
2,3-Diaminopropanols Obtained from d-Serine as Intermediates in the Synthesis of Protected 2,3-l-Diaminopropanoic Acid (l-Dap) Methyl Esters


 ,
, 
 ,
,  and
and
Abstract
1. Introduction
1.1. The Role of l-Dap in Natural and Synthetic Compounds
1.2. Synthetic Routes to l-Dap
2. Scope of the Work
3. Results and Discussion
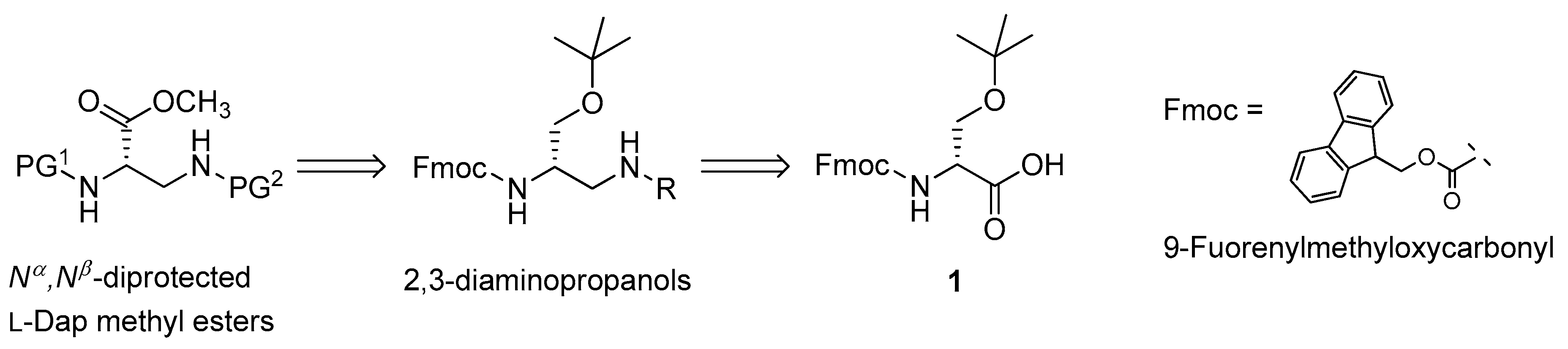
3.1. Retrosynthetic Analysis
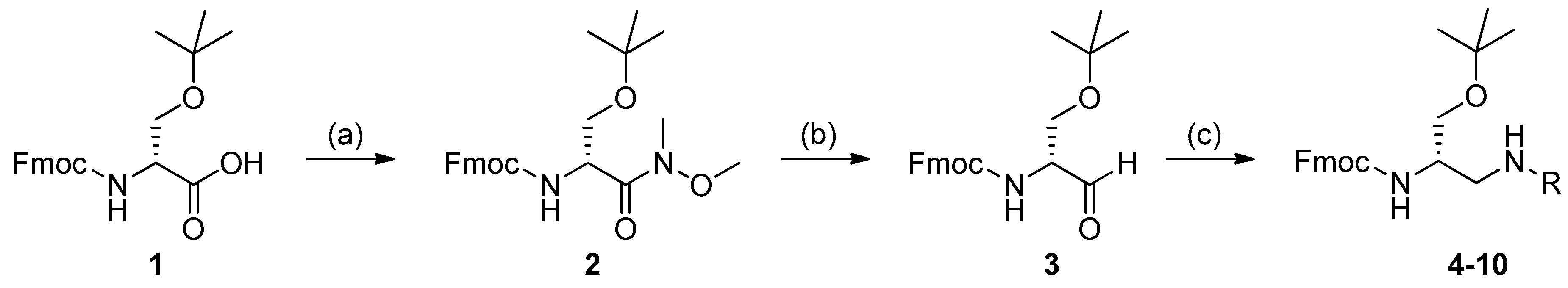
3.2. Synthesis of the 2,3-Diaminopropanols
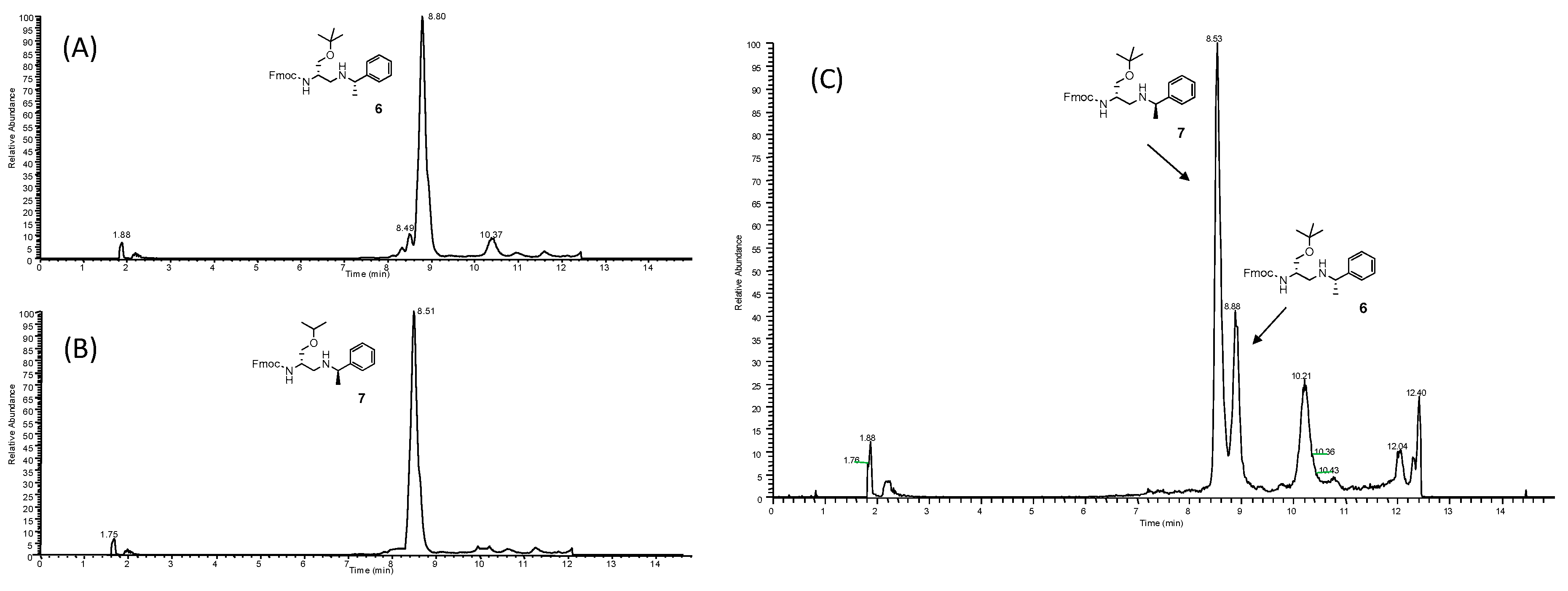
3.3. Analysis of Racemization
3.4. Reductive Amination of the Aldehyde 3 with Sulfonamides
3.5. Synthesis of the l-Dap methyl Ester 14
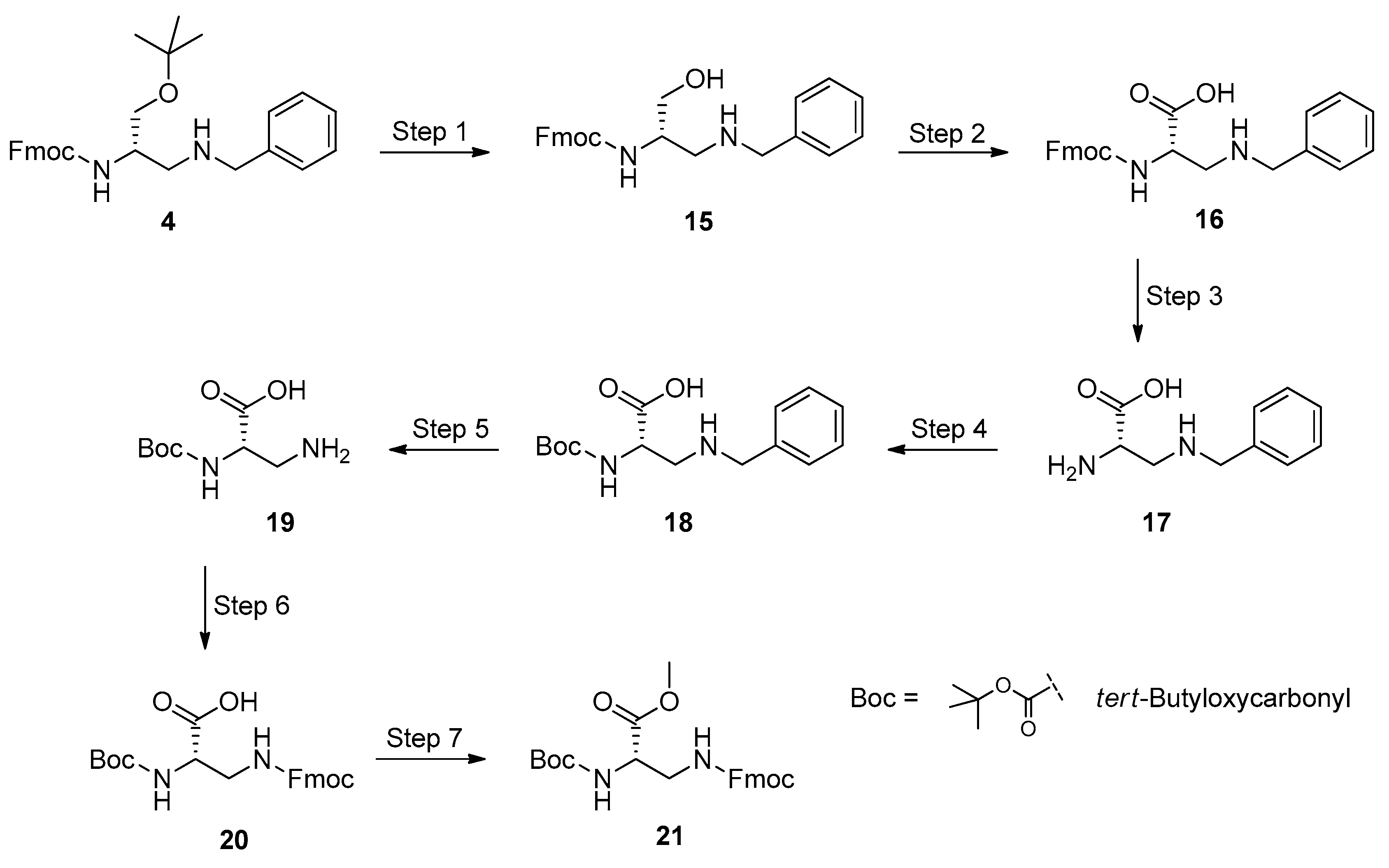
3.6. Synthesis of the l-Dap Methyl Ester 21
3.7. Couplings of l-Dap Methyl Ester 21
4. Materials and Methods
4.1. Synthesis of the Weinreb–Nahm Amide 2
4.2. Synthesis of the Aldehyde 3
4.3. Synthesis of 4–10. General Procedure for the Reductive Amination
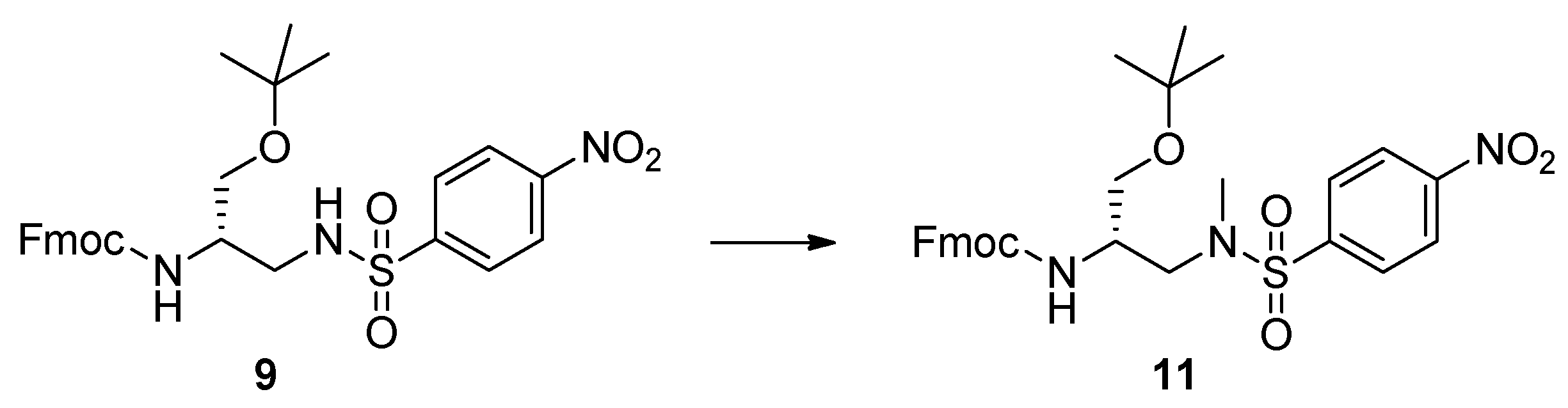
4.4. Methylation of 9. Synthesis of 2,3-diaminopropanol 11
4.5. Synthesis of 2,3-diaminopropanol 12
4.6. Preparation of Fmoc-l-Dap(Ts)-OCH3 (14)
4.7. Synthesis of Boc-l-Dap(Fmoc)-OH (20): Multistep Procedure
4.8. Methylation of 20. Synthesis of Fmoc-L-Dap(Boc)-OCH3 (21)
4.9. Coupling of 21 with Fmoc-l-Ala-OH and Fmoc-d-Ala-OH. Synthesis of diastereomers 23 and 24
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Xu, Z.; Sun, Z.; Li, S.; Xu, Z.; Cao, C.; Xu, Z.; Feng, X.; Xu, H. Systematic unravelling of the biosynthesis of poly (L-diaminopropionic acid) in Streptomyces albulus PD-1. Sci. Rep. 2015, 5, 17400. [Google Scholar] [CrossRef]
- Dobrovinskaya, N.A.; Archer, I.; Hulme, A.N. Chemoenzymatic and chemical routes to the non-proteinaceous amino acid Albizziine and its amide derivative. Synlett 2008, 4, 513–516. [Google Scholar]
- Kjaer, A.; Olesen Larsen, P. Amino acid studies. Part II. Structure and synthesis of Albizziine (L-2-amino-3-ureidopropionic acid), an amino acid from higher plants. Acta Chem. Scand. 1959, 13, 1565–1574. [Google Scholar] [CrossRef]
- Otsuka, M.; Kittaka, A.; Iimori, T.; Yamashita, H.; Kobayashi, S.; Ohno, M. Synthetic studies on an antitumor antibiotic, Bleomycin. XII. Preparation of an l-2,3-diaminopropionic acid derivative as a synthetic intermediate. Chem. Pharm. Bull. 1985, 33, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Oshitari, T.; Shibasaki, M.; Yoshizawa, T.; Tomita, M.; Takao, K.; Kobayashi, S. Synthesis of 2-O-(3-O-carbamoyl-α-D-mannopyranosyl)-L-gulopyranose: Sugar moiety of antitumor antibiotic Bleomycin. Tetrahedron 1997, 53, 10993–11006. [Google Scholar] [CrossRef]
- Highfield, J.A.; Mehta, L.K.; Parrick, J.; Wardman, P. Synthesis, hydroxyl radical production and cytotoxicity of analogues of Bleomycin. Bioorg. Med. Chem. 2000, 8, 1065–1073. [Google Scholar] [CrossRef]
