Major Contribution of Caspase-9 to Honokiol-Induced Apoptotic Insults to Human Drug-Resistant Glioblastoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Higher Expression of Caspase-9 in Human Glioblastomas
2.2. Preparation of Human Drug-Resistant Glioblastoma Cells
2.3. Honokiol Lessened Proliferation and Viability of Human Drug-Resistant Glioblastoma Cells
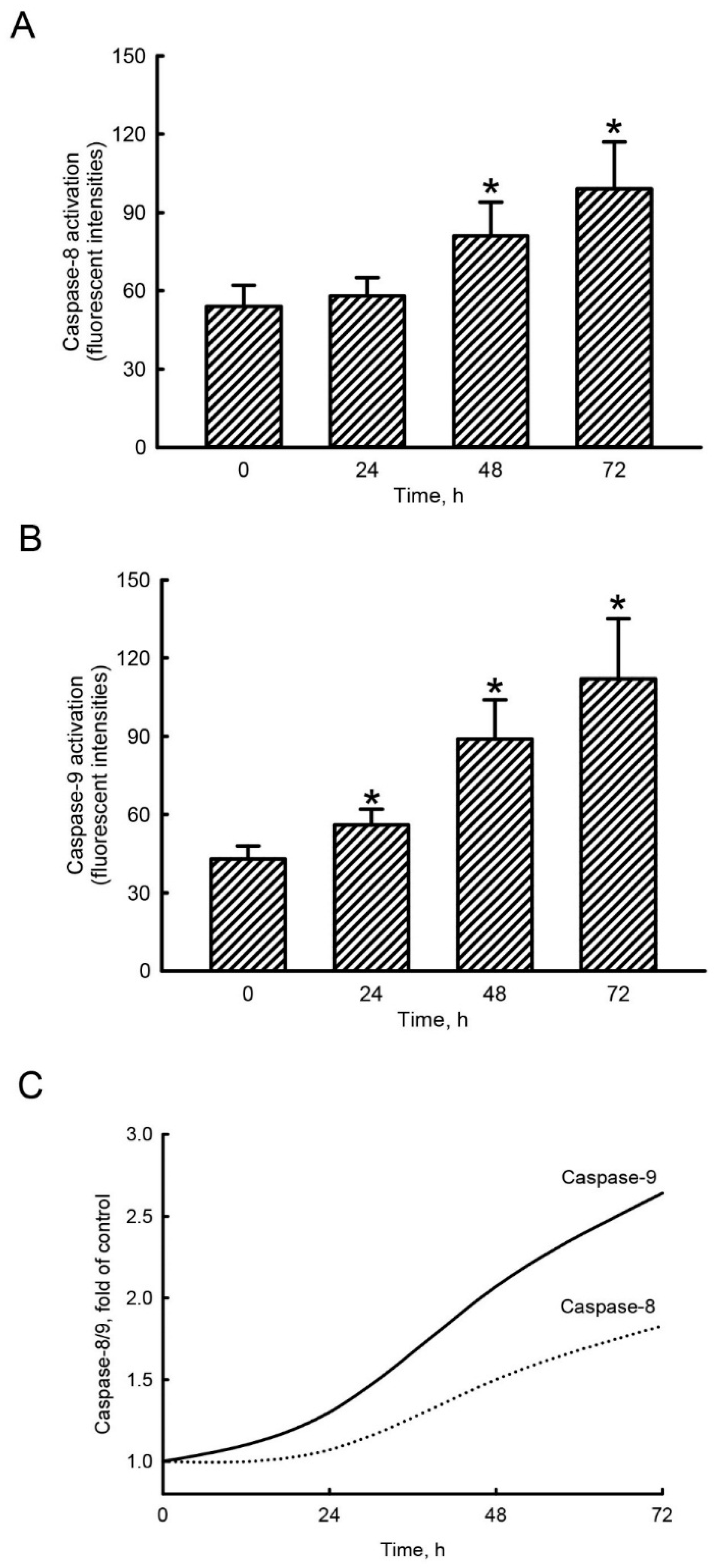
2.4. Honokiol Mainly Triggered Caspase-9 Activation in Human Drug-Resistant Glioblastoma Cells
2.5. Honokiol Sequentially Stimulated Cascade Activation of Caspase-3 and Caspase-6 in Human Drug-Resistant Glioblastoma Cells
2.6. Honokiol Selectively Induced DNA Fragmentation and Cell Apoptosis in Human Drug-Resistant Glioblastoma Cells
2.7. Caspase-9 Contributes to Honokiol-Induced Caspase-6 Activation, DNA Fragmentation, and Cell Apoptosis in Human Drug-Resistant Glioblastoma Cells
3. Discussion
4. Materials and Methods
4.1. Data Downloading and Preprocessing
4.2. Immunohistochemical Analyses of Caspase-8 and Caspase-9
4.3. Cell Culture
4.4. Preparation of Human Drug-Resistant Glioblastoma Cells and Drug Treatment
4.5. Analysis of Cell Proliferation
4.6. Assay of Cell Viability
4.7. Assay of Caspase Activities
4.8. Immunoblotting Analyses
4.9. Quantification of DNA Fragmentation
4.10. Analysis of Apoptotic Cells
4.11. Quantification of Necrotic Cells
4.12. Inhibition Assay of Caspase-9 Activity
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the extent of resection with survival in glioblastoma: A systematic review and meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Kageji, T.; Nagahiro, S.; Mizobuchi, Y.; Matsuzaki, K.; Nakagawa, Y.; Kumada, H. Boron neutron capture therapy (BNCT) for newly-diagnosed glioblastoma: Comparison of clinical results obtained with BNCT and conventional treatment. J. Med. Investig. 2014, 61, 254–263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Friedman, H.S.; Kerby, T.; Calvert, H. Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 2000, 6, 2585–2597. [Google Scholar] [PubMed]
- Gerson, S.L. Clinical relevance of MGMT in the treatment of cancer. J. Clin. Oncol. 2002, 20, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.E.; Arbiser, J.L. Honokiol, a multifunctional antiangiogenic and antitumor agent. Antioxid. Redox Signal. 2009, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Zotova, J.; Baschieri, A.; Valgimigli, L. Antioxidant activity of magnolol and honokiol: Kinetic and mechanistic investigations of their reaction with peroxyl radicals. J. Org. Chem. 2015, 80, 10651–10659. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Deng, Y.; Li, L.; He, H.; Sun, J.; Xu, D. Honokiol synergizes chemotherapy drugs in multidrug resistant breast cancer cells via enhanced apoptosis and additional programmed necrotic death. Int. J. Oncol. 2013, 42, 721–732. [Google Scholar] [CrossRef]
