Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications

 ,
, 
 , and
, and
Abstract
1. Introduction
2. Results
2.1. CS-g-CNO Characterization
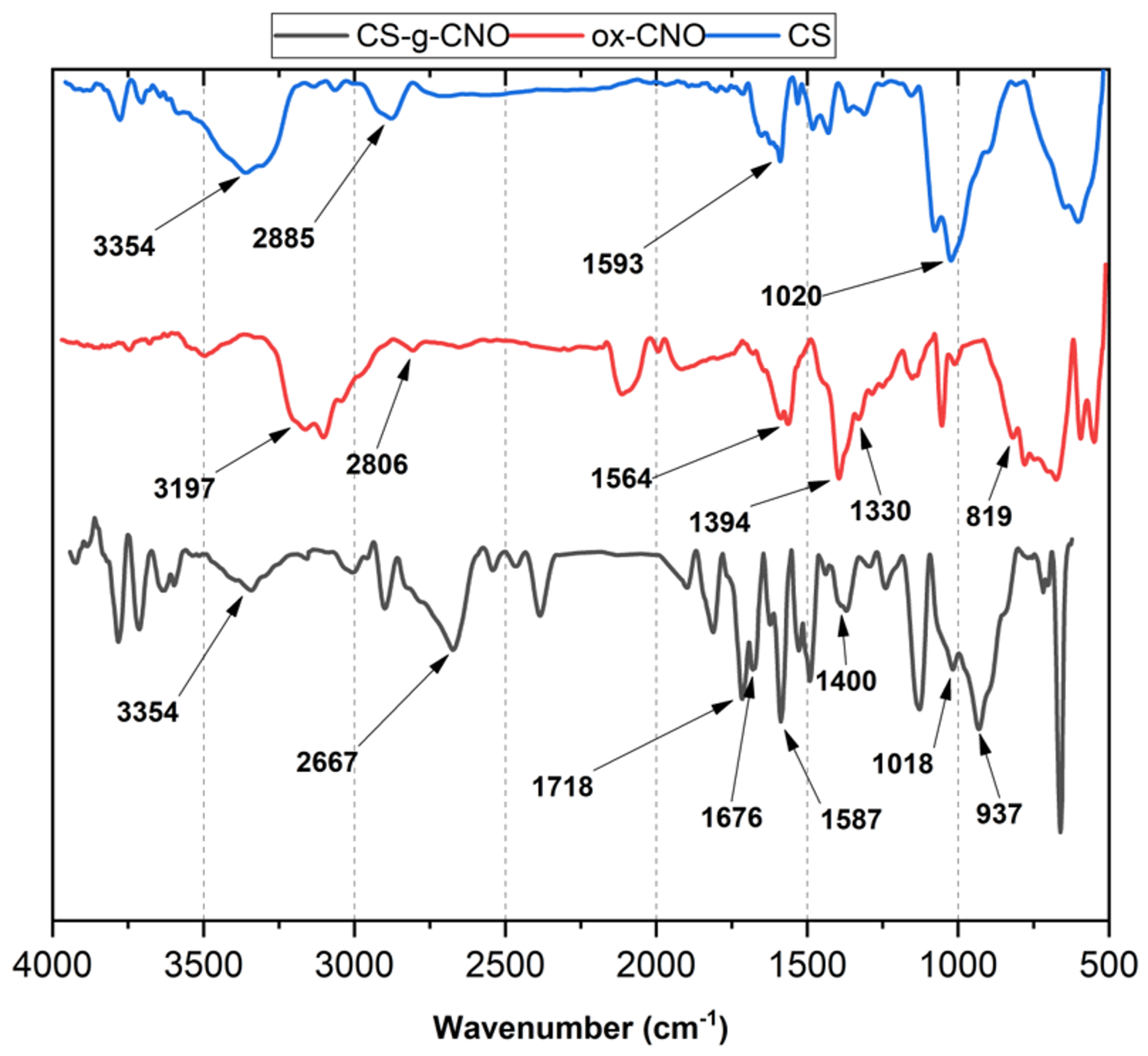
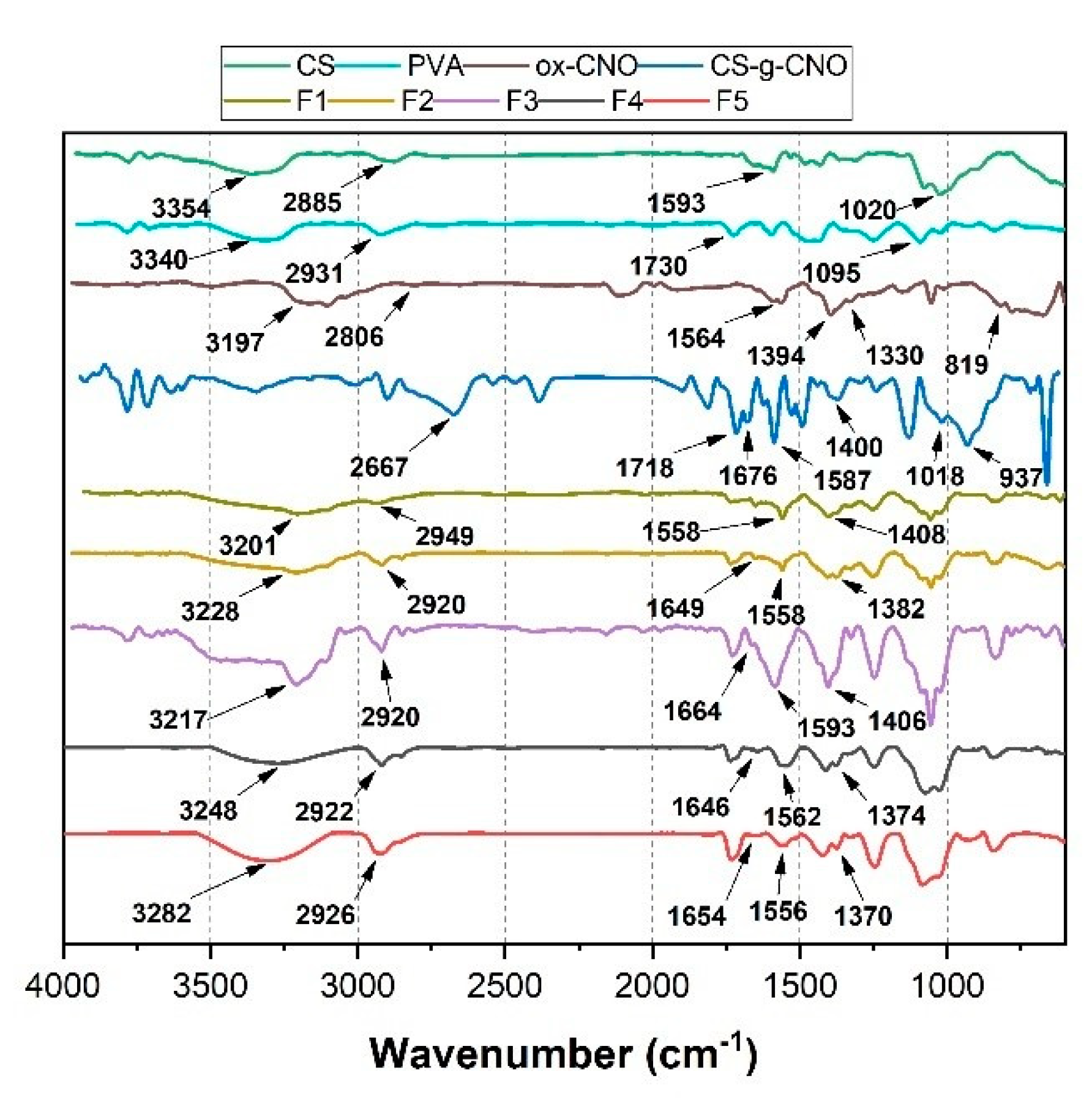
2.1.1. Fourier-Transform Infrared Spectroscopy (FTIR)