- Sugiyama, M.; Kumagai, T. Molecular and structural biology of Bleomycin and its resistance determinants. J. Biosci. Bioeng. 2002, 93, 105–116. [Google Scholar] [CrossRef]
- Xu, Z.-D.; Wang, M.; Xiao, S.-L.; Liu, C.-L.; Yang, M. Synthesis, biological evaluation and DNA binding properties of novel Bleomycin analogues. Bioorg. Med. Chem. Lett. 2003, 13, 2595–2599. [Google Scholar] [CrossRef]
- Schmidt, U.; Mundinger, K.; Mangold, R.; Lieberknecht, A. Lavendomycin: Total synthesis and assignment of configuration. J. Chem. Soc. Chem. Commun. 1990, 18, 1216–1219. [Google Scholar] [CrossRef]
- Wang, M.; Gould, S.J. Biosynthesis of Capreomycin. 2. Incorporation of L-serine, L-alanine, and L-2,3-diaminopropionic acid. J. Org. Chem. 1993, 58, 5176–5180. [Google Scholar] [CrossRef]
- Bastiaans, H.M.M.; van der Baan, J.L.; Ottenheijm, H.C.J. Flexible and convergent total synthesis of cyclotheonamide B. J. Org. Chem. 1997, 62, 3880–3889. [Google Scholar] [CrossRef]
- Gopalsamy, A.; Yang, H.; Ellingboe, J.W.; Kees, K.L.; Yoon, J.; Murrills, R. Parallel solid-phase synthesis of Vitronectin receptor (αvβ3) inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 1715–1718. [Google Scholar] [CrossRef]
- Stokowa-Sołtys, K.; Dzyhovskyi, V.; Wieczorek, R.; Jeżowska-Bojczuk, M. Phleomycin complex—Coordination mode and in vitro cleavage of DNA. J. Inorg. Biochem. 2019, 195, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Noden, M.; Moreira, R.; Huang, E.; Yousef, A.; Palmer, M.; Taylor, S.D. Total synthesis of Paenibacterin and its analogues. J. Org. Chem. 2019, 84, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Mant, C.T.; Jiang, Z.; Gera, L.; Davis, T.; Nelson, K.L.; Bevers, S.; Hodges, R.S. De novo designed amphipathic α-helical antimicrobial peptides incorporating Dab and Dap residues on the polar face to treat the Gram-negative pathogen, Acinetobacter baumannii. J. Med. Chem. 2019, 62, 3354–3366. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Sieckman, G.L.; Owen, N.K.; Hayes, D.L.; Mazuru, D.G.; Kannan, R.; Volkert, W.A.; Hoffman, T.J. Radiochemical investigations of Gastrin-releasing Peptide Receptor-specific [99mTc(X)(CO)3-Dpr-Ser-Ser-Ser-Gln-Trp-Ala-Val-Gly-His-Leu-Met-(NH2)] in PC-3, tumor-bearing, rodent models: Syntheses, radiolabeling, and in vitro/in vivo studies where Dpr = 2,3-Diaminopropionic acid and X = H2O or P(CH2OH)3. Cancer Res. 2003, 63, 4082–4088. [Google Scholar]
- Audic, N.; Potier, G.; Sasaki, N.A. New 2,3-diaminopropionic acid inhibitors of AGE and ALE formation. Org. Biomol. Chem. 2013, 11, 773–780. [Google Scholar] [CrossRef]
- Fujisawa, T.; Katakura, S.-I.; Odake, S.; Morita, Y.; Yasuda, J.; Yasumatsu, I.; Morikawa, T. Design and synthesis of carboxylate inhibitors for matrix metalloproteinases. Chem. Pharm. Bull. 2001, 49, 1272–1279. [Google Scholar] [CrossRef]
