
Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability


Abstract
1. Introduction
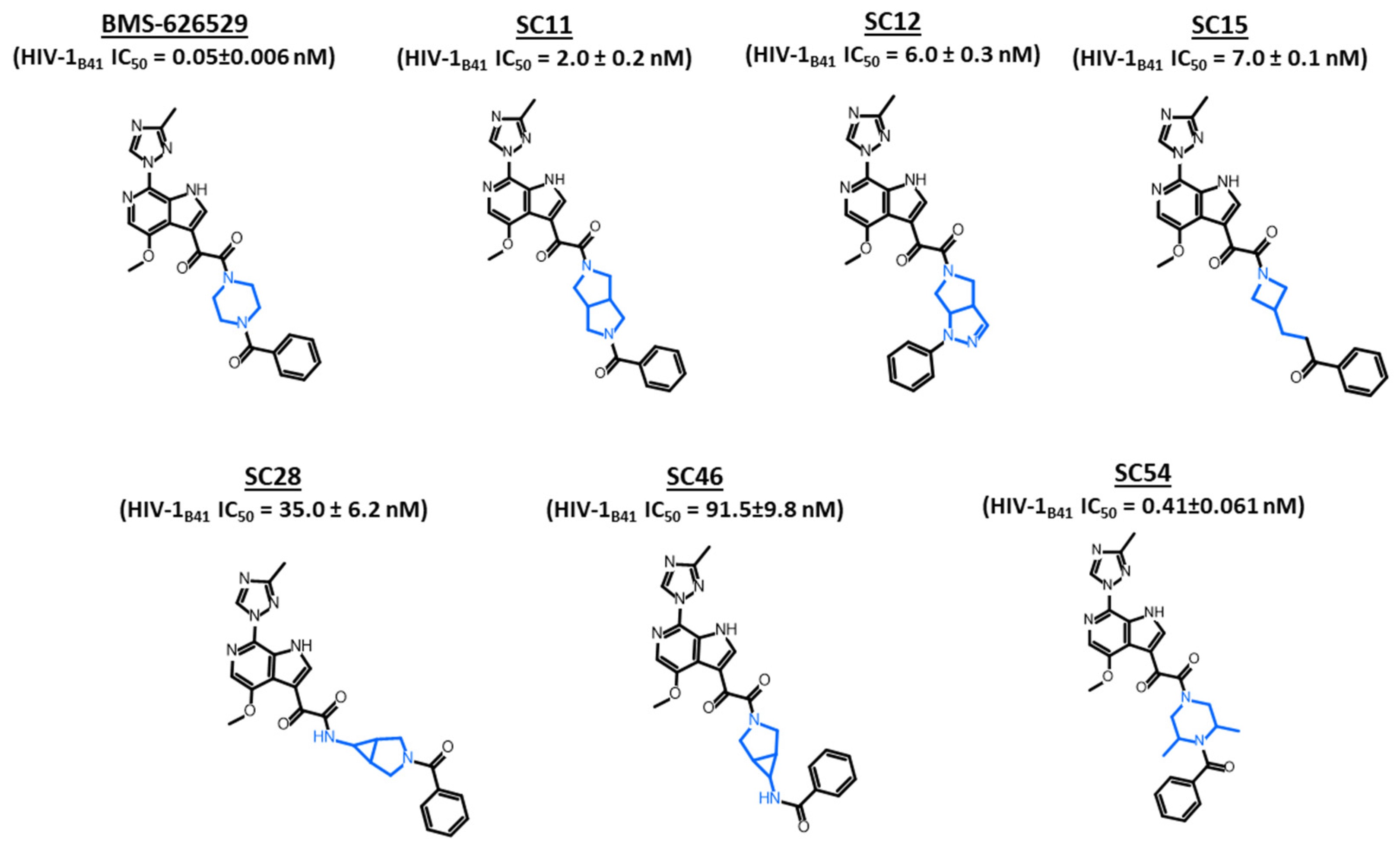
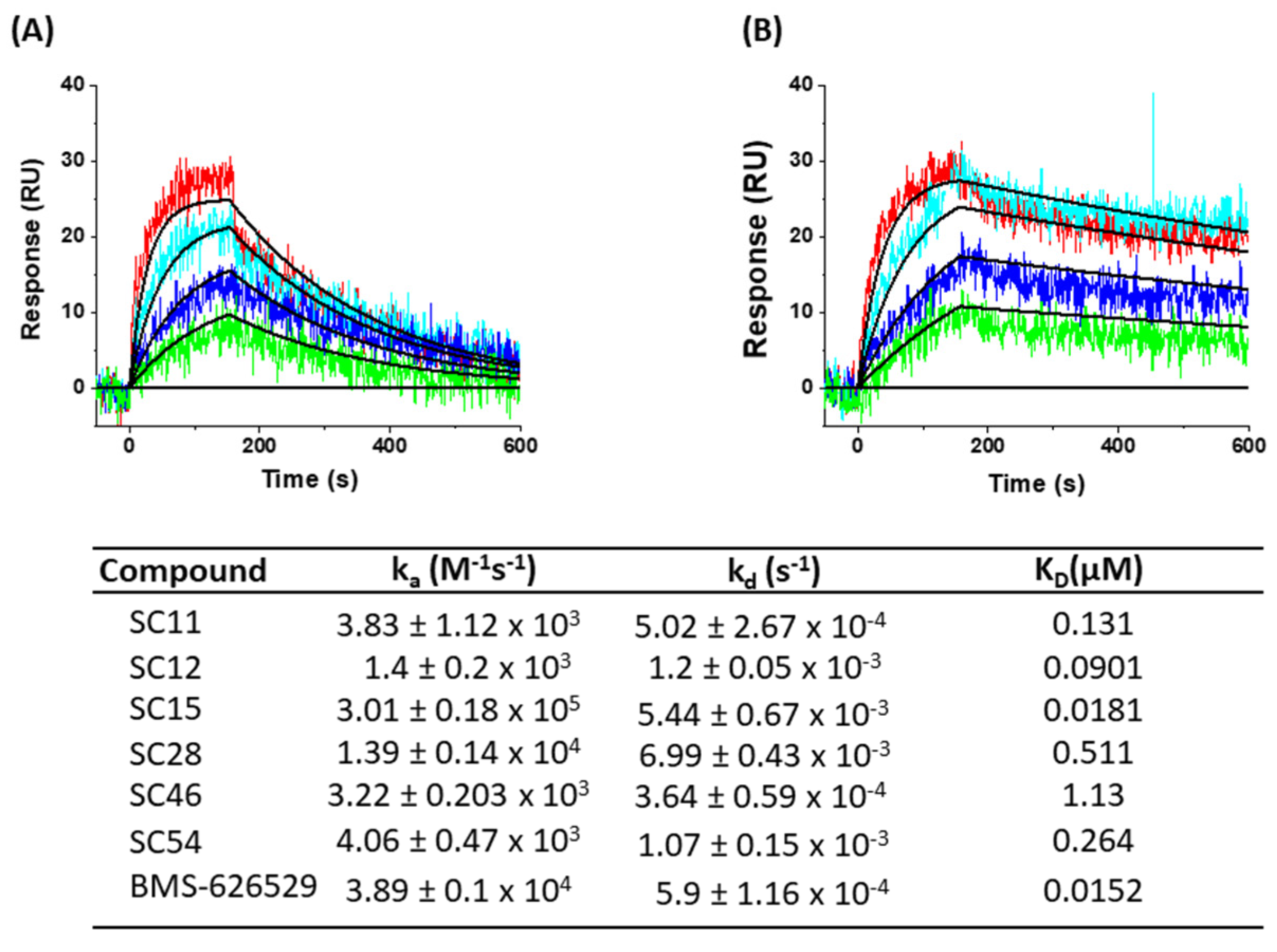
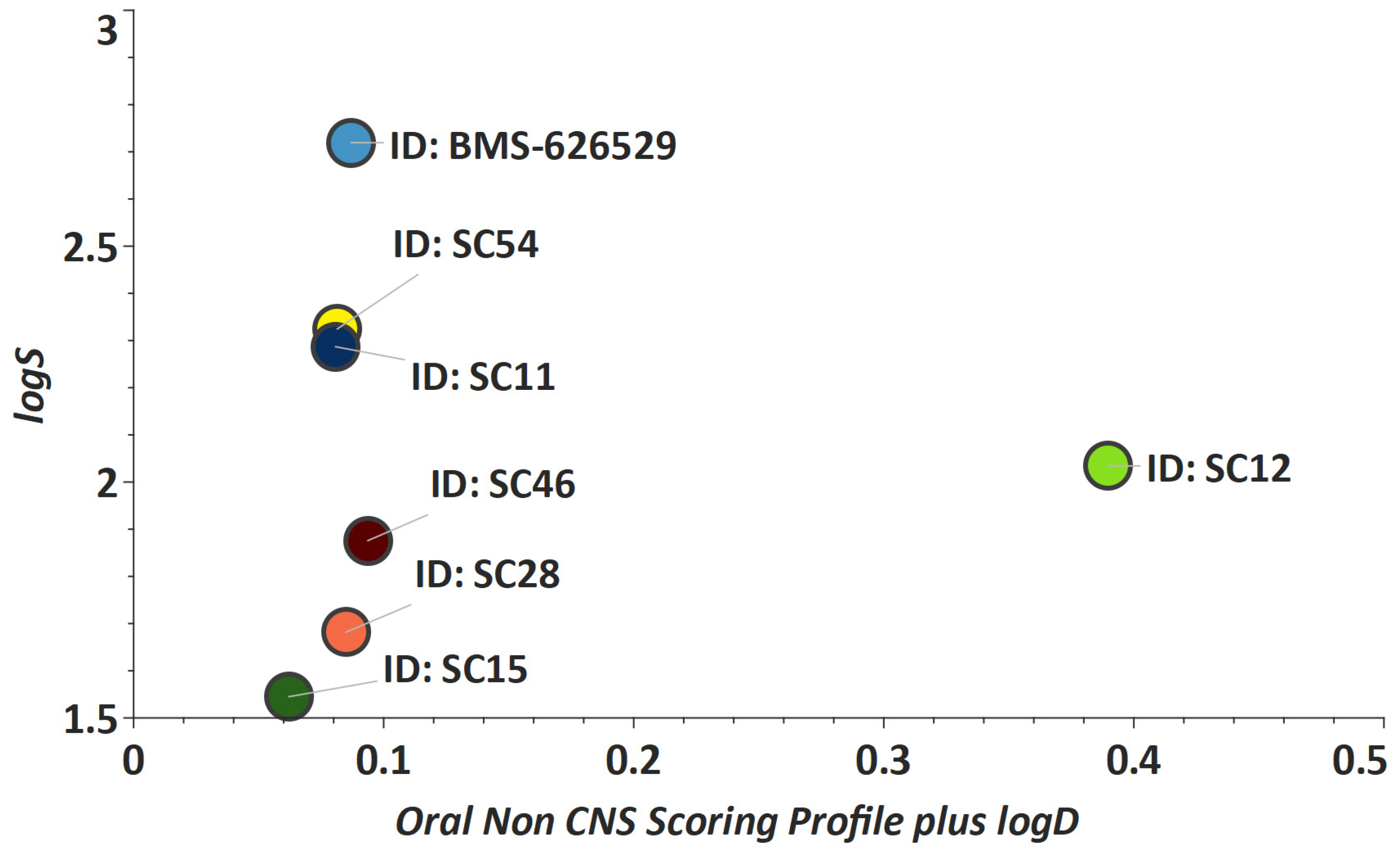
2. Results and Discussion
3. Materials and Methods
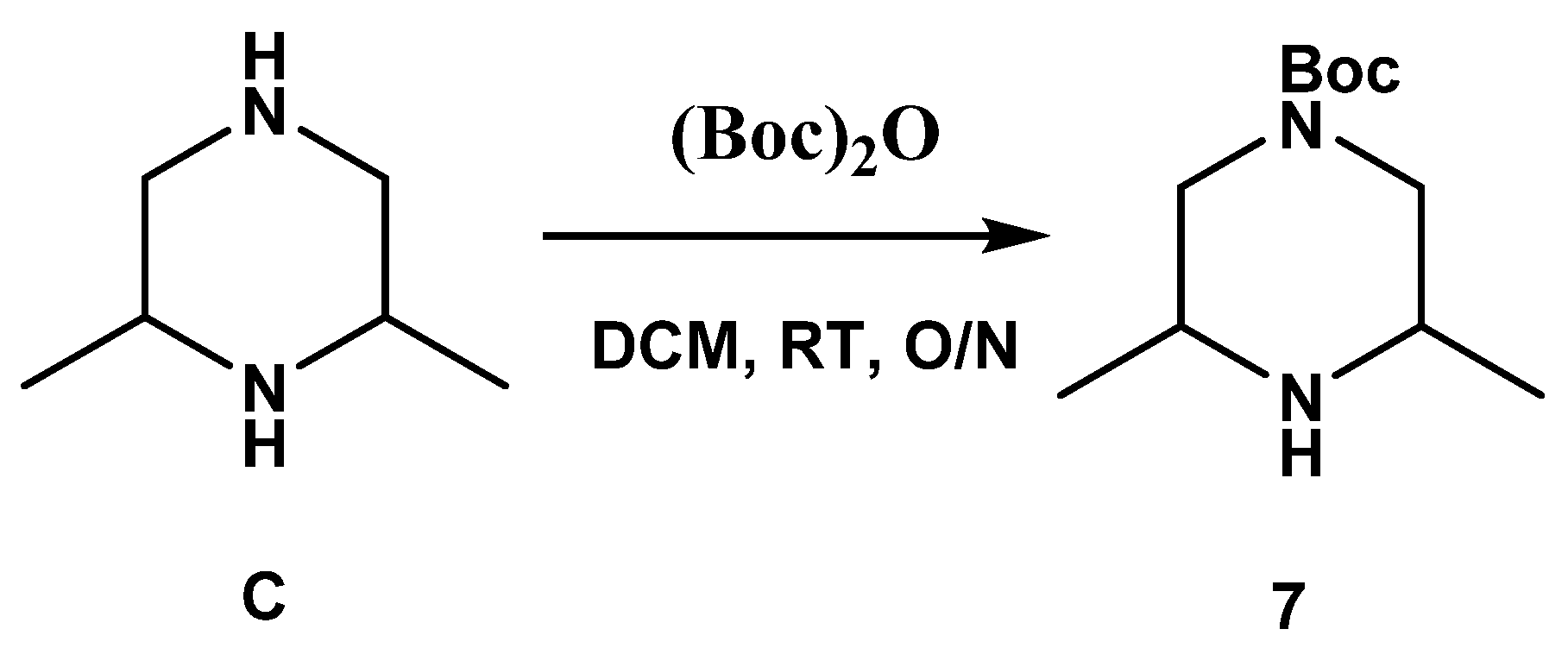
3.1. Compounds
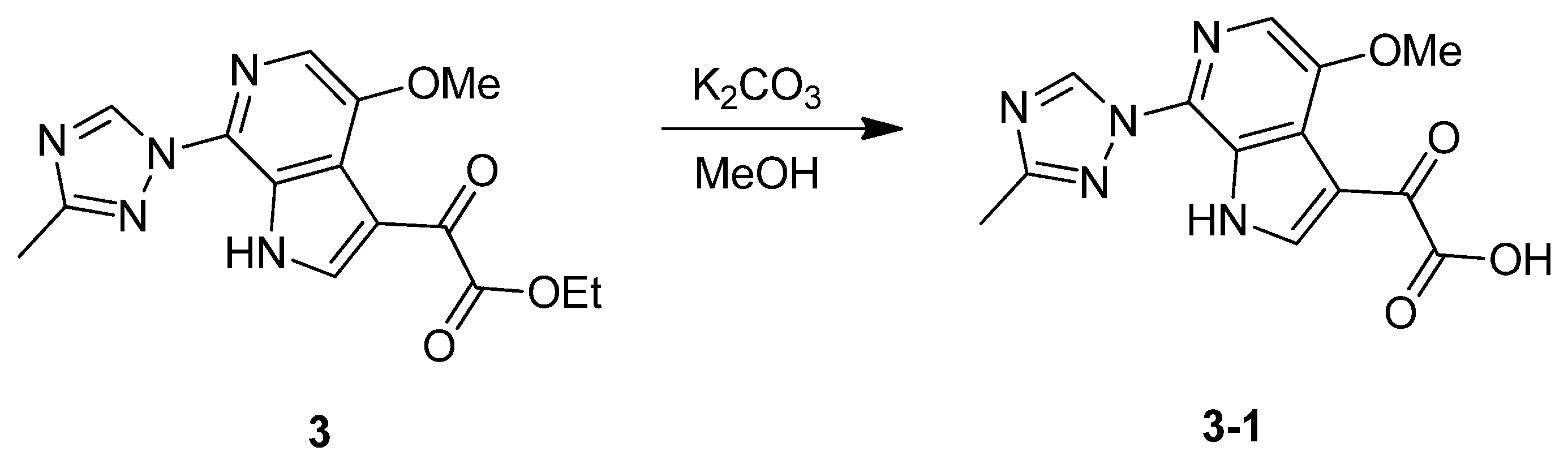
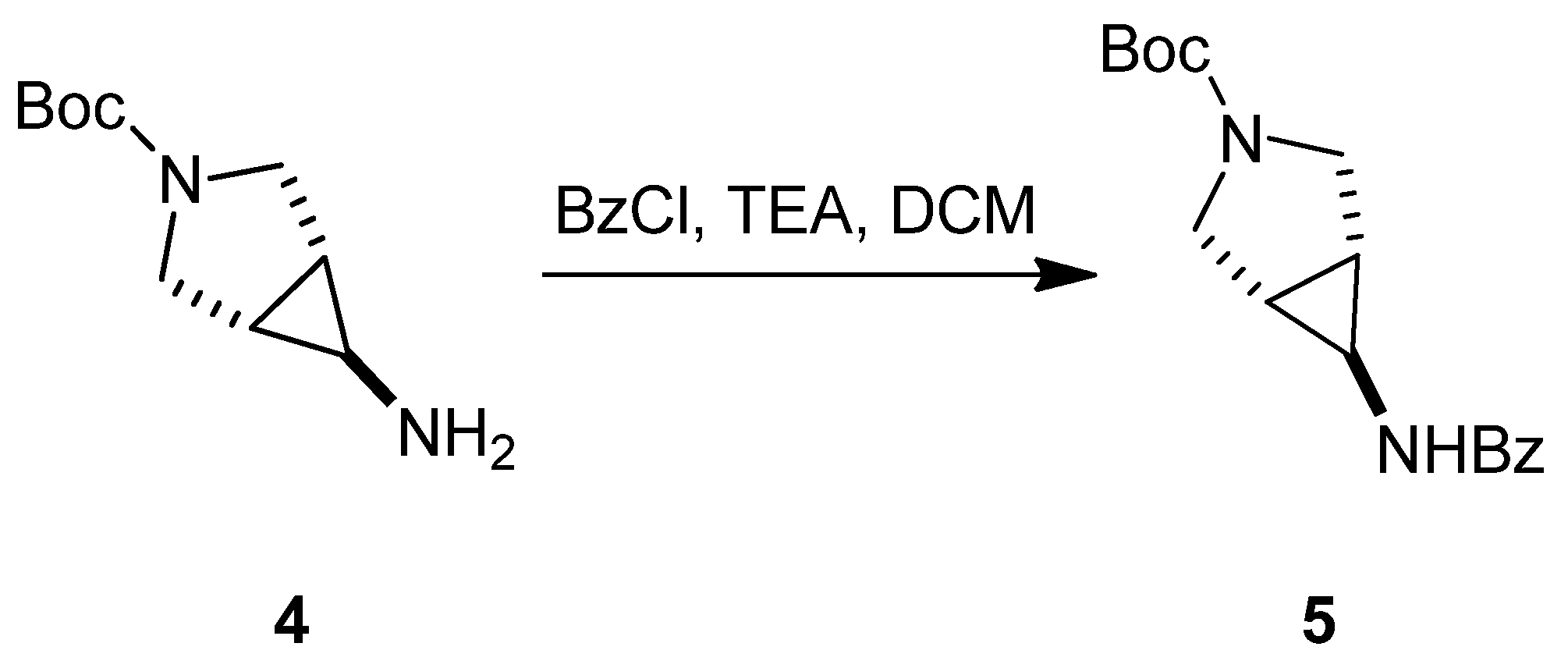
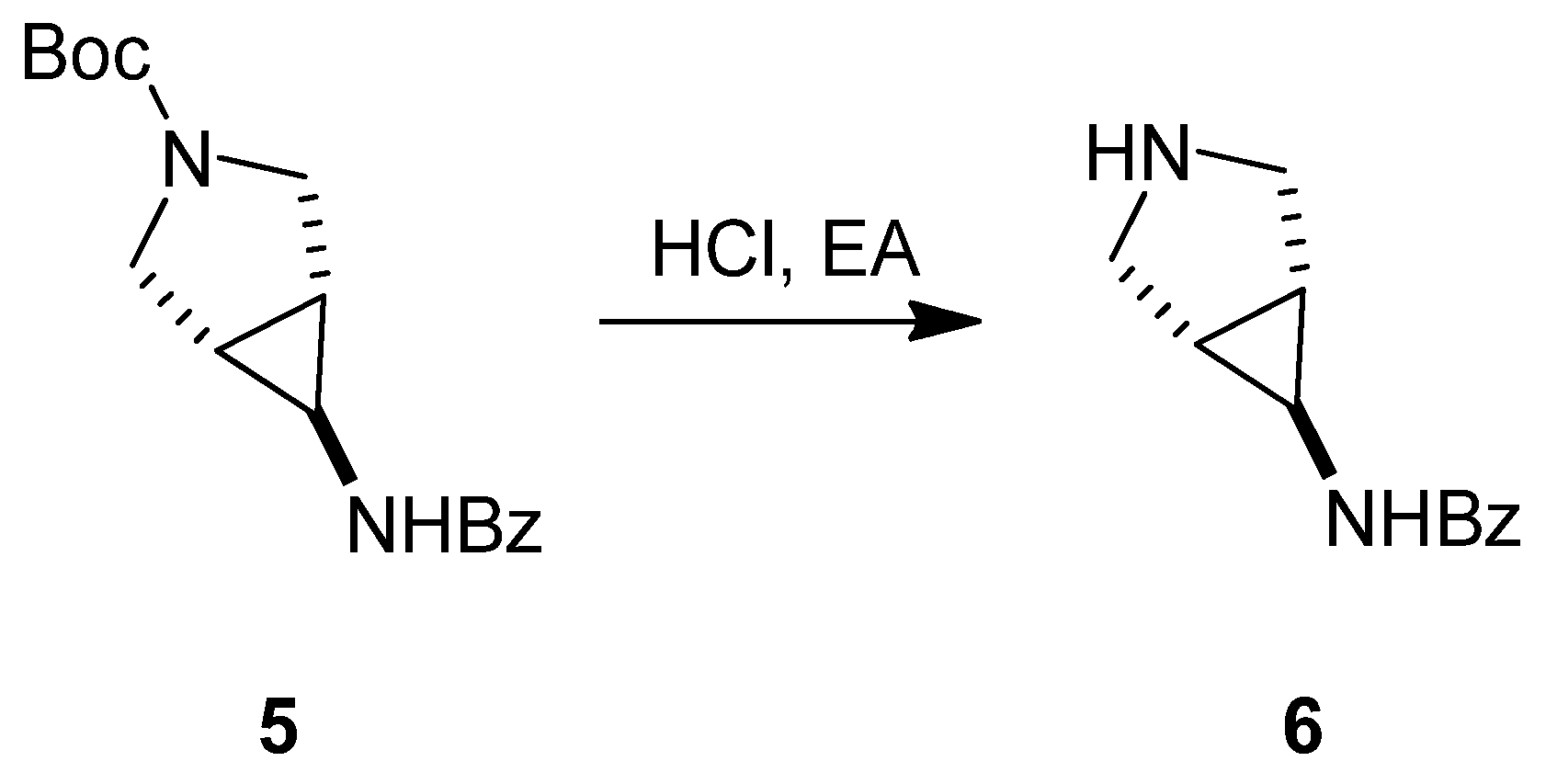
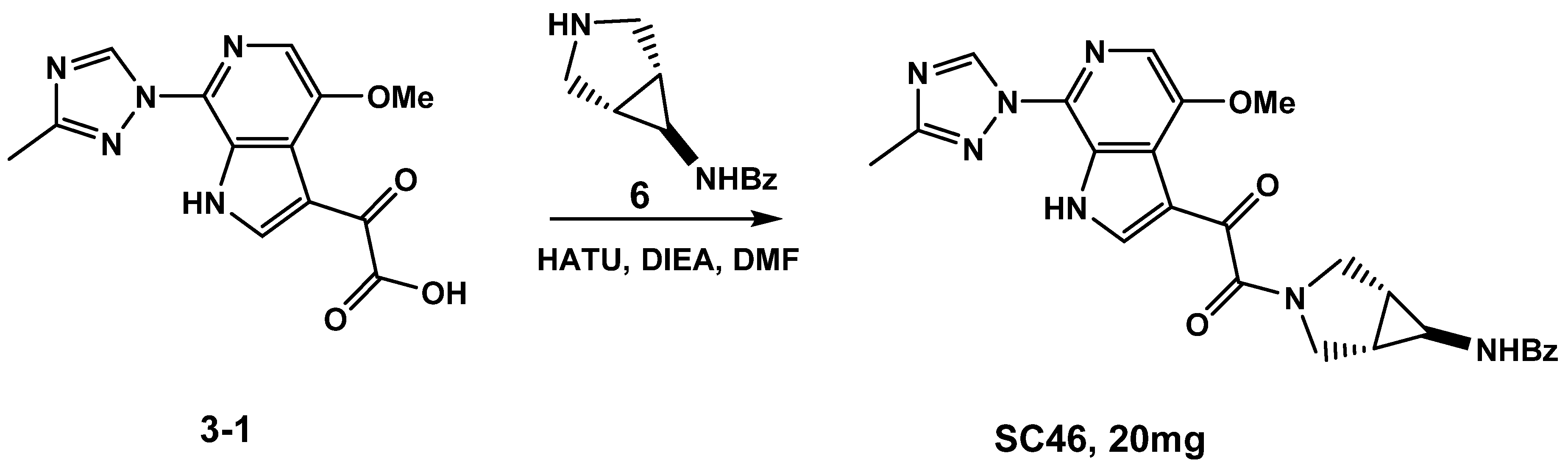
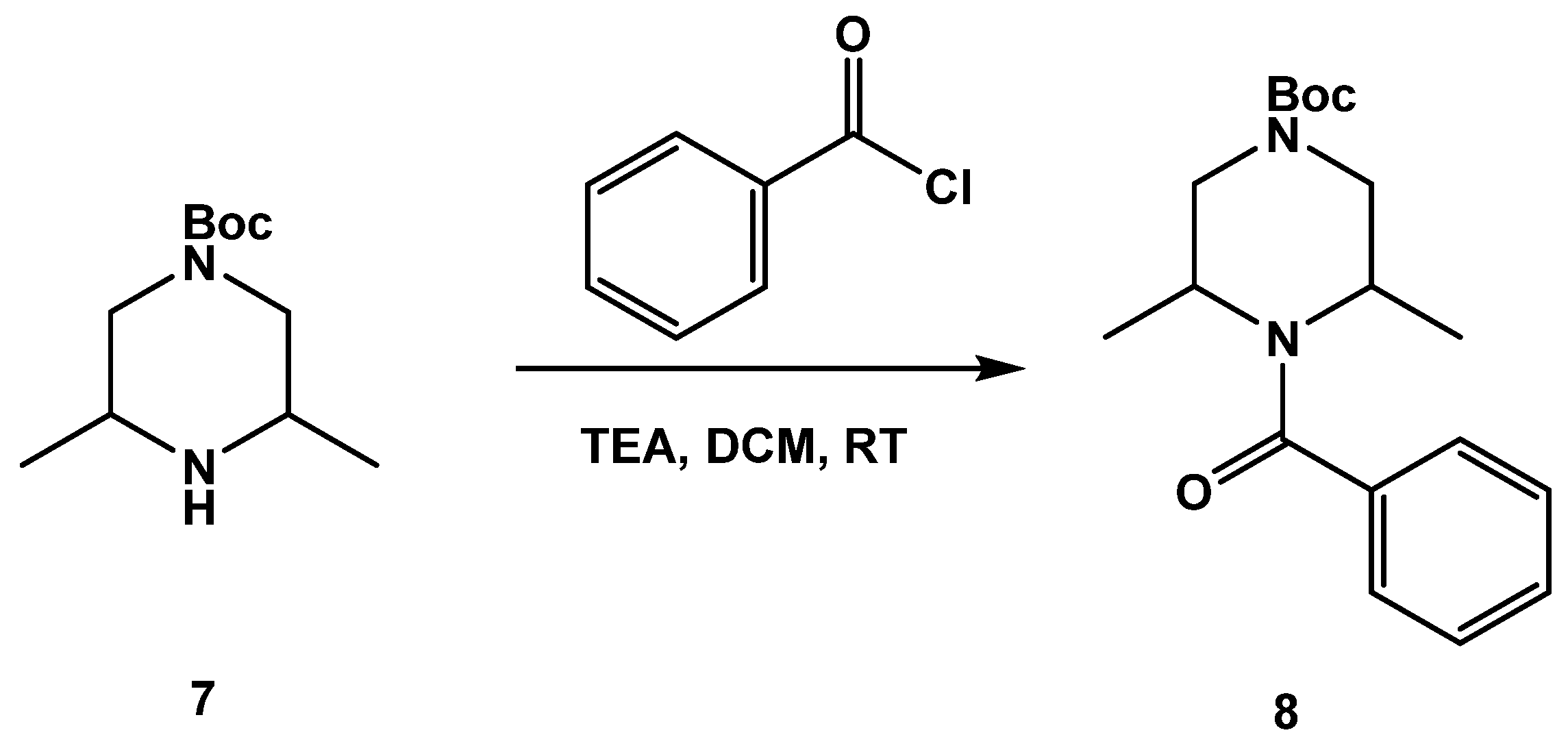
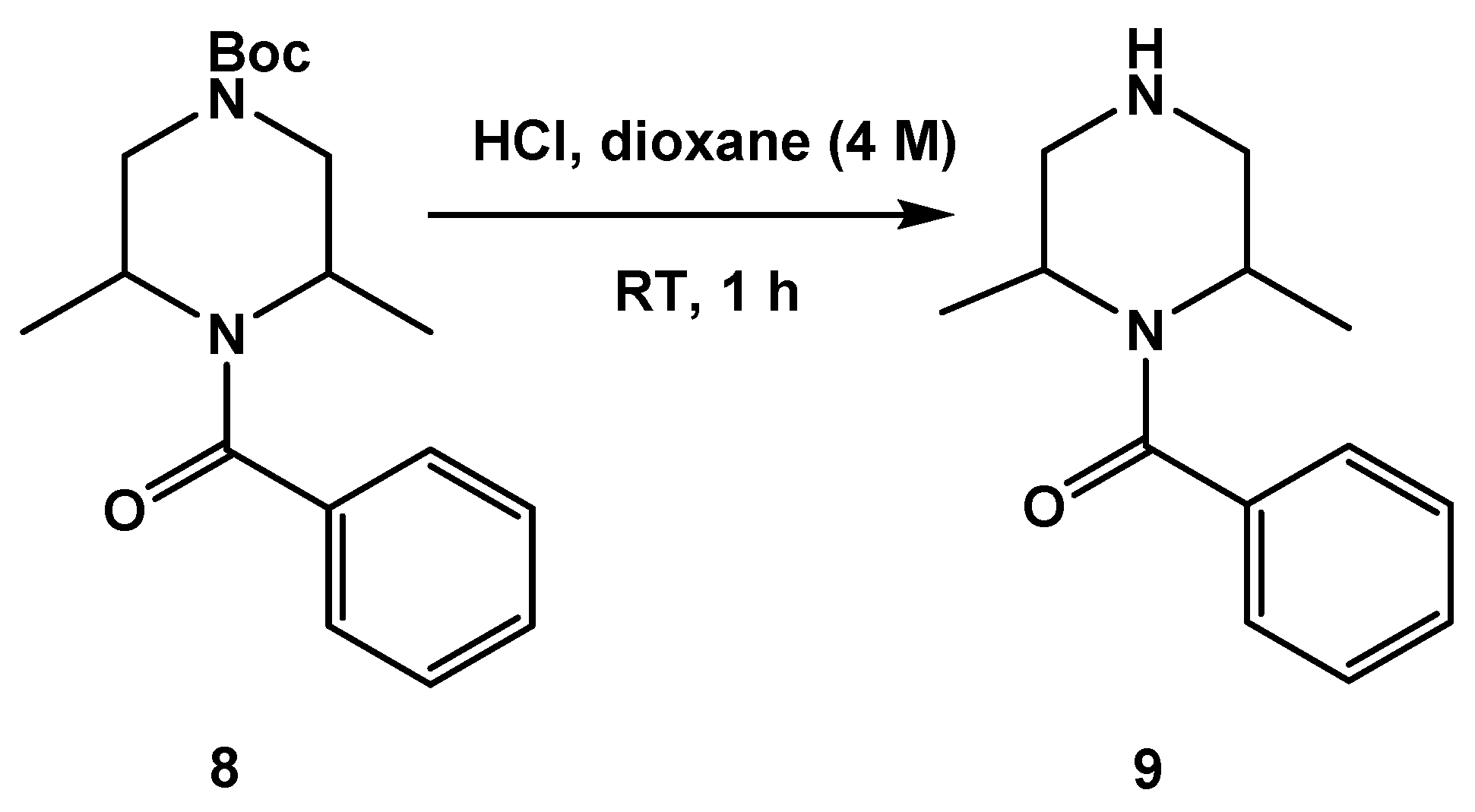
3.1.1. Synthesis of SC46
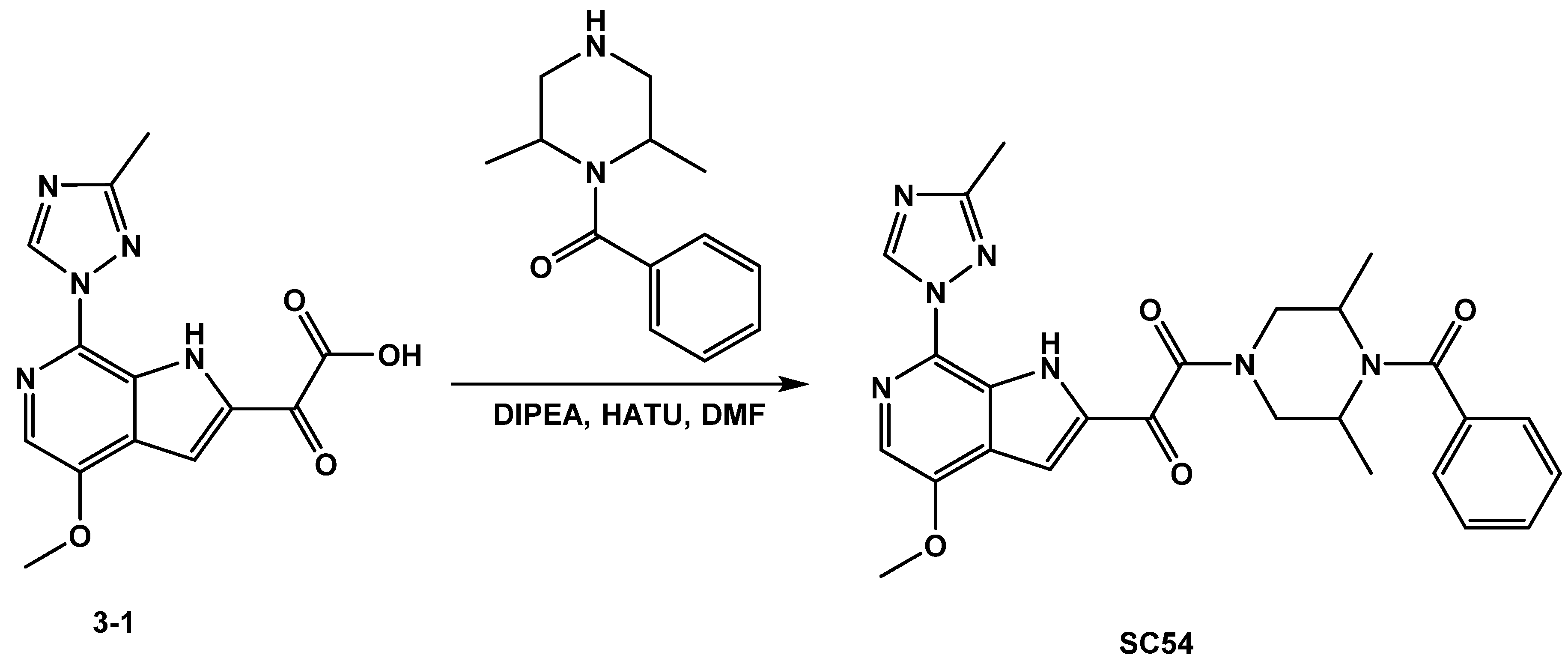
3.1.2. Synthesis of SC54
3.2. Cells
3.3. Proteins
3.4. Production of Pseudotyped Viruses
3.5. ELISA-Based Quantification of p24 Content
3.6. Single-Round Infection Assay
3.7. SPR Direct Interaction Analysis
3.7.1. Immobilization of Env Constructs
3.7.2. Direct Binding Analysis
