R.; Cini, M.; Caltavuturo, L. A general method for the extraction of citrus leaf proteins and separation by 2D electrophoresis: A follow up. J. Chromatogr. B 2007, 849, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Maserti, B.E.; Del Carratore, R.; Della Croce, C.M.; Podda, A.; Migheli, Q.; Froelicher, Y.; Luro, F.; Morillon, R.; Ollitrault, P.; Talon, M.; et al. Comparative analysis of proteome changes induced by the two spotted spider mite Tetranychus urticae and methyl jasmonate in citrus leaves. J. Plant Physiol. 2011, 168, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Lliso, I.; Tadeo, F.R.; Phinney, B.S.; Wilkerson, C.G.; Talon, M. Protein changes in the albedo of citrus fruits on post-harvesting storage. J. Agric. Food Chem. 2007, 55, 9047–9053. [Google Scholar] [CrossRef] [PubMed]
- Cantú, M.D.; Mariano, A.G.; Palma, M.S.; Carrilho, E.; Wulff, N.A. Proteomic analysis reveals suppression of bark chitinases and proteinase inhibitors in Citrus plants affected by the Citrus Sudden Death Disease. Phytopathology 2008, 98, 1084–1092. [Google Scholar] [CrossRef]
- Shi, J.X.; Chen, S.; Gollop, N.; Goren RGoldschmidt, E.E.; Porat, R. Effects of anaerobic stress on the proteome of citrus fruit. Plant Sci. 2008, 175, 478–486. [Google Scholar] [CrossRef]
- Napoli, A.; Aiello, D.; Di Donna, L.; Prendushi, H.; Sindona, G. Exploitation of endogenous protease activity in raw mastitic milk by MALDI-TOF/TOF. Anal. Chem. 2007, 79, 5941–5948. [Google Scholar] [CrossRef]
- Aiello, D.; Materazzi, S.; Risoluti, R.; Thangavel, H.; Di Donna, L.; Mazzotti, F.; Casadonte, F.; Siciliano, C.; Sindona, G.; Napoli, A. Major allergen in rainbow trout (Oncorhynchus mykiss): Complete sequences of Parvalbumin by MALDI tandem mass spectrometry. Mol. BioSyst. 2015, 11, 2373–2381. [Google Scholar] [CrossRef]
- Aiello, D.; Casadonte, F.; Terracciano, R.; Damiano, R.; Savino, R.; Sindona, G.; Napoli, A. Targeted proteomics approach in prostatic tissue: A panel of potential biomarkers for cancer detection. Oncoscience 2016, 3, 220–241. [Google Scholar]
- Aiello, D.; Cardiano, P.; Cigala, R.M.; Gans, P.; Giacobello, F.; Giuffrè, O.; Napoli, A.; Sammartano, S. Sequestering ability of oligophosphate ligands toward Al3+ in Aqueous Solution. J. Chem. Eng. Data 2017, 62, 3981–3990. [Google Scholar] [CrossRef]
- Napoli, A.; Athanassopoulos, C.M.; Moschidis, P.; Aiello, D.; Di Donna, D.; Mazzotti, F.; Sindona, G. Solid phase isobaric mass tag reagent for simultaneous protein identification and assay. Anal. Chem. 2010, 82, 5552–5560. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, F.; Di Donna, L.; Napoli, A.; Aiello, D.; Siciliano, C.; Athanassopoulos, C.M.; Sindona, G. N-hydroxysuccinimidyl p-methoxybenzoate as suitable derivative reagent for isotopic dilution assay of biogenic amines in food. J. Mass Spectrom. 2014, 49, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.E.; Albanito, L.; De Marco, P.; Aiello, D.; Maggiolini, M.; Napoli, A.; Musti, A.M. Multisite phosphorylation of c-Jun at threonine 91/93/95 triggers the onset of c-Jun pro-apoptotic activity in cerebellar granule neurons. Cell Death Disease 2013, 4, e852. [Google Scholar] [CrossRef]