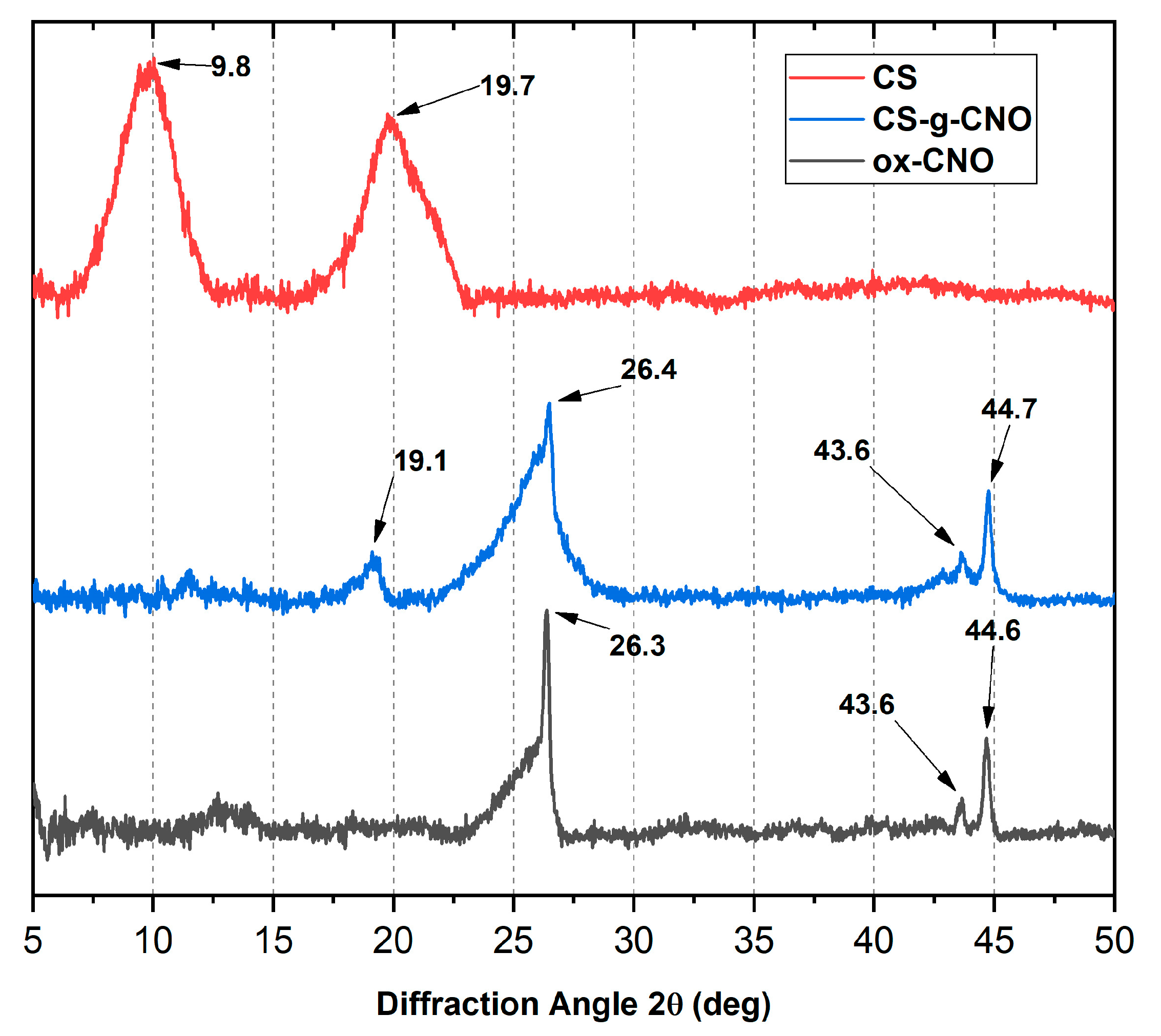
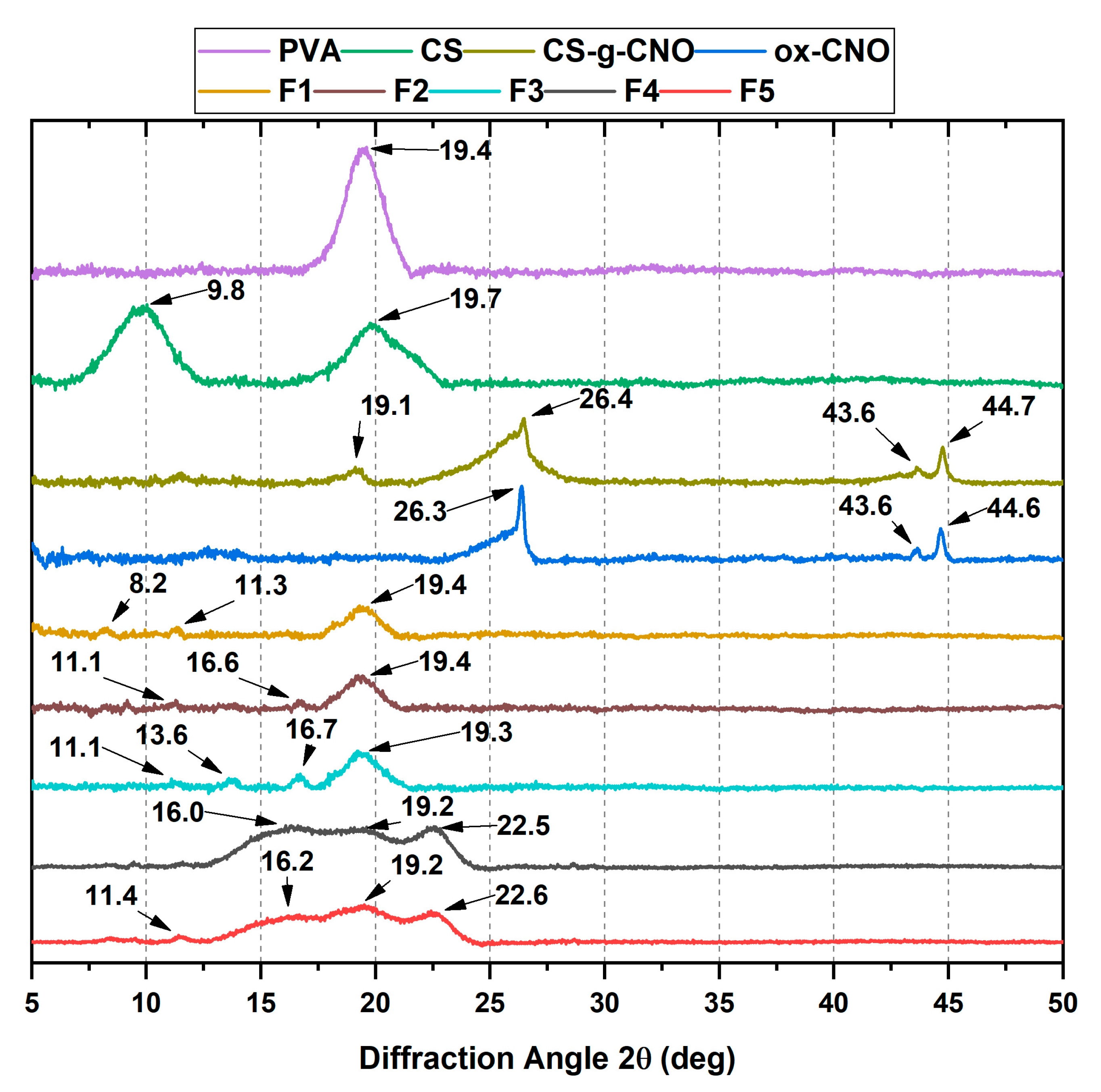
2.1.2. X-ray Diffraction (XRD)