- Saeed, M.; Kuete, V.; Kadioglu, O.; Börtzler, J.; Khalid, H.; Greten, H.J.; Efferth, T. Cytotoxicity of the bisphenolic honokiol from Magnolia officinalis against multiple drug-resistant tumor cells as determined by pharmacogenomics and molecular docking. Phytomedicine 2014, 21, 1525–1533. [Google Scholar] [CrossRef]
- Pearson, H.E.; Iida, M.; Orbuch, R.A.; McDaniel, N.K.; Nickel, K.P.; Kimple, R.J.; Arbiser, J.L.; Wheeler, D.L. Overcoming Resistance to Cetuximab with Honokiol, A Small-Molecule Polyphenol. Mol. Cancer Ther. 2018, 17, 204–214. [Google Scholar] [CrossRef]
- Lin, J.W.; Chen, J.T.; Hong, C.Y.; Lin, Y.L.; Wang, K.T.; Yao, C.J.; Lai, G.M.; Chen, R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic Bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro-oncology 2012, 14, 302–314. [Google Scholar] [CrossRef]
- Doolittle, N.D.; Anderson, C.P.; Bleyer, W.A.; Cairncross, J.G.; Cloughesy, T.; Eck, S.L.; Guastadisegni, P.; Hall, W.A.; Muldoon, L.L.; Patel, S.J.; et al. Importance of dose intensity in neuro-oncology clinical trials: Summary report of the Sixth Annual Meeting of the Blood-Brain Barrier Disruption Consortium. Neuro-oncology 2001, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chang, Y.A.; Lin, Y.L.; Liu, S.H.; Chang, C.K.; Chen, R.M. Preclinical effects of honokiol on treating glioblastoma multiforme via G1 phase arrest and cell apoptosis. Phytomedicine 2016, 23, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.L.; Tseng, Y.Y.; Wu, G.J.; Hsieh, M.H.; Lin, Y.W.; Chen, R.M. Honokiol induces autophagic cell death in malignant glioma through reactive oxygen species-mediated regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol. Appl. Pharmacol. 2016, 304, 59–69. [Google Scholar] [CrossRef]
- Yeh, P.S.; Wang, W.; Chang, Y.A.; Lin, C.J.; Wang, J.J.; Chen, R.M. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.C.; Lin, Y.W.; Tseng, Y.Y.; Lee, Y.W.; Chen, J.T.; Chen, R.M. Honokiol induces autophagy and successive apoptotic insults to neuroblastomas via a p53-dependent mechanism. Am. J. Chin. Med. 2019, 47, 859–912. [Google Scholar] [CrossRef]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells & drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Huang, J.S.; Yao, C.J.; Chuang, S.E.; Yeh, C.T.; Lee, L.M.; Chen, R.M.; Chao, W.J.; Whang-Peng, J.; Lai, G.M. Honokiol inhibits sphere formation and xenograft growth of oral cancer side population cells accompanied with JAK/STAT signaling pathway suppression and apoptosis induction. BMC Cancer 2016, 16, 245. [Google Scholar] [CrossRef]
- Chang, M.T.; Lee, S.P.; Fang, C.Y.; Hsieh, P.L.; Liao, Y.W.; Lu, M.Y.; Tsai, L.L.; Yu, C.C.; Liu, C.M. Chemosensitizing effect of honokiol in oral carcinoma stem cells via regulation of IL-6/Stat3 signaling. Environ. Toxicol. 2018, 33, 1105–1112. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, M.K.; No, J.H.; Chung, H.H.; Song, Y.S. Metabolic approaches to overcoming chemoresistance in ovarian cancer. Ann. N. Y. Acad. Sci. 2011, 1229, 53–60. [Google Scholar] [CrossRef]