- Jahouh, F.; Saksena, R.; Aiello, D.; Napoli, A.; Sindona, G.; Kovàc, P.; Banoub, J.H. Glycation sites in neoglycoglycoconjugates from the terminal monosaccharide antigen of the O-PS of Vibrio cholerae O1; serotype Ogawa; and BSA revealed by matrix-assisted laser desorption–ionization tandem mass spectrometry. J. Mass. Spectrom. 2010, 45, 1148–1159. [Google Scholar] [CrossRef]
- Van Kampen, J.J.; Burgers, P.C.; de Groot, R.; Gruters, R.A.; Luider, T.M. Biomedical application of MALDI mass spectrometry for small-molecule analysis. Mass Spectrom. Rev. 2011, 30, 101–120. [Google Scholar] [CrossRef]
- Persike, M.; Karas, M. Rapid simultaneous quantitative determination of different small pharmaceutical drugs using a conventional matrix-assisted laser desorption/ionization time-of-flight mass spectrometry system. Rapid Commun. Mass Spectrom. 2009, 23, 3555–3562. [Google Scholar] [CrossRef]
- Persike, M.; Zimmermann, M.; Klein, J.; Karas, M. Quantitative Determination of Acetylcholine and Choline in Microdialysis Samples by MALDI-TOF MS. Anal. Chem. 2010, 82, 922–929. [Google Scholar] [CrossRef]
- Meesters, R.J.; Hooff, G.P.; Gruters, R.; van Kampen, J.J.; Luider, T.M. Incurred sample reanalysis comparison of dried blood spots and plasma samples on the measurement of lopinavir in clinical samples. Bioanalysis 2012, 4, 237–240. [Google Scholar] [CrossRef]
- Van Kampen, J.J.; Burgers, P.C.; de Groot, R.; Luider, T.M. Qualitative and quantitative analysis of pharmaceutical compounds by MALDI-TOF mass spectrometry. Anal. Chem. 2006, 78, 5403–5411. [Google Scholar] [CrossRef][Green Version]
- Aiello, D.; Giambona, A.; Leto, F.; Passarello, C.; Damiani, G.; Maggio, A.; Siciliano, C.; Napoli, A. Human coelomic fluid investigation: A MS-based analytical approach to prenatal screening. Sci. Rep. 2018, 8, 10973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.H.; Zhao, J.F.; Song, Y.; Zhang, C.J.; Guo, Y. Identification of an Apoplastic Protein Involved in the Initial Phase of Salt Stress Response in Rice Root by Two-Dimensional Electrophoresis. Plant Physiol. 2009, 149, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Ghahremani, M.; Stigter, K.A.; Plaxton, W. Extraction and characterization of extracellular proteins and their post-translational modifications from arabidopsis thaliana suspension cell cultures and seedlings: A critical review. Proteomes 2016, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Albenne, C.; Canut, H.; Jamet, E. Plant cell wall proteomics: The leadership of arabidopsis thaliana. Front. Plant Sci. 2013, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Malmirchegini, G.R.; Clubb, R.T.; Loo, J.A. Native top-down mass spectrometry for the structural characterization of human hemoglobin. Eur. J. Mass Spectrom. 2015, 21, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.Y.; Shi, W.Y.; Bryant, S.H.J. Open mass spectrometry search algorithm. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Han, X.M.; Jin, M.; Breuker, K.; McLafferty, F.W. Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons. Science 2006, 314, 109–112. [Google Scholar] [CrossRef]
- Risoluti, R.; Piazzese, D.; Napoli, A.; Materazzi, S. Study of [2-(2′-pyridyl) imidazole] complexes to confirm two main characteristic thermoanalytical behaviors of transition metal complexes based on imidazole derivatives. J. Anal. Appl. Pyrolysis 2016, 117, 82–87. [Google Scholar] [CrossRef]