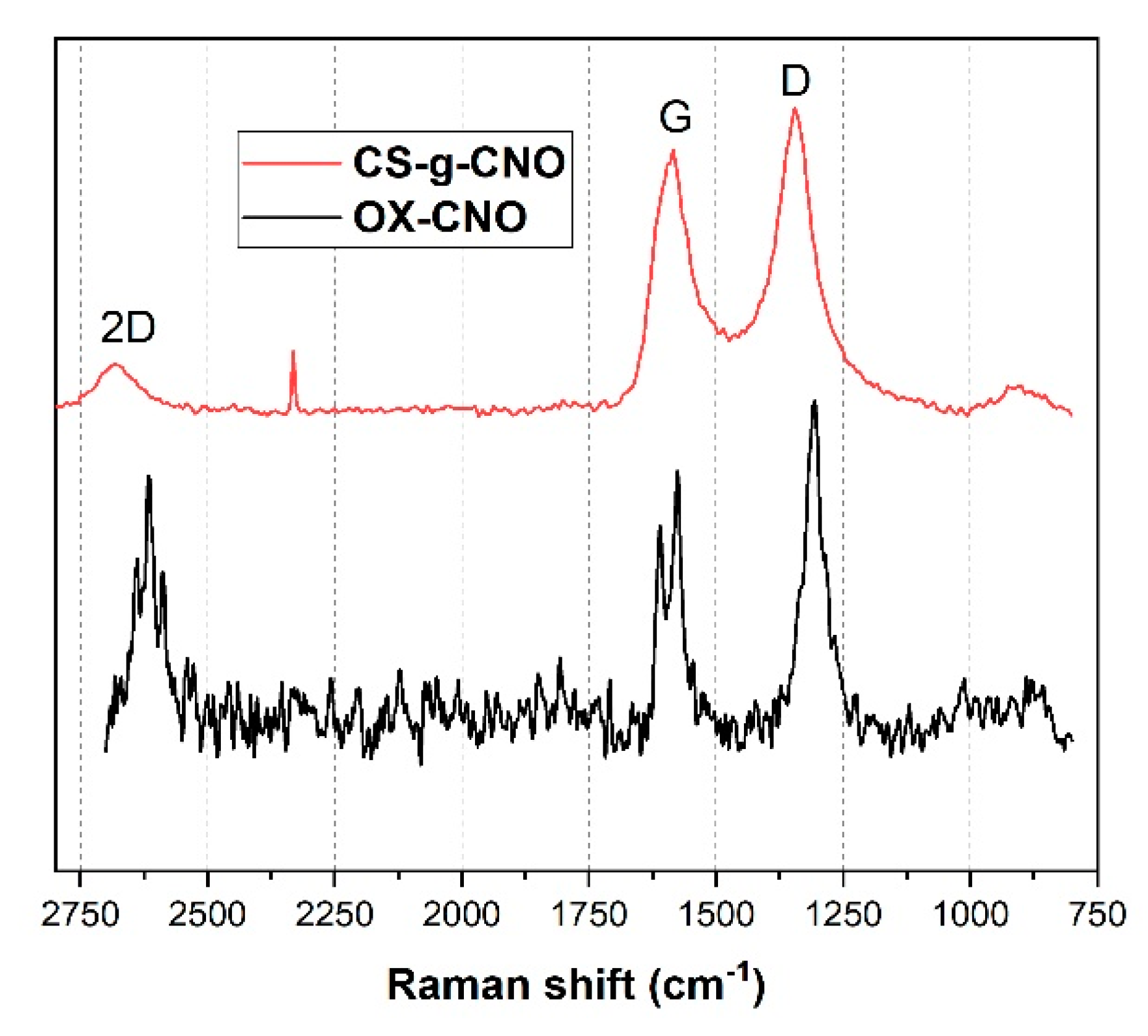
2.1.3. Raman Spectroscopy
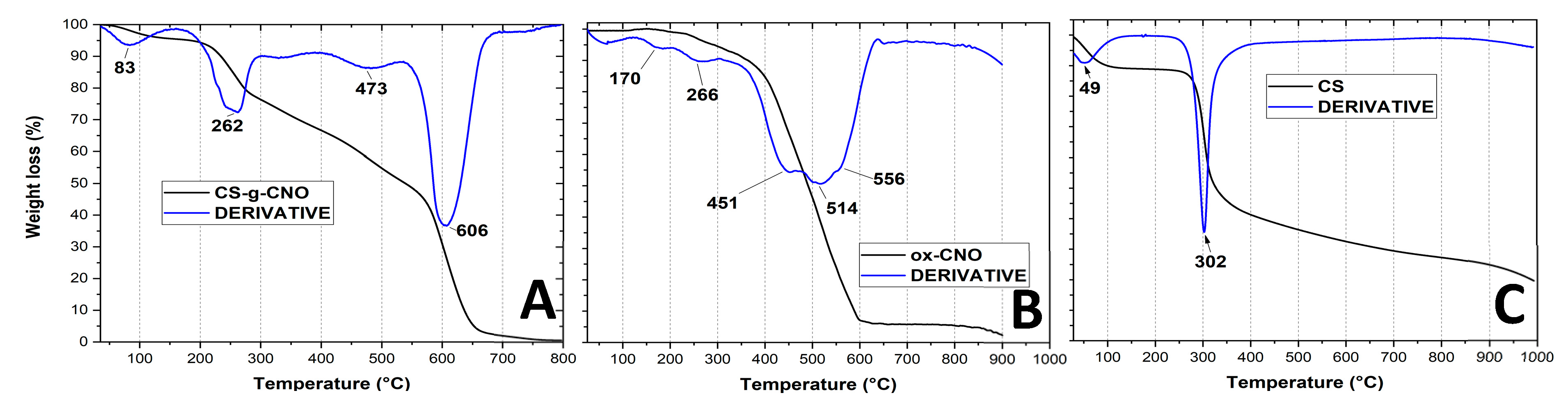
2.1.4. Thermogravimetric Analysis of CS-g-CNO
