
An Enzymatic Flow-Based Preparative Route to Vidarabine


 ,
,  ,
,  ,
,  and
and
Abstract
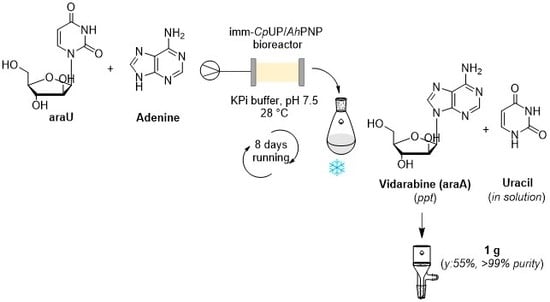
1. Introduction
2. Results and Discussion
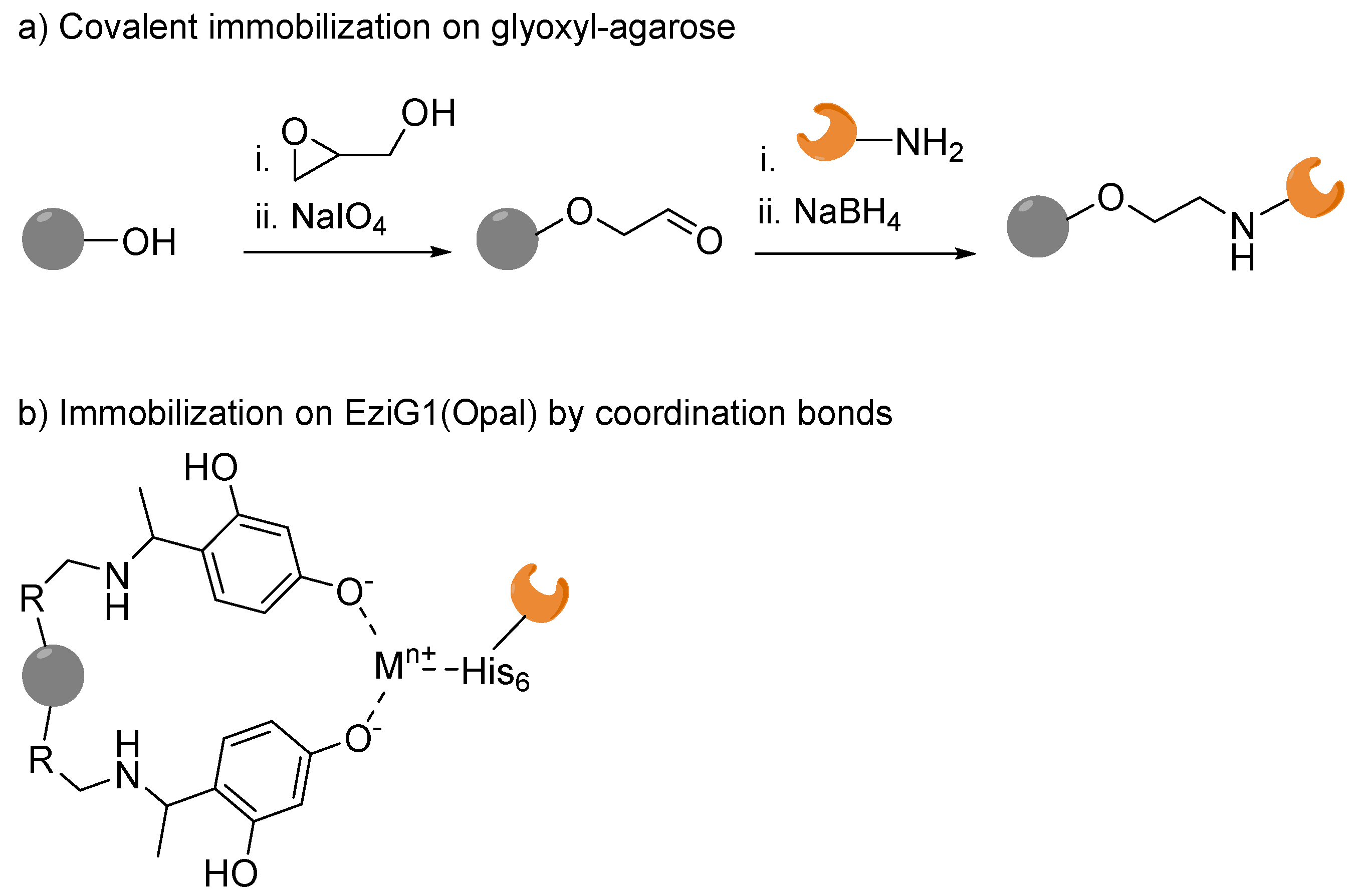
2.1. Immobilization on Glyoxyl–Agarose under Continuous Flow
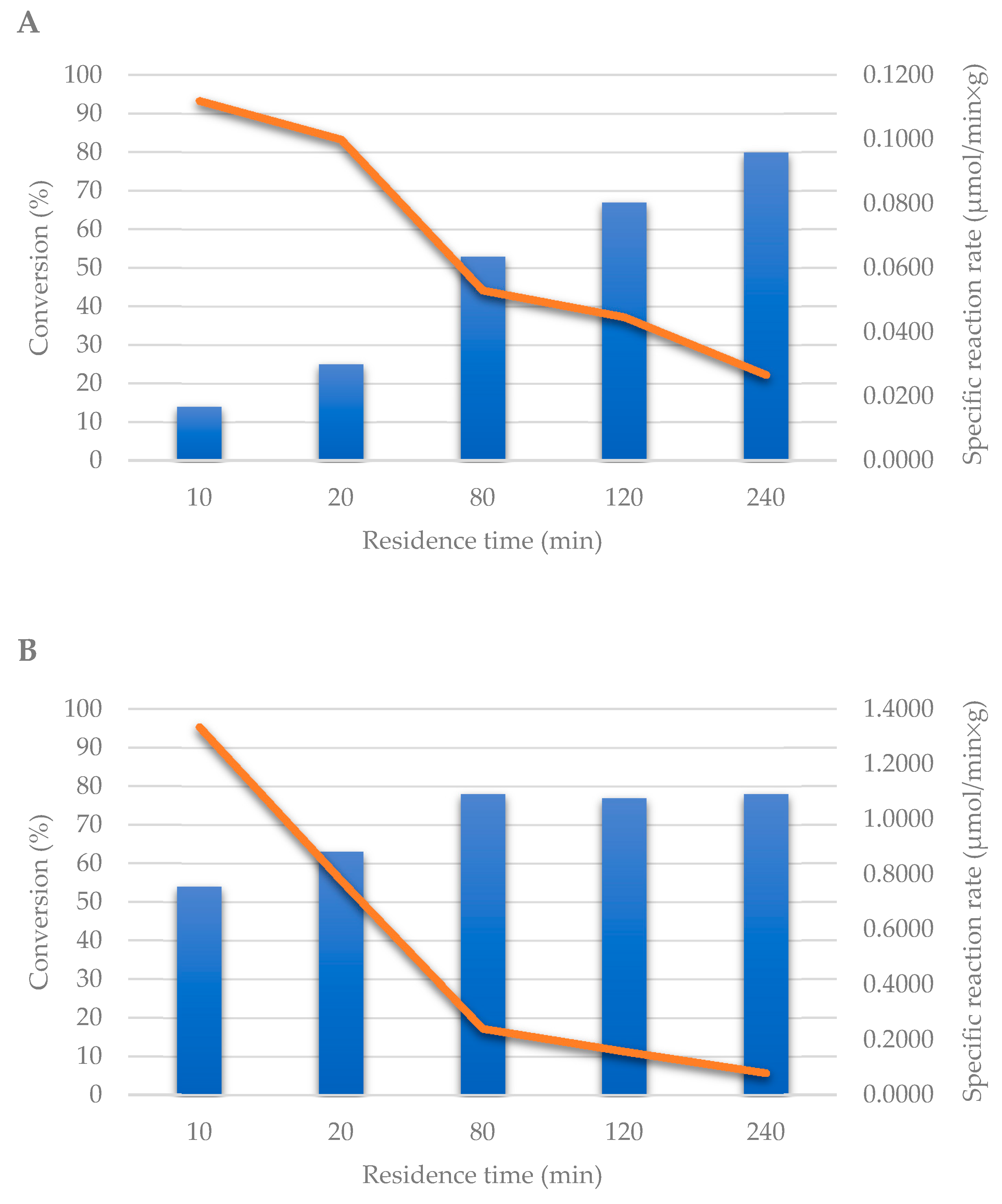
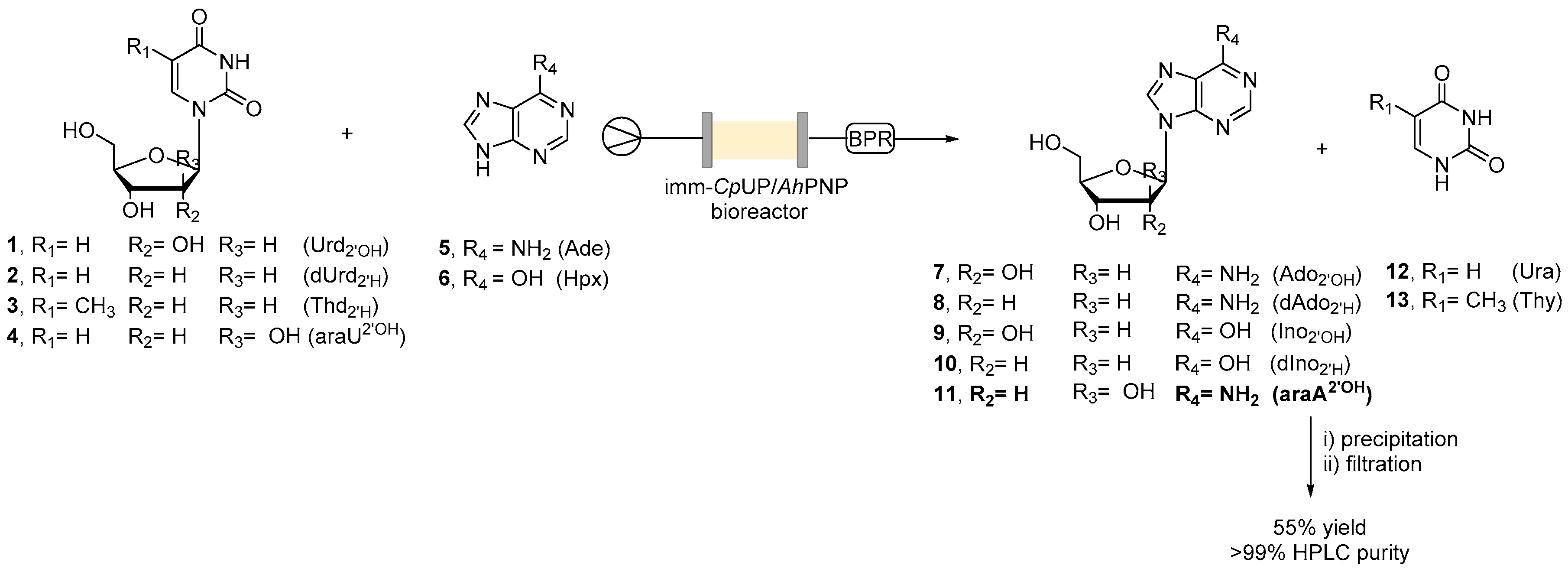
2.2. Transglycosylation Reaction Using Glyoxyl–Agarose-Based CpUP/AhPNP-Bioreactor
2.3. Co-Immobilization on EziGTM Carriers in Continuous Flow
2.4. Continuous Flow Synthesis of Ara-A (11) and Product Isolation
3. Materials and Methods
3.1. Analytical Methods
3.2. Immobilization Yields
3.3. Co-Immobilization of CpUP and AhPNP on Glyoxyl–Agarose under Flow Conditions
3.4. Co-Immobilization of CpUP and AhPNP on EziGTM1 (Opal) under Flow Conditions
3.5. Standard Activity Assay of CpUP
3.6. Standard Activity Assay of AhPNP
3.7. Flow Activity Assay of AhPNP
3.8. Flow Activity Assay of CpUP
3.9. General Procedure for the Flow Transglycosylation Reaction
3.10. Synthesis of Vidarabine (11)
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Truppo, M.D. Biocatalysis in the pharmaceutical industry: The need for speed. ACS Med. Chem. Lett. 2017, 8, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Alcántara, A.R. Biocatalysis and pharmaceuticals: A smart tool for sustainable development. Catalysts 2019, 9, 792. [Google Scholar] [CrossRef]
- Woodley, J.M. Accelerating the implementation of biocatalysis in industry. Appl. Microbiol. Biotechnol. 2019, 103, 4733–4739. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandez, P.; Paradisi, F.; Molinari, F. Flow bioreactors as complementary tools for biocatalytic process intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Contente, M.L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Flow-based enzymatic synthesis of melatonin and other high value tryptamine derivatives: A five-minute intensified process. Green Chem. 2019, 21, 3263–3266. [Google Scholar] [CrossRef]
- De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.L.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Bioprocess intensification using flow reactors: Stereoselective oxidation of chiral 1,3-diols with immobilized Acetobacter aceti. Catalysts 2019, 9, 208. [Google Scholar] [CrossRef]
- Contente, M.L.; Dall’Oglio, F.; Tamborini, L.; Molinari, F.; Paradisi, F. Highly efficient oxidation of amines to aldehydes via flow-based biocatalysis. ChemCatChem 2017, 9, 3843–3848. [Google Scholar] [CrossRef]
- De Vitis, V.; Dall’Oglio, F.; Pinto, A.; De Micheli, C.; Molinari, F.; Conti, P.; Romano, D.; Tamborini, L. Chemoenzymatic synthesis in flow reactors: A rapid and convenient preparation of Captopril. Chem. Open 2017, 6, 668–673. [Google Scholar] [CrossRef]
- Devine, P.N.; Howard, R.M.; Kumar, R.; Thompson, M.P.; Truppo, M.D.; Turner, N.J. Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem. 2018, 2, 409–421. [Google Scholar] [CrossRef]
- Vorbrueggen, H.; Ruh-Pohlenz, C. Synthesis of nucleosides. Org. React. 2000, 55. [Google Scholar] [CrossRef]
- Mikhailopulo, I.; Miroshnikov, A.I. Biologically important nucleosides: Modern trends in biotechnology and application. Mendeleev Commun. 2011, 21, 57–68. [Google Scholar] [CrossRef]