- Zubarev, R.A.; Kelleher, N.L.; McLafferty, F.W. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc. 1998, 120, 3265–3266. [Google Scholar] [CrossRef]
- Syka, J.E.P.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef]
- Pitteri, S.J.; Chrisman, P.A.; Hogan, J.M.; McLuckey, S.A. Electron-Transfer Ion/Ion Reactions of Doubly Protonated Peptides: Effect of Elevated Bath Gas Temperature. Anal. Chem. 2005, 77, 1831–1839. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Little, D.P.; Speir, J.P.; Senko, M.W.; Oconnor, P.B.; McLafferty, F.W. Infrared Multiphoton Dissociation of Large Multiply Charged Ions for Biomolecule Sequencing. Anal. Chem. 1994, 66, 2809–2815. [Google Scholar] [CrossRef] [PubMed]
- Laskin, J.; Futrell, J.H. Activation of large ions in FT-ICR mass spectrometry. Mass Spectrom. Rev. 2005, 24, 135–167. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2019, 29. [Google Scholar] [CrossRef]
- Brick, D.J.; Brumlik, M.J.; Buckley, J.T.; Cao, J.X.; Davies, P.C.; Misra, S.; Tranbarger, T.J.; Upton, C. A new family of lipolytic plant enzymes with members in rice; arabidopsis and maize. FEBS Lett. 1995, 377, 475–480. [Google Scholar]
- Lee, D.S.; Kim, B.K.; Kwon, S.J.; Jin, H.C.; Park, O.K. Arabidopsis GDSL lipase 2 plays a role in pathogen defense via negative regulation of auxin signaling. Biochem. Biophys. Res. Commun. 2009, 379, 1038–1042. [Google Scholar] [CrossRef]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL family of serine esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef]
- Martinez-Esteso, M.J.; Selles-Marchart, S.; Vera-Urbina, J.C.; Pedreno, M.A.; Bru-Martinez, R. Changes of defense proteins in the extracellular proteome of grapevine (Vitis vinifera cv. Gamay) cell cultures in response to elicitors. J. Proteom. 2009, 73, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Englert, M.; Beier, H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res. 2005, 33, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, P.; Cui, D.; Wu, L.; Parkn, I.; Saberianfar, R.; Menassa, R.; Pan, H.; Westcottn, M.Y.; Gruber, M.Y. The Arabidopsis tt19-4 mutant differentially accumulates proanthocyanidin and anthocyanin through a 3’ amino acid substitution in glutathione S-transferase. Plant Cell Environ. 2011, 34, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Roxas, V.P.; Smith, R.K., Jr.; Allen, E.R.; Allen, R.D. Overexpression of glutathione S-transferase/glutathione peroxidase enhances the growth of transgenic tobacco seedlings during stress. Nat. Biotechnol. 1997, 15, 988–991. [Google Scholar] [CrossRef]
- Held, K.; Pascaud, F.; Eckert, C.; Gajdanowicz, P.; Hashimoto, K.; Corratgé-Faillie, C.; Offenborn, J.N.; Lacombe, B.; Dreyer, I.; Thibaud, J.B.; et al. Calcium-dependent modulation and plasma membrane targeting of the AKT2 potassium channel by the CBL4/CIPK6 calcium sensor/protein kinase complex. Cell Res. 2011, 21, 1116–1130. [Google Scholar] [CrossRef]
- Romeis, T.; Ludwig, A.A.; Martin, R.; Jones, J.D. Calcium-dependent protein kinases play an essential role in a plant defence response. EMBO J. 2001, 20, 5556–5567. [Google Scholar] [CrossRef]