- Beasley, F.C.; Cheung, J.; Heinrichs, D.E. Mutation of L-2,3-diaminopropionic acid synthase genes blocks staphyloferrin B synthesis in Staphylococcus aureus. BMC Microbiol. 2011, 11, 1–12. [Google Scholar] [CrossRef]
- Kobylarz, M.J.; Grigg, J.C.; Takayama, S.-I.J.; Rai, D.K.; Heinrichs, D.E.; Murphy, M.E.P. Synthesis of l-2,3-diaminopropionic acid, a siderophore and antibiotic precursor. Chem. Biol. 2014, 21, 379–388. [Google Scholar] [CrossRef]
- Liu, Y.; Oliveira, B.L.; Correia, J.D.G.; Santos, I.C.; Santos, I.; Spingler, B.; Alberto, R. Syntheses of bifunctional 2,3-diamino propionic acid-based chelators as small and strong tripod ligands for the labelling of biomolecules with 99mTc. Org. Biomol. Chem. 2010, 8, 2829–2839. [Google Scholar] [CrossRef]
- Sengupta, S.; Krishnan, M.A.; Dudhe, P.; Reddy, R.B.; Giri, B.; Chattopadhyay, S.; Chelvam, V. Novel solid-phase strategy for the synthesis of ligand-targeted fluorescent-labelled chelating peptide conjugates as a theranostic tool for cancer. Beilstein J. Org. Chem. 2018, 14, 2665–2679. [Google Scholar] [CrossRef] [PubMed]
- Szyrwiel, Ł.; Shimura, M.; Setner, B.; Szewczuk, Z.; Malec, K.; Malinka, W.; Brasun, J.; Pap, J.S. SOD-Like activity of copper(II) containing metallopeptides branched by 2,3-diaminopropionic acid: What the N-termini elevate, the C-terminus ruins. Int. J. Pept. Res. Ther. 2019, 25, 711–717. [Google Scholar] [CrossRef]
- Batoon, P.; Oomens, J.; Berden, G.; Ren, J. Conformations of protonated AlaDap and DapAla characterized by IRMPD spectroscopy and molecular modeling. J. Phys. Chem. B 2018, 122, 2191–2202. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.-B.; Roderick, J.; Jackson, S.; Rafalski, M.; Rockwell, A.; Mousa, S.; Olson, R.E.; DeGrado, W.F. Design, synthesis, and in vitro activities of benzamide-core glycoprotein IIb/IIIa antagonists: 2,3-diaminopropionic acid derivatives as surrogates of aspartic acid. Bioorg. Med. Chem. 1997, 5, 693–705. [Google Scholar] [CrossRef]
- Quan, K.; Li, G.; Tao, L.; Xie, Q.; Yuan, Q.; Wang, X. Diaminopropionic acid reinforced graphene sponge and its use for hemostasis. ACS Appl. Mater. Interfaces 2016, 8, 7666–7673. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, Y.; Kang, S.; Waki, M.; Kato, T.; Izumiya, N. Synthesis of derivatives on N2-acetyl-N3-glycyl-l-2,3-diaminopropionic acid and their hydrolyses by trypsin. Bull. Chem. Soc. Jpn. 1981, 54, 297–298. [Google Scholar] [CrossRef]
- Huguenin-Dezot, N.; Alonzo, D.A.; Heberlig, G.W.; Mahesh, M.; Nguyen, D.P.; Dornan, M.H.; Boddy, C.N.; Schmeing, T.M.; Chin, J.W. Trapping biosynthetic acyl-enzyme intermediates with encoded 2,3-diaminopropionic acid. Nature 2019, 565, 112–117. [Google Scholar] [CrossRef]
- Maillard, N.; Darbre, T.; Reymond, J.-L. Identification of catalytic peptide dendrimers by “off-bead” in silica high-throughput screening of combinatorial libraries. J. Comb. Chem. 2009, 11, 667–675. [Google Scholar] [CrossRef]