- Ubiali, D.; Serra, C.D.; Serra, I.; Morelli, C.F.; Terreni, M.; Albertini, A.M.; Manitto, P.; Speranza, G. Produc-tion, characterization and synthetic application of a purine nucleoside phosphorylase from Aeromonas hy-drophila. Adv. Synth. Catal. 2012, 354, 96–104. [Google Scholar] [CrossRef]
- Zhou, X.; Szeker, K.; Jiao, L.-Y.; Oestreich, M.; Mikhailopulo, I.A.; Neubauer, P. Synthesis of 2,6-dihalogenated purine nucleosides by thermostable nucleoside phosphorylases. Adv. Synth. Catal. 2015, 357, 1237–1244. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, W.; Zhang, C.; Yang, Z.; Neubauer, P.; Mikhailopulo, I.A.; Huang, Z. Biocatalytic synthesis of seleno-, thio- and chloro-nucleobase modified nucleosides by thermostable nucleoside phosphorylases. Catal. Commun. 2019, 121, 32–37. [Google Scholar] [CrossRef]
- Rabuffetti, M.; Bavaro, T.; Semproli, R.; Cattaneo, G.; Massone, M.; Morelli, C.F.; Speranza, G.; Ubiali, D. Synthesis of Ribavirin, Tecadenoson, and Cladribine by enzymatic transglycosylation. Catalysts 2019, 9, 355. [Google Scholar] [CrossRef]
- Fernández-Lucas, J.; Camarasa Rius, M.J. Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives, 1st ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018; pp. 1–325. [Google Scholar]
- Serra, I.; Daly, S.; Alcantara, A.R.; Bianchi, D.; Terreni, M.; Ubiali, D. Redesigning the synthesis of Vidarabine via a multienzymatic reaction catalyzed by immobilized nucleoside phosphorylases. RSC Adv. 2015, 5, 23569–23577. [Google Scholar] [CrossRef]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A multi-enzymatic cascade reaction for the synthesis of vidarabine 5′-monophosphate. Catalysts 2020, 9, 60. [Google Scholar] [CrossRef]