- Yang, C.W.; Gonzàlez-Lamothe, R.; Ewan, R.A.; Rowland, O.; Yoshioka, H.; Shenton, M.; Ye, H.; O’Donnell, E.; Jones, J.D.G.; Sadanandoma, A. The E3 ubiquitin ligase activity of arabidopsis PLANT U-BOX17 and its functional tobacco homolog ACRE276 are required for cell death and defense. Plant Cell 2006, 18, 1084–1098. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Jwa, N.S.; Lebrun, M.H.; Job, D.; Rakwal, R. Plant secretome: Unlocking secrets of the secreted proteins. Proteomics 2010, 10, 799–827. [Google Scholar] [CrossRef]
- Rose, J.K.C.; Lee, S.J. Straying off the highway: Trafficking of secreted plant proteins and complexity in the plant cell wall proteome. Plant Physiol. Biochem. 2010, 153, 433–436. [Google Scholar] [CrossRef]
- Zhu, J.M.; Chen, S.X.; Alvarez, S.; Asirvatham, V.S.; Schachtman, D.P.; Wu, Y.J.; Sharp, R.E. Cell wall proteome in the maize primary root elongation zone. I. Extraction and identification of water-soluble and lightly ionically bound proteins. Plant Physiol. 2006, 140, 311–325. [Google Scholar] [CrossRef]
- De Miccolis Angelini, R.M.; Rotolo, C.; Gerin, D.; Abate, D.; Pollastro, S.; Faretra, F. Global transcriptome analysis and differentially expressed genes in grapevine after application of the yeast-derived defense inducer cerevisane. Pest Manag. Sci. 2019, 75, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.S.; Park, A.R.; Bae, M.S.; Kwon, S.J.; Kim, Y.S.; Lee, J.E.; Kang, N.Y.; Lee, S.; Cheong, H.; Park, O.K. Secretome analysis reveals an Arabidopsis lipase involved in defence against Alternaria brassicicola. Plant Cell 2005, 17, 2832–2847. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.L.; Yan, S.Z.; Tan, R.K.; Zhang, Z.Y.; Wang, Z.; Chen, J. Characterization and expression of a GDSL-like lipase gene from Brassica napus in Nicotiana benthamiana. Protein J. 2014, 33, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.L.; Mounet, F.; Lemaire-Chamley, M.; Gaillard, C.; Elmorjani, K.; Vivancos, J.; Runavot, J.L.; Quemener, B.; Petit, J.; Germain, V.; et al. Tomato GDSL1 is required for cutin deposition in the fruit cuticle. Plant Cell 2012, 24, 3119–3134. [Google Scholar] [CrossRef]
- Neilson, E.H.; Goodger, J.Q.D.; Woodrow, I.E.; Lindberg Møller, B. Plant chemical defense: At what cost? Trends Plant Sci. 2013, 18, 250–258. [Google Scholar] [CrossRef]
- Höller, K.; Király, L.; Künstler, A.; Müller, M.; Gullner, G.; Fattinger, M.; Zechmann, B. Enhanced glutathione metabolism is correlated with sulfur-induced resistance in tobacco mosaic virus-infected genetically susceptible Nicotiana tabacum plants. Mol. Plant Microbe Interact. 2010, 23, 1448–1459. [Google Scholar] [CrossRef]
- Lo Piero, A.R.; Puglisi, I.; Petrone, G. Gene isolation; analysis of expression and invitro synthesis of a glutathione S-transferase from orange fruit. [Citrus sinensis L. (Osbeck)]. J. Agric. Food Chem. 2006, 54, 9227–9233. [Google Scholar] [CrossRef]
- Mauch, F.; Dudler, R. Differential induction of distinct glutathione transferases of wheat by xenobiotics and by pathogen attack. Plant Physiol. 1993, 102, 1193–1201. [Google Scholar] [CrossRef]
- Moons, A. Osgstu3 and osgtu4; encoding tau class glutathione S-transferases; are heavy metal and hypoxic stress-induced and differentially salt stress-responsive in rice roots. FEBS Lett. 2003, 553, 427–432. [Google Scholar] [CrossRef]