- Featherston, A.L.; Shugrue, C.R.; Mercado, B.Q.; Miller, S.J. Phosphothreonine (pThr)-based multifunctional peptide catalysis for asymmetric Baeyer−Villiger oxidations of cyclobutanones. ACS Catal. 2019, 9, 242–252. [Google Scholar] [CrossRef]
- Abbate, V.; Liang, W.; Patel, J.; Lan, Y.; Capriotti, L.; Iacobucci, V.; Bui, T.T.; Chaudhuri, P.; Kudsiova, L.; Vermeer, L.S.; et al. Manipulating the pH response of 2,3-diaminopropionic acid rich peptides to mediate highly effective gene silencing with low-toxicity. J. Control. Release 2013, 172, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Roviello, G.N.; Musumeci, D.; Bucci, E.M.; Pedone, C. Synthesis of a diaminopropanoic acid-based nucleoamino acid and assembly of cationic nucleopeptides for biomedical applications. Amino Acids 2012, 43, 2537–2543. [Google Scholar] [CrossRef] [PubMed]
- Meierhenrich, U.J.; Muñoz Caro, G.M.; Bredehöft, J.H.; Jessberger, E.K.; Thiemann, W.H.P. Identification of diamino acids in the Murchison meteorite. Proc. Natl. Acad. Sci. USA 2004, 101, 9182–9186. [Google Scholar] [CrossRef] [PubMed]
- Frenkel-Pintera, M.; Haynesa, J.W.; Ca, M.; Petrova, A.S.; Burcara, B.T.; Krishnamurthya, R.; Huda, N.V.; Lemana, L.J.; Williams, L.D. Selective incorporation of proteinaceous over nonproteinaceous cationic amino acids in model prebiotic oligomerization reactions. Proc. Natl. Acad. Sci. USA 2019, 116, 16338–16346. [Google Scholar] [CrossRef]
- Nadir, U.K.; Vijaya Krishna, R.; Singh, A. A new and facile route for the synthesis of chiral 1,2-diamines and 2,3-diamino acids. Tetrahedron Lett. 2005, 46, 479–482. [Google Scholar] [CrossRef]
- Kucharczyk, N.; Badet, B.; Le Goffic, F. Quantitative synthesis of L or D N2-tert-butoxycarbonyl-2,3-diaminopropanoic acid from protected L or d-serine-β–lactone. Synth. Commun. 1989, 19, 1603–1609. [Google Scholar] [CrossRef]
- Stanley, M.S. Orthogonally protected N3-(carboxymethyl)-l-2,3-diaminopropanoic acids and O-(carboxymethyl)-l-serines on solid-phase peptide synthesis. J. Org. Chem. 1992, 57, 6421–6430. [Google Scholar] [CrossRef]
- Couturier, C.; Blanchet, J.; Schlama, T.; Zhu, J. Aziridinium from N,N-dibenzyl serine methyl ester: Synthesis of enantiomerically pure β-amino and α,β-diamino esters. Org. Lett. 2006, 8, 2183–2186. [Google Scholar] [CrossRef] [PubMed]
- Benoiton, L. Conversion of p-chloro-l-alanine to α-carbobenzoxy-DL-diaminopropionic acid. Can. J. Chem. 1968, 46, 1449–1552. [Google Scholar] [CrossRef]
- Lucet, D.; Le Gall, T.; Mioskowski, C. The chemistry of vicinal diamines. Angew. Chem. Int. Ed. 1998, 37, 2580–2627. [Google Scholar] [CrossRef]
- Davoli, P.; Forni, A.; Moretti, I.; Prati, F. Stereochemistry of nucleophilic ring-opening reactions of optically active N-acetyl-2-methoxycarbonylaziridine. Tetrahedron Asymmetry 1995, 6, 2011–2016. [Google Scholar] [CrossRef]