3.8. Metabolic Stability Evaluation of Compounds in Human Liver Microsomes.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, T.; Zhang, Z.; Wallace, O.B.; Deshpande, M.; Fang, H.; Yang, Z.; Zadjura, L.M.; Tweedie, D.L.; Huang, S.; Zhao, F.; et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H-pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2-(R)-methylpiperazine (BMS-378806): A novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J. Med. Chem. 2003, 46, 4236–4239. [Google Scholar] [CrossRef] [PubMed]
- Nettles, R.E.; Schurmann, D.; Zhu, L.; Stonier, M.; Huang, S.P.; Chang, I.; Chien, C.; Krystal, M.; Wind-Rotolo, M.; Ray, N.; et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J. Infect. Dis. 2012, 206, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorganic Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef] [PubMed]
- Tuyishime, M.; Lawrence, R.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorganic Med. Chem. Lett. 2016, 26, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Meuser, M.E.; Rashad, A.A.; Ozorowski, G.; Dick, A.; Ward, A.B.; Cocklin, S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules 2019, 24, 1581. [Google Scholar] [CrossRef] [PubMed]
- Pugach, P.; Ozorowski, G.; Cupo, A.; Ringe, R.; Yasmeen, A.; de Val, N.; Derking, R.; Kim, H.J.; Korzun, J.; Golabek, M.; et al. A Native-Like SOSIP.664 Trimer Based on an HIV-1 Subtype B env Gene. J. Virol. 2015, 89, 3380–3395. [Google Scholar] [CrossRef]
- Segall, M.; Champness, E.; Obrezanova, O.; Leeding, C. Beyond Profiling: Using ADMET Models to Guide Decisions. Chem. Biodivers. 2009, 6, 2144–2151. [Google Scholar] [CrossRef]
- Segall, M.D. Multi-Parameter Optimization: Identifying High Quality Compounds with a Balance of Properties. Curr. Pharm. Des. 2012, 18, 1292–1310. [Google Scholar] [CrossRef]