- Bianchi, M.W.; Roux, C.; Vartanian, N. Drought regulation of GST8; encoding the Arabidopsis homologue of ParC/Nt107 glutathione transferase/peroxidase. Physiol. Plant 2002, 116, 96–105. [Google Scholar] [CrossRef]
- Edwards, R.; Dixon, D.P.; Walbot, V. Plant glutathione transferases: Enzymes with multiple functions in sickness and in health. Trends Plant Sci. 2000, 5, 193–198. [Google Scholar] [CrossRef]
- Lo Piero, A.R.; Mercurio, V.; Puglisi, I.; Petrone, G. Gene isolation and expression analysis of two distinct sweet orange [Citrus sinensis L. (Osbeck)] tau-type glutathione transferases. Gene 2009, 443, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Axarli, A.; Rigden, D.J.; Labrou, N.E. Characterization of the ligandin site of maize glutathione transferase I. Biochem. J. 2004, 382, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, K.; Tommasini, R.; Martinoia, E. Herbicide detoxification in plants. Plant Physiol. 1996, 111, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Napoli, A.; Aiello, D.; Aiello, G.; Cappello, M.S.; Di Donna, L.; Mazzotti, F.; Materazzi, S.; Fiorillo, M.; Sindona, G. Mass spectrometry-based proteomic approach in Oenococcus oeni enological starter. J. Prot. Res. 2014, 13, 2856–2866. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP; SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Mak, M.-W.; Kun, S.-Y. mGOASVM: Multi-label protein subcellular localization based on gene ontology and support vector machines. BMC Bioinform. 2012, 13, 290. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes; a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession a | Protein Name a | Specie a | Functions and Domains a | MW a | |
|---|---|---|---|---|---|
| 1 | A0A067FLL5 | Alanine--tRNA ligase | c. s. | mitochondrial alanyl-tRNA aminoacylation, ATP binding and protein biosynthesis | 80.538 |
| 2 | A0A067FXS4 | Alanine--tRNA ligase | c. s. | mitochondrial alanyl-tRNA aminoacylation, ATP binding and protein biosynthesis | 85.228 |
| 3 | A0A067EAN5 | Belongs to the zinc-containing alcohol dehydrogenase family. | c. s. | oxidoreductase activity, zinc ion binding | 40.377 |
| 4 | Q3HM93 | Glutathione S-transferase | c. s. | transferase activity | 24.239 |
| 5 | A0A067F884 | Non-specific serine/threonine protein kinase | c. s. | ATP binding and protein serine/threonine kinase activity | 49.609 |
| 6 | A0A067FQ29 | Probable alanine--tRNA ligase, chloroplastic | c. s. | Aminoacyl-tRNA synthetase, Ligase, RNA-binding, tRNA-binding | 104.431 |
| 7 | A0A067GDZ1 | RING-type E3 ubiquitin transferase | c. s. | ubiquitin-protein transferase activity | 78.497 |
| 8 | A7U3F5 | RNA polymerase B (Fragment) | c. a. | DNA binding | 17.157 |
| 9 | C6KK63 | RpoB (Fragment) | c. a. | DNA binding | 13.966 |
| 10 | A0A067EE86 | Similar to Putative alcohol dehydrogenase | c. s. | oxidation-reduction process | 27.319 |
| 11 | A0A067H5U9 | Sodium/hydrogen exchanger | c. s. | sodium:proton antiporter activity | 57.964 |
| 12 | A0A067DAD8 | Uncharacterized protein | c. s. | Hypotetical member of ribonuclease H-like superfamily | 42.202 |
| 13 | A0A067DDE4 | Uncharacterized protein | c. s. | protein kinase activity | 50.12 |