- Luo, Y.; Blaskovich, M.A.; Lajoie, G.A. Stereoselective synthesis of α-substituted α,β-diamino acids from α-hydroxy amino acids. J. Org. Chem. 1999, 64, 6106–6111. [Google Scholar] [CrossRef]
- Kretsinger, J.K.; Schneider, J.P. Design and application of basic amino acids displaying enhanced hydrophobicity. J. Am. Chem. Soc. 2003, 125, 7907–7913. [Google Scholar] [CrossRef]
- Carrasco, M.R.; Brown, R.T.; Doan, V.H.; Kandel, S.M.; Lee, F.C. 2-(N-Fmoc)-3-(N-Boc-N-methoxy)-diaminopropanoic acid, an amino acid for the synthesis of mimics of O-linked glycopeptides. Biopolymers 2006, 84, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.R.; Tantry, S.J.; Suresh Babu, V.V. Practical and efficient synthesis of orthogonally protected α-2,3-diaminopropionic acid (2,3-Dap), 2,4-diaminobutanoic acid (2,4-Dab), and their N-methylated derivatives. Synth. Commun. 2006, 36, 2901–2912. [Google Scholar] [CrossRef]
- Barghash, R.F.; Massi, A.; Dondoni, A. Synthesis of thiourea-tethered C-glycosyl amino acids via isothiocyanate–amine coupling. Org. Biomol. Chem. 2009, 7, 3319–3330. [Google Scholar] [CrossRef]
- Banerjee, S.; Wiggins, W.J.; Geoghegan, J.L.; Anthony, C.T.; Woltering, E.A.; Masterson, D.S. Novel synthesis of various orthogonally protected Cα-methyllysine analogues and biological evaluation of a vapreotide analogue containing (S)-α-methyllysine. Org. Biomol. Chem. 2013, 11, 6307–6319. [Google Scholar] [CrossRef]
- Lindahl, F.; Hoang, H.N.; Fairlie, D.P.; Cooper, M.A. Facile synthesis of mono- and bis-methylated Fmoc-Dap, -Dab and -Orn amino acids. Chem. Commun. 2015, 51, 4496–4498. [Google Scholar] [CrossRef]
- Choi, D.; Kahn, H. Expedient syntheses of racemic 2,3-diaminopropanoic acid derivative. Tetrahedron Lett. 1995, 36, 7371–7374. [Google Scholar] [CrossRef]
- Egbertson, M.S.; Homnick, C.F.; Hartman, G.D. A selective protection of 2,3-diaminopropionic acid. Synth. Commun. 1993, 23, 703–709. [Google Scholar] [CrossRef]
- Englund, E.A.; Gopi, H.N.; Appella, D.H. An efficient synthesis of a probe for protein function: 2,3-diaminopropionic acid with orthogonal protecting groups. Org. Lett. 2004, 6, 213–215. [Google Scholar] [CrossRef]
- Kumaraswamy, G.; Pitchaiah, A. Highly enantioselective synthesis of orthogonally protected (2S)-2,3-diaminopropanoates through catalytic phase-transfer Aza-Henry reaction. Helv. Chim. Acta 2011, 94, 1543–1550. [Google Scholar] [CrossRef]
- Cativiela, C.; Díaz-de-Villegas, M.D.; Gálvez, J.A. Diastereoselective strecker reaction of imines derived from D-glyceraldehyde. A new route to β-hydroxy-α-amino acids. Tetrahedron Lett. 1995, 36, 2859–2860. [Google Scholar] [CrossRef]