- Engelmark Cassimjee, K.; Federsel, H.-J. EziG: A universal platform for enzyme immobilization. In Biocatalysis: An Industrial Perspective, 1st ed.; De Gonzalo, G., Domínguez de María, P., Eds.; RSC Publishing: Cambridge, UK, 2018; pp. 345–362. [Google Scholar] [CrossRef]
- Thompson, M.P.; Peñafiel, I.; Cosgrove, S.C.; Turner, N.J. Biocatalysis using immobilized enzymes in continuous flow for the synthesis of fine chemicals. Org. Process. Res. Dev. 2019, 23, 9–18. [Google Scholar] [CrossRef]
- Serra, I.; Ubiali, D.; Piškur, J.; Christoffersen, S.; Lewkowicz, E.S.; Iribarren, A.M.; Albertini, A.M.; Terreni, M. Developing a collection of immobilized nucleoside phosphorylases for the preparation of nucleoside analogues: Enzymatic synthesis of arabinosyladenine and 2′,3′-dideoxyinosine. ChemPlusChem 2013, 78, 157–165. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 27, 248–254. [Google Scholar] [CrossRef]
- Zambelli, P.; Tamborini, L.; Cazzamalli, S.; Pinto, A.; Arioli, S.; Balzaretti, S.; Plou, F.J.; Fernandez-Arrojo, L.; Molinari, F.; Conti, P.; et al. An efficient continuous flow process for the synthesis of a non-conventional mixture of fructooligosaccharides. Food Chem. 2016, 190, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Guisàn, J.M. Aldehyde-agarose gels as activated supports for immobilization-stabilization of enzymes. Enzym. Microb. Technol. 1988, 10, 375–382. [Google Scholar] [CrossRef]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of vidarabine are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Residence Time (min) | [Urd, 1] (mM) | [Ade, 5] (mM) | Conversion (%) a |
|---|---|---|---|---|
| 1 | 5 | 4 | 2 | 85 |