2.2. Film Characterization
2.2.1. Fourier-Transform Infrared Spectroscopy (FTIR)
2.2.2. X-ray Diffraction (XRD)
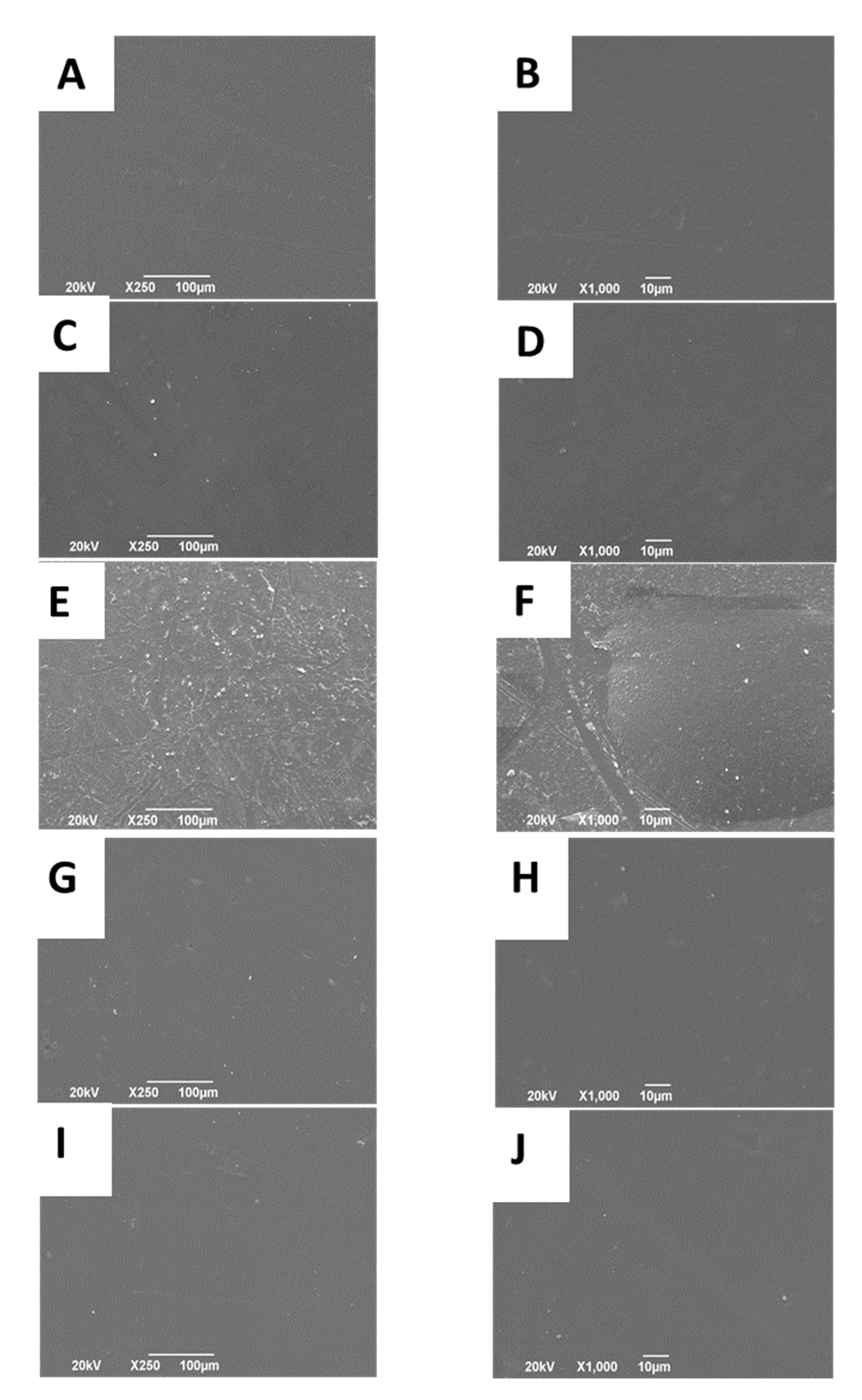
2.2.3. Scanning Electron Microscope (SEM)
2.2.4. The Tensile Strength of the Films
2.2.5. Thermal Studies