- Cativiela, C.; Diaz-de-Villegas, M.D.; Gálvez, J. Stereoselective Synthesis of α-Hydroxy-β-amino Acids Using D-Glyceraldehyde as the Homochiral Source. Tetrahedron Asymmetry 1996, 7, 529–536. [Google Scholar] [CrossRef]
- Mattson, R.J.; Pham, K.M.; Leuck, D.J.; Cowen, K.A. An improved method for reductive alkylation of amines using titanium (IV) isopropoxide and sodium cyanoborohydride. J. Org. Chem. 1990, 55, 2552–2554. [Google Scholar] [CrossRef]
- Zhao, M.M.; Li, J.; Mano, E.; Song, Z.J.; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. Oxidation of primary alcohols to carboxylic acids with sodium chlorite catalyzed by TEMPO and bleach. J. Org. Chem. 1999, 64, 2564–2566. [Google Scholar] [CrossRef]
- Huang, L.; Teumelsan, N.; Huang, X. A facile method for oxidation of primary alcohols to carboxylic acids and its application in glycosaminoglycan syntheses. Chem. Eur. J. 2006, 12, 5246–5252. [Google Scholar] [CrossRef]
- Siciliano, C.; Barattucci, A.; Bonaccorsi, P.; Di Gioia, M.L.; Leggio, A.; Minuti, L.; Romio, E.; Temperini, A. Synthesis of D-erythro-sphinganine through serine-derived α-amino epoxides. J. Org. Chem. 2014, 79, 5320–5326. [Google Scholar] [CrossRef]
- Borch, R.F.; Bernstein, M.D.; Dupont Durst, H. The cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; Le Pera, A.; Liguori, A.; Siciliano, C. Highly selective conversion of aryl peptidyl ketones into the corresponding peptidyl alcohols. Eur. J. Org. Chem. 2004, 3, 463–467. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; Le Pera, A.; Liguori, A.; Pitrelli, A.F.; Siciliano, C. A convenient method for the stereoselective conversion of aryl peptidyl ketones into the corresponding aryl aminomethin derivatives, a novel class of modified peptides. Protein Pept. Lett. 2005, 12, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A preparation of N-Fmoc-N-methyl-α-amino acids and N-Nosyl-N-methyl-α-amino acids. Amino Acids 2010, 38, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Stewart, W.E.; Siddall, T.H. Nuclear magnetic resonance studies of amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]
- Marcovici-Mizrahi, D.; Gottlieb, H.E.; Marks, V.; Nudelman, A. On the stabilization of the syn-rotamer of amino acid carbamate derivatives by hydrogen bonding. J. Org. Chem. 1996, 61, 8402–8406. [Google Scholar] [CrossRef]
- Modarresi-Alam, A.R.; Najaf, P.; Rostamizadeh, M.; Keykha, H.; Bijanzadeh, H.-R.; Kleinpeter, E. Dynamic 1H-NMR study of the barrier to rotationabout the C-N bond in primary carbamates and its solvent dependence. J. Org. Chem. 2007, 72, 2208–2211. [Google Scholar] [CrossRef]
- Deetz, M.J.; Forbes, C.C.; Jonas, M.; Malerich, J.P.; Smith, B.D.; Wiest, O. Unusually low barrier to carbamate C-N rotation. J. Org. Chem. 2002, 67, 3949–3952. [Google Scholar] [CrossRef]
- Ernani, A.; Basso, E.A.; Pontes, R.M. Further studies on the rotational barriers of Carbamates. An NMR and DFT analysis of the solvent effect for cyclohexyl N,N-dimethylcarbamate. J. Mol. Struct. Theochem. 2002, 594, 199–206. [Google Scholar]