- Hunt, P.A.; Segall, M.D.; Tyzack, J.D. WhichP450: A multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J. Comput. Aided Mol. Des. 2018, 32, 537–546. [Google Scholar] [CrossRef]
- Moore, K.P.; Mageau, A.S.; Magee, M.; Gorycki, P.D.; Ackerman, P.; Llamoso, C. Fostemsavir Drug–Drug Interaction Profile, an Attachment Inhibitor and Oral Prodrug of Temsavir, for Heavily Treatment Experienced HIV-1-Infected Patients. Open Forum Infect. Dis. 2019, 6, S867. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Hunt, P.A.; Segall, M.D. Predicting Regioselectivity and Lability of Cytochrome P450 Metabolism Using Quantum Mechanical Simulations. J. Chem. Inf. Model. 2016, 56, 2180–2193. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Chamberlin, A.R.; Poulos, T.L.; Sevrioukova, I.F. Structure-Based Inhibitor Design for Evaluation of a CYP3A4 Pharmacophore Model. J. Med. Chem. 2016, 59, 4210–4220. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Li, P.; Gallegos, R.; Uttamsingh, V.; Xia, C.Q.; Miwa, G.T.; Balani, S.K.; Gan, L.S. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: Evaluation of free fraction and uptake in hepatocytes. Drug Metab. Dispos. 2006, 34, 1600–1605. [Google Scholar] [CrossRef]
- Bjorndal, A.; Deng, H.; Jansson, M.; Fiore, J.R.; Colognesi, C.; Karlsson, A.; Albert, J.; Scarlatti, G.; Littman, D.R.; Fenyo, E.M. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J. Virol. 1997, 71, 7478–7487. [Google Scholar] [CrossRef]
- Zentner, I.; Sierra, L.J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martin-Garcia, J.; Cocklin, S. Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. ChemMedChem 2013, 8, 426–432. [Google Scholar] [CrossRef]
- Kortagere, S.; Madani, N.; Mankowski, M.K.; Schon, A.; Zentner, I.; Swaminathan, G.; Princiotto, A.; Anthony, K.; Oza, A.; Sierra, L.J.; et al. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J. Virol. 2012, 86, 8472–8481. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef]