- Chen, Q.M.; Tu, V.C. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am. J. Cardiovas. Drugs 2002, 2, 43–57. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Chen, K.Y.; Chuang, J.Y.; Liu, C.C.; Liu, S.H.; Chen, R.M. Improved effects of honokiol on temozolomide-induced autophagy and apoptosis of drug-sensitive and -tolerant glioma cells. BMC Cancer 2018, 18, 379. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol enhances temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondria-dependent pathway. Phytomedicine 2018, 49, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N.; Krammer, P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012, 19, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Wallace, M.; Aldape, K.D.; Berger, M.S.; Bettegowda, C.; Brat, D.J.; Cahill, D.P.; Cloughesy, T.; Haas-Kogan, D.A.; Marra, M.; et al. Where are we now? And where are we going? A report from the accelerate brain cancer cure (ABC2) low-grade glioma research workshop. Neuro-oncology 2014, 16, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514. [Google Scholar] [CrossRef]
- Semenza, G.L. Surviving ischemia: Adaptive responses mediated by hypoxia-inducible factor 1. J. Clin. Investig. 2000, 106, 809–812. [Google Scholar] [CrossRef]
- Cheng, B.C.; Chen, J.T.; Yang, S.T.; Chio, C.C.; Liu, S.H.; Chen, R.M. Cobalt chloride treatment induces autophagic apoptosis in human glioma cells via a p53-dependent pathway. Int. J. Oncol. 2017, 50, 964–974. [Google Scholar] [CrossRef]
- Li, W.; Winters, A.; Poteet, E.; Ryou, M.G.; Lin, S.; Hao, S.; Wu, Z.; Yuan, F.; Hatanpaa, K.J.; Simpkins, J.W.; et al. Involvement of estrogen receptor β5 in the progression of glioma. Brain Res. 2013, 1503, 97–107. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef]
- Rao, L.; Perez, D.; White, E. Lamin proteolysis facilitates nuclear events during apoptosis. J. Cell Biol. 1996, 135, 1441–1455. [Google Scholar] [CrossRef]
- Brandsma, D.; Stalpers, L.; Taal, W.; Sminia, P.; van den Bent, M.J. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 2008, 9, 453–461. [Google Scholar] [CrossRef]
- Jeong, J.J.; Lee, J.H.; Chang, K.C.; Kim, H.J. Honokiol exerts an anticancer effect in T98G human glioblastoma cells through the induction of apoptosis and the regulation of adhesion molecules. Int. J. Oncol. 2012, 41, 1358–1364. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, X.; Shi, M.; Jiang, Z.; Wang, H.; Su, Q.; Liu, G.; Li, Q.; Jiang, G. Downregulation of STAT3 and activation of MAPK are involved in the induction of apoptosis by HNK in glioblastoma cell line U87. Oncol. Rep. 2004, 32, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Wei, L.; Chen, T.G.; Lin, C.M.; Shieh, J.P.; Yeh, P.S.; Chen, R.M. Neuron-derived orphan receptor 1 transduces survival signals in neuronal cells in response to hypoxia-induced apoptotic insults. J. Neurosurg. 2016, 124, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Lo, W.L.; Ko, C.Y.; Chou, S.Y.; Chen, R.M.; Chang, K.Y.; Hung, J.J.; Su, W.C.; Chang, W.C.; Hsu, T.I. Upregulation of CYP17A1 by Sp1-mediated DNA demethylation confers temozolomide resistance through DHEA-mediated protection in glioma. Oncogenesis 2017, 6, e339. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Andreieva, S.V.; Korets, K.V.; Mykytenko, D.O.; Baklaushev, V.P.; Huleyuk, N.L.; Kovalova, O.A.; Kotsarenko, K.V.; Chekhonin, V.P.; Vassetzky, Y.S.; et al. Temozolomide promotes genomic and phenotypic changes in glioblastoma cells. Cancer Cell Int. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.Y.; Chen, T.L.; Cherng, Y.G.; Tai, Y.T.; Chen, T.G.; Chen, R.M. Lipopolysaccharide induces apoptotic insults to human alveolar epithelial A549 cells through reactive oxygen species-mediated activation of an intrinsic mitochondrion-dependent pathway. Arch. Toxicol. 2011, 85, 209–218. [Google Scholar] [CrossRef]
- Wu, G.J.; Wang, W.; Lin, Y.L.; Liu, S.H.; Chen, R.M. Oxidative stress-induced apoptotic insults to rat osteoblasts are attenuated by nitric oxide pretreatment via GATA-5-involved regulation of Bcl-XL gene expression and protein translocation. Arch. Toxicol. 2016, 90, 905–916. [Google Scholar] [CrossRef]
- Lin, C.J.; Lin, Y.L.; Luh, F.; Yen, Y.; Chen, R.M. Preclinical effects of CRLX101, an investigational camptothecin-containing nanoparticle drug conjugate, on treating glioblastoma multiforme via apoptosis and antiangiogenesis. Oncotarget 2016, 7, 42408–42421. [Google Scholar] [CrossRef]
- Chio, C.C.; Lin, J.W.; Cheng, H.A.; Chiu, W.T.; Wang, Y.H.; Wang, J.J.; Hsing, C.H.; Chen, R.M. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch. Toxicol. 2013, 87, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.H.; Lin, P.I.; Ho, W.P.; Chan, W.P.; Chen, T.L.; Chen, R.M. Participation of GATA-3 in regulation of bone healing through transcriptionally upregulating bcl-xL gene expression. Exp. Mol. Med. 2017, 49, e398. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.M.; Lin, Y.L.; Chou, C.W. GATA-3 transduces survival signals in osteoblasts through upregulation of bcl-xL gene expression. J. Bone Min. Res. 2010, 25, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.-J.; Yang, S.-T.; Chen, R.-M. Major Contribution of Caspase-9 to Honokiol-Induced Apoptotic Insults to Human Drug-Resistant Glioblastoma Cells. Molecules 2020, 25, 1450. https://doi.org/10.3390/molecules25061450
Wu G-J, Yang S-T, Chen R-M. Major Contribution of Caspase-9 to Honokiol-Induced Apoptotic Insults to Human Drug-Resistant Glioblastoma Cells. Molecules. 2020; 25(6):1450. https://doi.org/10.3390/molecules25061450
Chicago/Turabian StyleWu, Gong-Jhe, Sun-Ta Yang, and Ruei-Ming Chen. 2020. "Major Contribution of Caspase-9 to Honokiol-Induced Apoptotic Insults to Human Drug-Resistant Glioblastoma Cells" Molecules 25, no. 6: 1450. https://doi.org/10.3390/molecules25061450
APA StyleWu, G.-J., Yang, S.-T., & Chen, R.-M. (2020). Major Contribution of Caspase-9 to Honokiol-Induced Apoptotic Insults to Human Drug-Resistant Glioblastoma Cells. Molecules, 25(6), 1450. https://doi.org/10.3390/molecules25061450