- Solomon, M.E.; Lynch, C.L.; Rich, D.H. Synthesis of Nα,Nβ-alkylated diaminopropionic acid analogs. Tetrahedron Lett. 1995, 36, 4955–4958. [Google Scholar] [CrossRef]
- Pitteloud, J.-P.; Bionda, N.; Cudi, P. Direct access to side chain N,N’-diaminoalkylated derivatives of basic amino acids suitable for solid-phase peptide synthesis. Amino Acids 2013, 44, 321–333. [Google Scholar] [CrossRef]
- Jurczak, J.; Golębiowski, A. Optically active N-protected α-amino aldehydes in organic synthesis. Chem. Rev. 1989, 89, 149–164. [Google Scholar] [CrossRef]
- Leggio, A.; Le Pera, A.; Liguori, A.; Napoli, A.; Romeo, C.; Siciliano, C.; Sindona, G. Highly stereoselective synthesis of optically pure C-aryl imines from α-L-amino acid methyl esters. Synth. Commun. 2003, 33, 4331–4338. [Google Scholar] [CrossRef]
- De Marco, R.; Di Gioia, M.L.; Liguori, A.; Perri, F.; Siciliano, C.; Spinella, M. N-alkylation of N-arylsulfonyl-α-amino acid methyl esters by trialkyloxonium tetrafluoroborates. Tetrahedron 2011, 67, 9708–9714. [Google Scholar] [CrossRef]
- You, S.-L.; Kelly, J.W. Highly efficient enantiospecific synthesis of imidazoline-containing amino acids using bis (triphenyl) oxodiphosphonium trifluoromethanesulfonate. Org. Lett. 2004, 6, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- Belsito, E.; Di Gioia, M.L.; Greco, A.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. N-methyl-N-nosyl-β3-amino acids. J. Org. Chem. 2007, 72, 4798–4802. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Kessler, H. Solid-phase synthesis of 1,3-azole-based peptides and peptidomimetics. Org. Lett. 2006, 8, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Anantharamaiah, G.M.; Sivanandaiah, K.M. Transfer hydrogenation; a convenient method for removal of some commonly used protecting groups in peptide synthesis. J. Chem. Soc. Perkin Trans. 1977, 1, 490–491. [Google Scholar] [CrossRef]
- Felix, A.M.; Heimer, E.P.; Lambros, T.J.; Tzougraki, C.; Meienhofer, J. Rapid removal of protecting groups from peptides by catalytic transfer hydrogenation with 1,4-cyclohexadiene. J. Org. Chem. 1978, 43, 4194–4196. [Google Scholar] [CrossRef]
- Siciliano, C.; De Marco, R.; Guidi, L.E.; Spinella, M.; Liguori, A. A one-pot procedure for the preparation of N-9-fluorenylmethyloxycarbonyl-α-amino diazoketones from α-amino acids. J. Org. Chem. 2012, 77, 10575–10582. [Google Scholar] [CrossRef]
- Aiello, D.; Furia, E.; Siciliano, C.; Bongiorno, D.; Napoli, A. Study of the coordination of ortho-tyrosine and trans-4-hydroxyproline with aluminum(III) and iron(III). J. Mol. Liq. 2018, 269, 387–397. [Google Scholar] [CrossRef]
- De Marco, R.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef]