- Hofmann, W.; Schubert, D.; LaBonte, J.; Munson, L.; Gibson, S.; Scammell, J.; Ferrigno, P.; Sodroski, J. Species-specific, postentry barriers to primate immunodeficiency virus infection. J. Virol. 1999, 73, 10020–10028. [Google Scholar] [CrossRef]
- Cocklin, S.; Gopi, H.; Querido, B.; Nimmagadda, M.; Kuriakose, S.; Cicala, C.; Ajith, S.; Baxter, S.; Arthos, J.; Martin-Garcia, J.; et al. Broad-spectrum anti-human immunodeficiency virus (HIV) potential of a peptide HIV type 1 entry inhibitor. J. Virol. 2007, 81, 3645–3648. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Choe, S.; Walker, R.; di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [PubMed]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CSL 3A4 | Labile Sites | Predicted 3A4 Affinity (M) |
|---|---|---|---|
| SC11 | 0.9514 | 3 | 0.004849324 |
| SC12 | 0.9627 | 4 | 0.083703353 |
| SC15 | 0.9564 | 3 | 0.003455543 |
| SC28 | 0.9516 | 2 | 0.767515588 |
| SC46 | 0.9474 | 3 | 0.166818977 |
| SC54 | 0.9396 | 3 | 0.006416669 |
| BMS-626529 | 0.9416 | 3 | 0.000802 |
| Compound ID | k | T1/2 | Clint | Clapp | Clh | Eh |
|---|---|---|---|---|---|---|
| (min) | (mL/min/mg) | (mL/min/kg) | (mL/min/kg) | (%) | ||
| Testosterone | 0.05134 | 13.5 | 0.1027 | 99.005 | 16.639 | 83.19 |
| Propranolol | 0.01305 | 53.1 | 0.0261 | 25.170 | 11.145 | 55.72 |
| Warfarin | 0.00352 | 196.6 | 0.0070 | 6.797 | 5.073 | 25.37 |
| SC11 | 0.07253 | 9.6 | 0.1451 | 139.887 | 17.498 | 87.49 |
| SC12 | 0.105828 | 6.548348 | 0.211656436 | 204.0972779 | 18.21506087 | 91.0753 |
| SC15 | 0.05268 | 13.2 | 0.1054 | 101.588 | 16.710 | 83.55 |
| SC28 | 0.00479 | 144.6 | 0.0096 | 9.245 | 6.322 | 31.61 |
| SC46 | 0.00760 | 91.2 | 0.0152 | 14.657 | 8.455 | 42.28 |
| SC54 | 0.022234 | 31.16817 | 0.04446845 | 42.8802911 | 13.63870629 | 68.19353 |
| BMS-626529 | 0.010075 | 68.78436 | 0.020149929 | 19.43028912 | 9.855514403 | 49.27757 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karadsheh, R.; Meuser, M.E.; Cocklin, S. Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability. Molecules 2020, 25, 1430. https://doi.org/10.3390/molecules25061430
Karadsheh R, Meuser ME, Cocklin S. Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability. Molecules. 2020; 25(6):1430. https://doi.org/10.3390/molecules25061430
Chicago/Turabian StyleKaradsheh, Rama, Megan E. Meuser, and Simon Cocklin. 2020. "Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability" Molecules 25, no. 6: 1430. https://doi.org/10.3390/molecules25061430
APA StyleKaradsheh, R., Meuser, M. E., & Cocklin, S. (2020). Composition and Orientation of the Core Region of Novel HIV-1 Entry Inhibitors Influences Metabolic Stability. Molecules, 25(6), 1430. https://doi.org/10.3390/molecules25061430