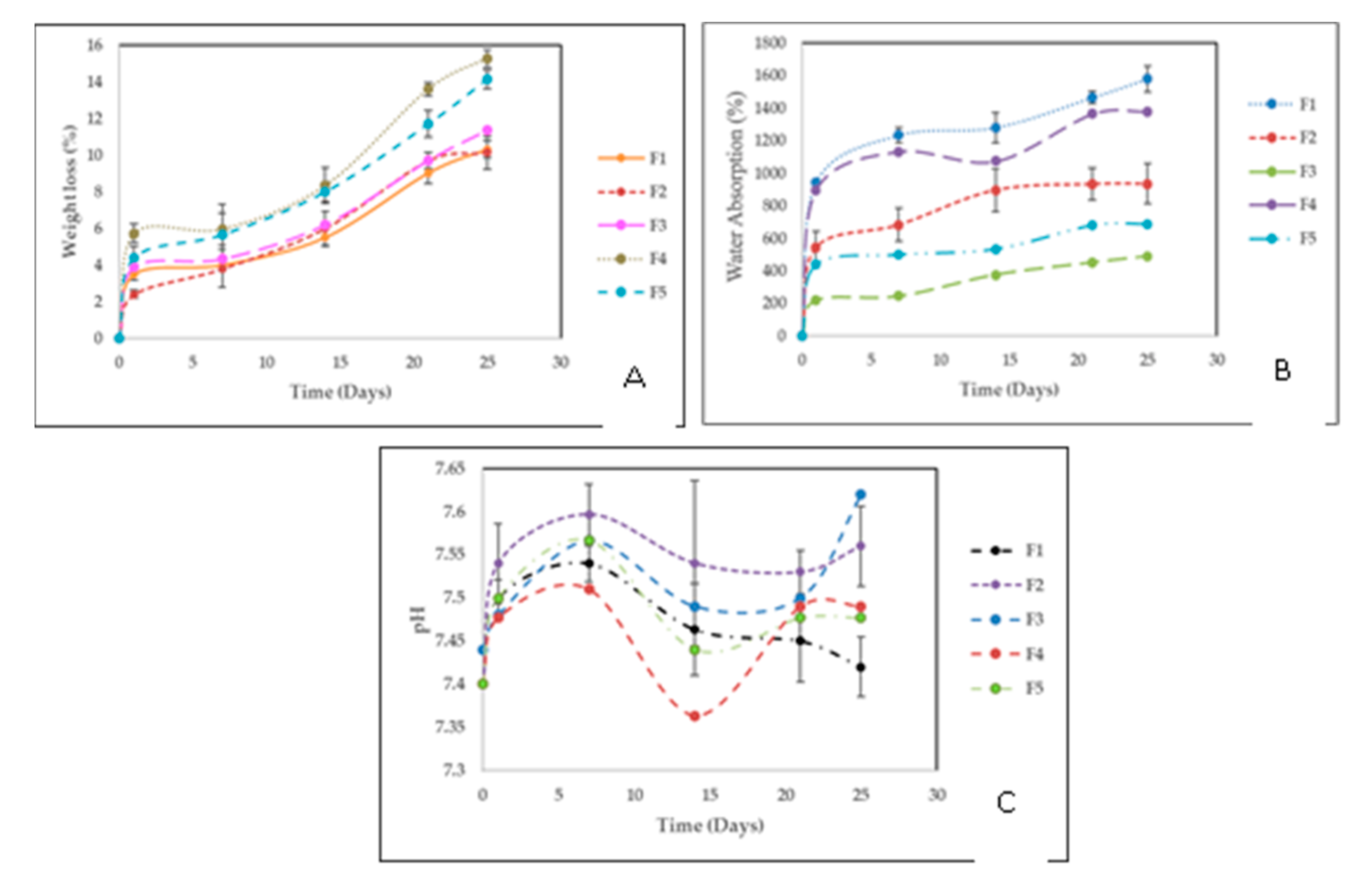
2.2.6. Degradation in a Simulated Biological Fluid (SBF)
Weight Loss
Water absorption