- Piazzolla, F.; Siciliano, C.; Minuti, L.; Temperini, A. Exploration of Synthetic strategies for the stereoselective preparation of novel tetrahydrofuran-containing biaryls: A high-pressure promoted Diels-Alder approach. Tetrahedron 2018, 74, 6534–6543. [Google Scholar] [CrossRef]
- Di Donna, L.; Benabdelkamel, H.; Taverna, D.; Indelicato, S.; Aiello, D.; Napoli, A.; Sindona, G.; Mazzotti, F. Determination of ketosteroid hormones in meat by liquid chromatography tandem mass spectrometry and derivatization chemistry. Anal. Chem. Biochem. 2015, 407, 5835–5842. [Google Scholar] [CrossRef] [PubMed]
- Di Donna, L.; Mazzotti, F.; Napoli, A.; Salerno, R.; Saijad, A.; Sindona, G. Secondary metabolism of olive secoiridoids. New microcomponents detected in drupes by electrospray ionization and high-resolution tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Aiello, D.; Giambona, A.; Leto, F.; Passarello, C.; Damiani, G.; Maggio, A.; Siciliano, C.; Napoli, A. Human coelomic fluid investigation: A MS approach to prenatal screening. Sci. Rep. 2018, 8, 10973. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.I.; Pepe, D.A.; Aiello, D.; Siciliano, C.; Athanassopoulos, C.M. Chemoselective protection of glutathione in the preparation of bioconjugates: The case of tripanothione disulfide. J. Org. Chem. 2016, 81, 4353–4358. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Product | 2,3-Diaminopropanol Structure | Yield (%) 1 |
|---|---|---|---|
 | 4 |  | 92 |
 | 5 |  | 90 |
 | 6 |  | 90 |
 | 7 |  | 91 |
 | 8 |  | 89 |
 | 9 |  | 82 |
 | 10 |  | 85 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Temperini, A.; Aiello, D.; Mazzotti, F.; Athanassopoulos, C.M.; De Luca, P.; Siciliano, C. 2,3-Diaminopropanols Obtained from d-Serine as Intermediates in the Synthesis of Protected 2,3-l-Diaminopropanoic Acid (l-Dap) Methyl Esters. Molecules 2020, 25, 1313. https://doi.org/10.3390/molecules25061313
Temperini A, Aiello D, Mazzotti F, Athanassopoulos CM, De Luca P, Siciliano C. 2,3-Diaminopropanols Obtained from d-Serine as Intermediates in the Synthesis of Protected 2,3-l-Diaminopropanoic Acid (l-Dap) Methyl Esters. Molecules. 2020; 25(6):1313. https://doi.org/10.3390/molecules25061313
Chicago/Turabian StyleTemperini, Andrea, Donatella Aiello, Fabio Mazzotti, Constantinos M. Athanassopoulos, Pierantonio De Luca, and Carlo Siciliano. 2020. "2,3-Diaminopropanols Obtained from d-Serine as Intermediates in the Synthesis of Protected 2,3-l-Diaminopropanoic Acid (l-Dap) Methyl Esters" Molecules 25, no. 6: 1313. https://doi.org/10.3390/molecules25061313
APA StyleTemperini, A., Aiello, D., Mazzotti, F., Athanassopoulos, C. M., De Luca, P., & Siciliano, C. (2020). 2,3-Diaminopropanols Obtained from d-Serine as Intermediates in the Synthesis of Protected 2,3-l-Diaminopropanoic Acid (l-Dap) Methyl Esters. Molecules, 25(6), 1313. https://doi.org/10.3390/molecules25061313