| 14 | A0A067DMF5 | Uncharacterized protein | c. s. | aspartic-type endopeptidase activity | 37.197 |
| 15 | A0A067DUQ6 | Uncharacterized protein | c. s. | aspartic-type endopeptidase activity | 42.258 |
| 16 | A0A067DV99 | Uncharacterized protein | c. s. | gene silencing by RNA, containig XS domain | 132.604 |
| 17 | A0A067DVX6 | Uncharacterized protein | c. s. | aspartic-type endopeptidase activity | 40.249 |
| 18 | A0A067DXK8 | Uncharacterized protein | c. s. | containing development and cell death domain | 66.65 |
| 19 | A0A067DYD3 | Uncharacterized protein | c. s. | oxidation-reduction process | 28.91 |
| 20 | A0A067DYR7 | Uncharacterized protein | c. s. | oxidation-reduction process | 36.107 |
| 21 | A0A067DZ88 | Uncharacterized protein | c. s. | 104.139 | |
| 22 | A0A067E608 | Uncharacterized protein | c. s. | containing development and cell death domain | 66.418 |
| 23 | A0A067EAX4 | Uncharacterized protein | c. s. | similar to Importin subunit alpha-6 (Arabidopsis thaliana), protein transporter activity | 61.989 |
| 24 | A0A067ECD2 | Uncharacterized protein | c. s. | DNA binding | 26.85 |
| 25 | A0A067ECH7 | Uncharacterized protein | c. s. | ATP binding and protein kinase activity | 86.835 |
| 26 | A0A067EGL9 | Uncharacterized protein | c. s. | oxidoreductase activity | 31.792 |
| 27 | A0A067EJ07 | Uncharacterized protein | c. s. | transcription factor activity, containig GATA-type domain | 34.845 |
| 28 | A0A067EJ84 | Uncharacterized protein | c. s. | methyltransferase activity | 38.231 |
| 29 | A0A067EKU4 | Uncharacterized protein | c. s. | DNA binding; protein containing SAND domain | 20.855 |
| 30 | A0A067EPP0 | Uncharacterized protein | c. s. | ATP binding and protein kinase activity | 113.792 |
| 31 | A0A067ES66 | Uncharacterized protein | c. s. | containing coiled coil domaina | 55.599 |
| 32 | A0A067EVC3 | Uncharacterized protein | c. s. | metal binding, containig zinc finger (Znf) domains | 31.557 |
| 33 | A0A067F275 | Uncharacterized protein | c. s. | similar to Glutathione S-transferase (C. S.) | 24.233 |
| 34 | A0A067FBM6 | Uncharacterized protein | c. s. | transcription factor activity, | 27.199 |
| 35 | A0A067FNX1 | Uncharacterized protein | c. s. | 17.715 | |
| 36 | A0A067FS06 | Uncharacterized protein | c. s. | containing 3 coiled coil domain | 98.755 |
| 37 | A0A067FYX5 | Uncharacterized protein | c. s. | aspartic-type endopeptidase activity | 41.826 |
| 38 | A0A067FZS8 | Uncharacterized protein | c. s. | protein serine/threonine phosphatase activity | 64.983 |
| 39 | A0A067G2U9 | Uncharacterized protein | c. s. | 54.933 | |
| 40 | A0A067G2Z9 | Uncharacterized protein | c. s. | 53.389 | |
| 41 | A0A067G6L7 | Uncharacterized protein | c. s. | O-methyltransferase activity | 105.692 |
| 42 | A0A067G9E6 | Uncharacterized protein | c. s. | O-methyltransferase activity | 105.779 |
| 43 | A0A067GBI2 | Uncharacterized protein | c. s. | protein serine/threonine phosphatase activity | 78.787 |
| 44 | A0A067GET1 | Uncharacterized protein | c. s. | 50.073 | |
| 45 | A0A067GIB5 | Uncharacterized protein | c. s. | DNA binding and regulation of transcription | 31.651 |
| 46 | A0A067GIK6 | Uncharacterized protein | c. s. | O-methyltransferase activity | 103.594 |
| 47 | A0A067GIV0 | Uncharacterized protein | c. s. | O-methyltransferase activity | 86.016 |