| 2 | 5 | 8 | 4 | 86 |
| 3 | 10 | 8 | 4 | 84 |
| 4 | 20 | 8 | 4 | 84 |
| 5 | 5 | 16 | 8 | 86 |
| 6 | 2.5 | 16 | 8 | 86 |
| 7 | 1 | 16 | 8 | 86 |
| 8 b | 5 | 32 | 16 | 88 |
| 9 b | 5 | 40 | 20 | 89 |
| 10 c | 5 | 100 | 50 | 89 |
| Residence Time (min) | Conversion (%) | Space Time Yield (g/day) | Catalyst Productivity (24 h) (mmolproduct/mgenzyme) |
|---|---|---|---|
| 1 | 85 | 1.8 | 0.98 |
| Sugar Donor | Sugar Acceptor | Product | Conversion a (%) |
|---|---|---|---|
| 1 (Urd) | 5 (Ade) | 7 (Ado) | 76 |
| 1 (Urd) | 6 (Hpx) | 9 (Ino) | 45 |
| 2 (dUrd) | 5 (Ade) | 8 (dAdo) | 70 |
| 2 (dUrd) | 6 (Hpx) | 10 (dIno) | 50 |
| 3 (Thd) | 5 (Ade) | 8 (dAdo) | 73 |
| 3 (Thd) | 6 (Hpx) | 10 (dIno) | 40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules 2020, 25, 1223. https://doi.org/10.3390/molecules25051223
Tamborini L, Previtali C, Annunziata F, Bavaro T, Terreni M, Calleri E, Rinaldi F, Pinto A, Speranza G, Ubiali D, et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules. 2020; 25(5):1223. https://doi.org/10.3390/molecules25051223
Chicago/Turabian StyleTamborini, Lucia, Clelia Previtali, Francesca Annunziata, Teodora Bavaro, Marco Terreni, Enrica Calleri, Francesca Rinaldi, Andrea Pinto, Giovanna Speranza, Daniela Ubiali, and et al. 2020. "An Enzymatic Flow-Based Preparative Route to Vidarabine" Molecules 25, no. 5: 1223. https://doi.org/10.3390/molecules25051223
APA StyleTamborini, L., Previtali, C., Annunziata, F., Bavaro, T., Terreni, M., Calleri, E., Rinaldi, F., Pinto, A., Speranza, G., Ubiali, D., & Conti, P. (2020). An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules, 25(5), 1223. https://doi.org/10.3390/molecules25051223