| 48 | A0A067GNR1 | Uncharacterized protein | c. s. | ubiquitin-protein transferase activity | 407.981 |
| 49 | A0A067GQL4 | Uncharacterized protein | c. s. | 71.588 | |
| 50 | A0A067GRF1 | Uncharacterized protein | c. s. | ubiquitin-protein transferase activity | 395.412 |
| 51 | A0A067GT43 | Uncharacterized protein | c. s. | containing Cir_N domain and coiled coil doman | 48.391 |
| 52 | A0A067GUC9 | Uncharacterized protein | c. s. | Potential transmembrane proteins | 30.548 |
| 53 | A0A067GUN6 | Uncharacterized protein | c. s. | Potential transmembrane proteins | 23.614 |
| 54 | A0A067GV48 | Uncharacterized protein | c. s. | 83.17 | |
| 55 | A0A067GVN8 | Uncharacterized protein | c. s. | DNA binding and regulation of transcription | 27.869 |
| 56 | A0A067GYR1 | Uncharacterized protein | c. s. | containing post-SET domain | 87.903 |
| 57 | A0A067H0N2 | Uncharacterized protein | c. s. | ubiquitin-protein transferase activity | 406.782 |
| 58 | A0A067H3Y3 | Uncharacterized protein | c. s. | pyridoxal phosphate binding | 51.821 |
| 59 | A0A067GNF9 | Uncharacterized protein | c. s. | ubiquitin-protein transferase activity | 407.805 |
| 60 | A0A067DIT7 | Uncharacterized protein | c. s. | aspartic-type endopeptidase activity | 45.31 |
| 61 | A0A067EBP6 | Uncharacterized protein | c. s. | hydrolase activity, acting on ester bonds. Belongs to the ‘GDSL’ lipolytic enzyme family. Signal Peptide (1-29). | 40.484 |
| 62 | A0A067EBA9 | Uncharacterized protein | c. s. | hydrolase activity, acting on ester bonds. Belongs to the ‘GDSL’ lipolytic enzyme family. Signal Peptide (1-29). | 37.88 |
| 63 | A0A067EF15 | Uncharacterized protein | c. s. | Signal Peptide (1-31); Lipase_GDSL domain (34 – 316. Hydrolase activity, acting on ester bonds. Belongs to the ‘GDSL’ lipolytic enzyme family. | 37.337 |
| 64 | A0A067ENI5 | Uncharacterized protein | c. s. | Lipase_GDSL domain (78-265). Hydrolase activity, acting on ester bonds. Belongs to the ‘GDSL’ lipolytic enzyme family | 32.421 |
| 65 | A0A067EMQ7 | Uncharacterized protein | c. s. | Lipase_GDSL domain (40 – 352). Hydrolase activity, acting on ester bonds. Belongs to the ‘GDSL’ lipolytic enzyme family | 41.18 |
| 66 | V4TXR3 | Uncharacterized protein | c. s | Lipase_GDSL domain (58-365). Hydrolase activity, acting on ester bonds. | 43.441 |
| 67 | A0A067FW02 | Uncharacterized protein | c. s. | Signal Peptide (1–20); Peptidase A1 domain (140-476). Aspartic-type endopeptidase activity. Belongs to the peptidase A1 family | 50.918 |
| 68 | A0A067FVB0 | Uncharacterized protein | c. s. | Signal Peptide (1–20); Peptidase A1 domain (140–476). Aspartic-type endopeptidase activity. Belongs to the peptidase A1 family | 48.178 |
| 69 | A0A067DCQ1 | Uncharacterized protein (Fragment) | c. s. | solute:proton antiporter activity | 84.525 |
| 70 | A0A067DDS7 | Uncharacterized protein (Fragment) | c. s. | 63.918 | |
| 71 | A0A067DW09 | Uncharacterized protein (Fragment) | c. s. | 13.294 | |
| 72 | A0A067DZ15 | Uncharacterized protein (Fragment) | c. s. | diacylglycerol O-acyltransferase activity | 50.24 |
| 73 | A0A067ED32 | Uncharacterized protein (Fragment) | c. s. | containing coiled coil domain | 13.593 |
| 74 | A0A067EZE8 | Uncharacterized protein (Fragment) | c. s. | containing domain of unknown function (DUF1995) | 36.936 |
| 75 | A0A067FVE2 | Uncharacterized protein (Fragment) | c. s. | containing 5 coiled coil domain | 124.974 |
| 76 | A0A067G352 | Uncharacterized protein (Fragment) | c. s. | containing 5 coiled coil domain | 121.041 |
| 77 | A0A067GCY0 | Uncharacterized protein (Fragment) | c. s. | microtubule binding | 68.001 |
| 78 | A0A067GQ70 | Uncharacterized protein (Fragment) | c. s. | catalytic activity | 38.307 |
| Sequence a | Mr found b | Mr calc b |
|---|---|---|
| YIISEYRK | 1071.59 | 1071.58 |
| QFSLPDYVK | 1096.58 | 1096.57 |
| QFTLPNYVK | 1109.61 | 1109.60 |
| MASSFVFGVR (1Acetyl) | 1142.58 | 1142.57 |
| mASSFVFGVR (1Acetyl) | 1158.57 | 1158.56 |
| GSNGGCSAELQR | 1178.53 | 1178.52 |
| VTALIGPQRTK | 1183.73 | 1183.72 |
| EKIIGDSCCSNK | 1296.61 | 1296.59 |
| KVLRKmYDLGAR | 1465.85 | 1465.83 |
| KLLmRLYELGAR | 1478.87 | 1478.85 |
| MSMAIATSSASVAMR | 1513.73 | 1513.72 |
| KLLmRLYELGARR | 1634.97 | 1634.95 |
| AMRGRNGQCAADLQR | 1646.81 | 1646.80 |
| VKYNTMASSFVFGVR | 1705.89 | 1705.87 |
| VSAVIGAQQARQLVNR | 1709.99 | 1709.98 |
| VLVTGTGPLGCVPAERAMR | 1927.04 | 1927.03 |
| 1Met-loss (-)MAVEPWPKLHSKLRFSR | 1951.12 | 1951.10 |
| ADAPPYGIDFPTHRPTGR | 1967.99 | 1967.97 |
| AVEPWPKLHSKLRFSR (1Acetyl) | 1993.13 | 1993.11 |
| ADSPPYGIDYPTRRPTGR | 2019.02 | 2019.00 |
| RVLVTGTGPLGCVPAELALR | 2022.17 | 2022.15 |
| TILGLVmALGALAPQAAEAAR | 2053.17 | 2053.15 |
| RVLVTGTGPLGCVPAERAMR | 2083.15 | 2083.13 |
| QFTLPNYVKYIISEYRK | 2162.19 | 2162.16 |
| YVISEYRKLLTRLHDLGAR | 2303.32 | 2303.30 |
| RVLVTGTGPLGCVPAERAmRGR | 2312.27 | 2312.24 |
| YVISEYRKLLTRLYDLGAR | 2329.33 | 2329.30 |
| FSRIRVKYNTMASSFVFGVR | 2365.28 | 2365.26 |
| QFSLPDYVKYVISEYRKLLTR | 2618.46 | 2618.43 |
| ALVLITVGGNDFVNNYYLVPYSAR | 2658.42 | 2658.39 |
| MASSFVFGVRTILGLVmALGALAPQAAEAAR | 3134.72 | 3134.68 |
| mYDLGARRVLVTGTGPmGCVPAELAQRSR | 3136.61 | 3136.58 |
| MFRQFEYFQEYQNRVTALIGPQRTK | 3150.63 | 3150.59 |
| mASSFVFGVRTILGLVMALGALAPQAAEAAR (1Acetyl) | 3176.73 | 3176.70 |
| DLNSQYGSEIFVAVNTGKMQYNFISNPR | 3192.57 | 3192.54 |
| FSNGLNIPDLISEHLGQESPMPYLSPMLKKDK | 3598.86 | 3598.83 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, D.; Siciliano, C.; Mazzotti, F.; Di Donna, L.; Risoluti, R.; Napoli, A. Protein Extraction, Enrichment and MALDI MS and MS/MS Analysis from Bitter Orange Leaves (Citrus aurantium). Molecules 2020, 25, 1485. https://doi.org/10.3390/molecules25071485
Aiello D, Siciliano C, Mazzotti F, Di Donna L, Risoluti R, Napoli A. Protein Extraction, Enrichment and MALDI MS and MS/MS Analysis from Bitter Orange Leaves (Citrus aurantium). Molecules. 2020; 25(7):1485. https://doi.org/10.3390/molecules25071485
Chicago/Turabian StyleAiello, Donatella, Carlo Siciliano, Fabio Mazzotti, Leonardo Di Donna, Roberta Risoluti, and Anna Napoli. 2020. "Protein Extraction, Enrichment and MALDI MS and MS/MS Analysis from Bitter Orange Leaves (Citrus aurantium)" Molecules 25, no. 7: 1485. https://doi.org/10.3390/molecules25071485
APA StyleAiello, D., Siciliano, C., Mazzotti, F., Di Donna, L., Risoluti, R., & Napoli, A. (2020). Protein Extraction, Enrichment and MALDI MS and MS/MS Analysis from Bitter Orange Leaves (Citrus aurantium). Molecules, 25(7), 1485. https://doi.org/10.3390/molecules25071485