pH Changes
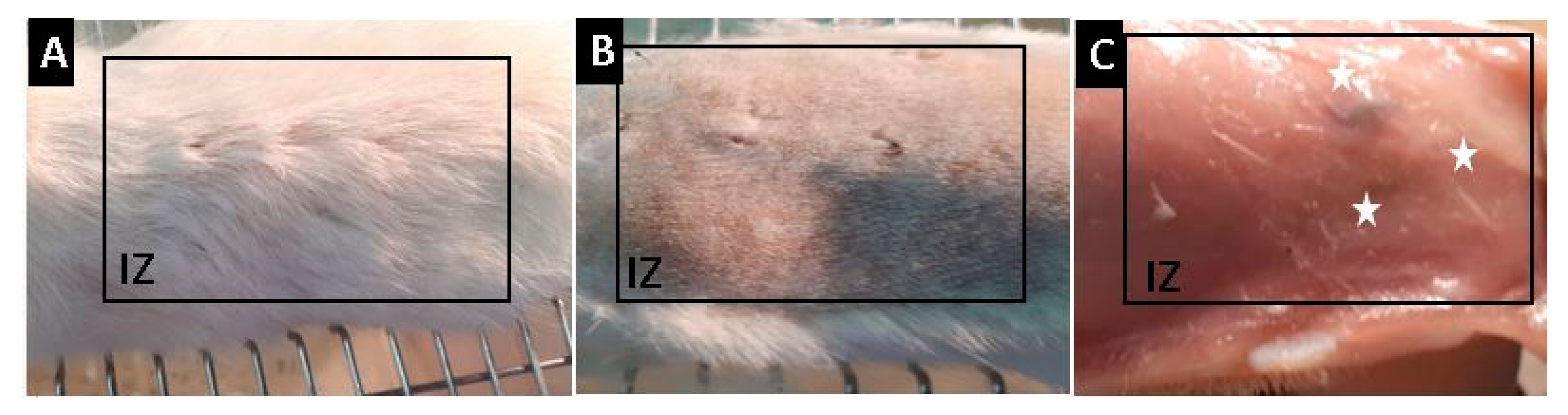
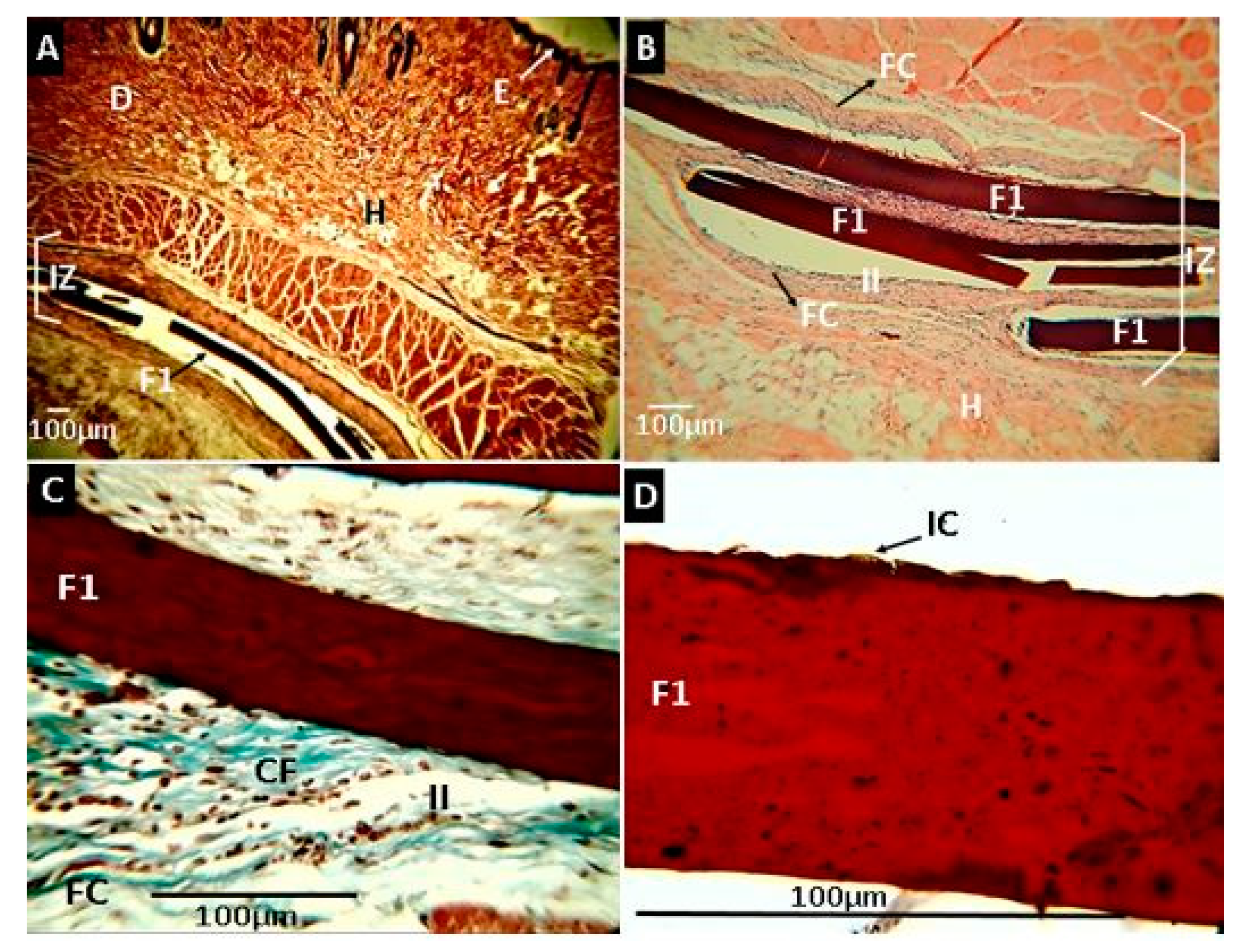
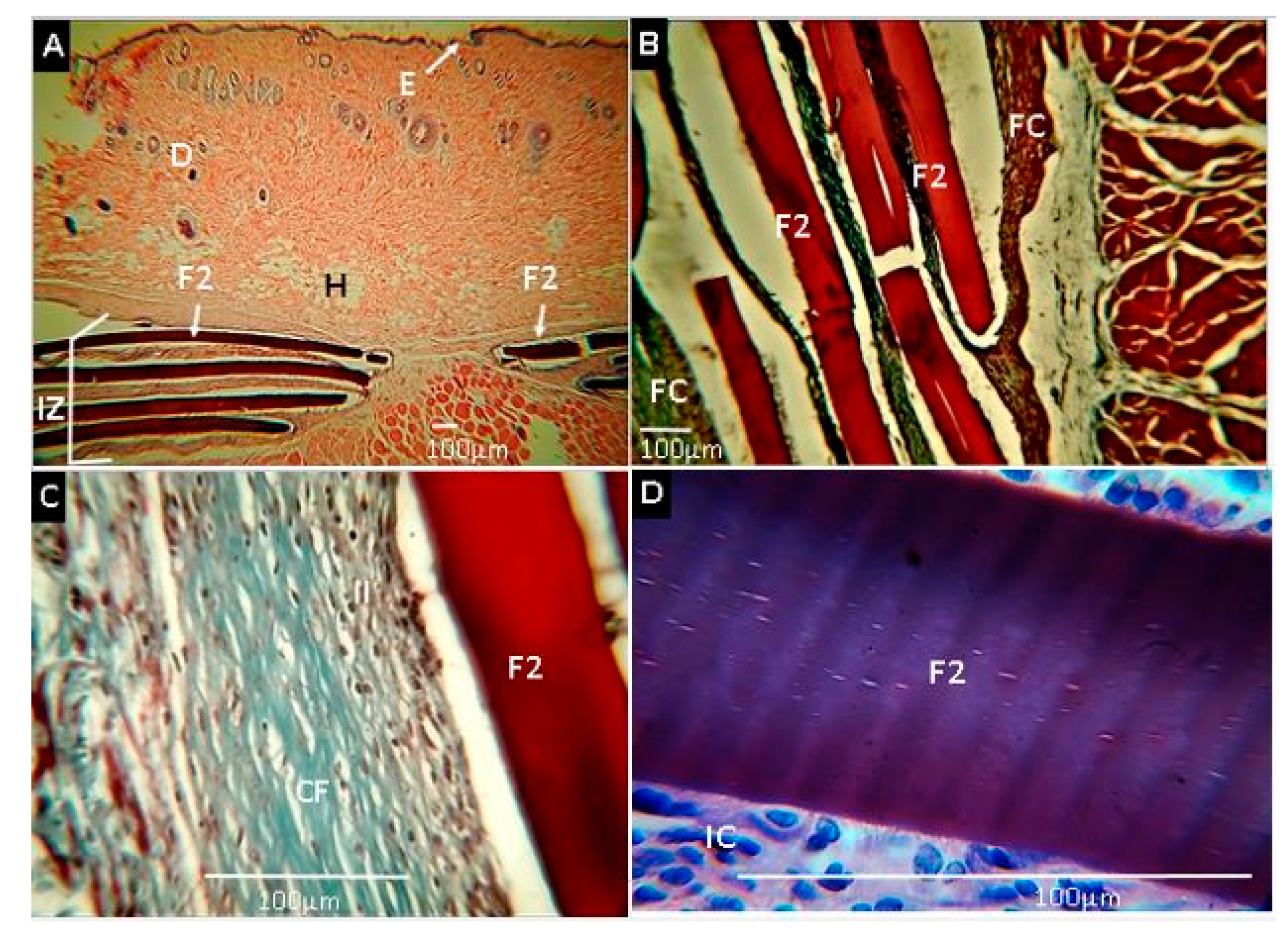
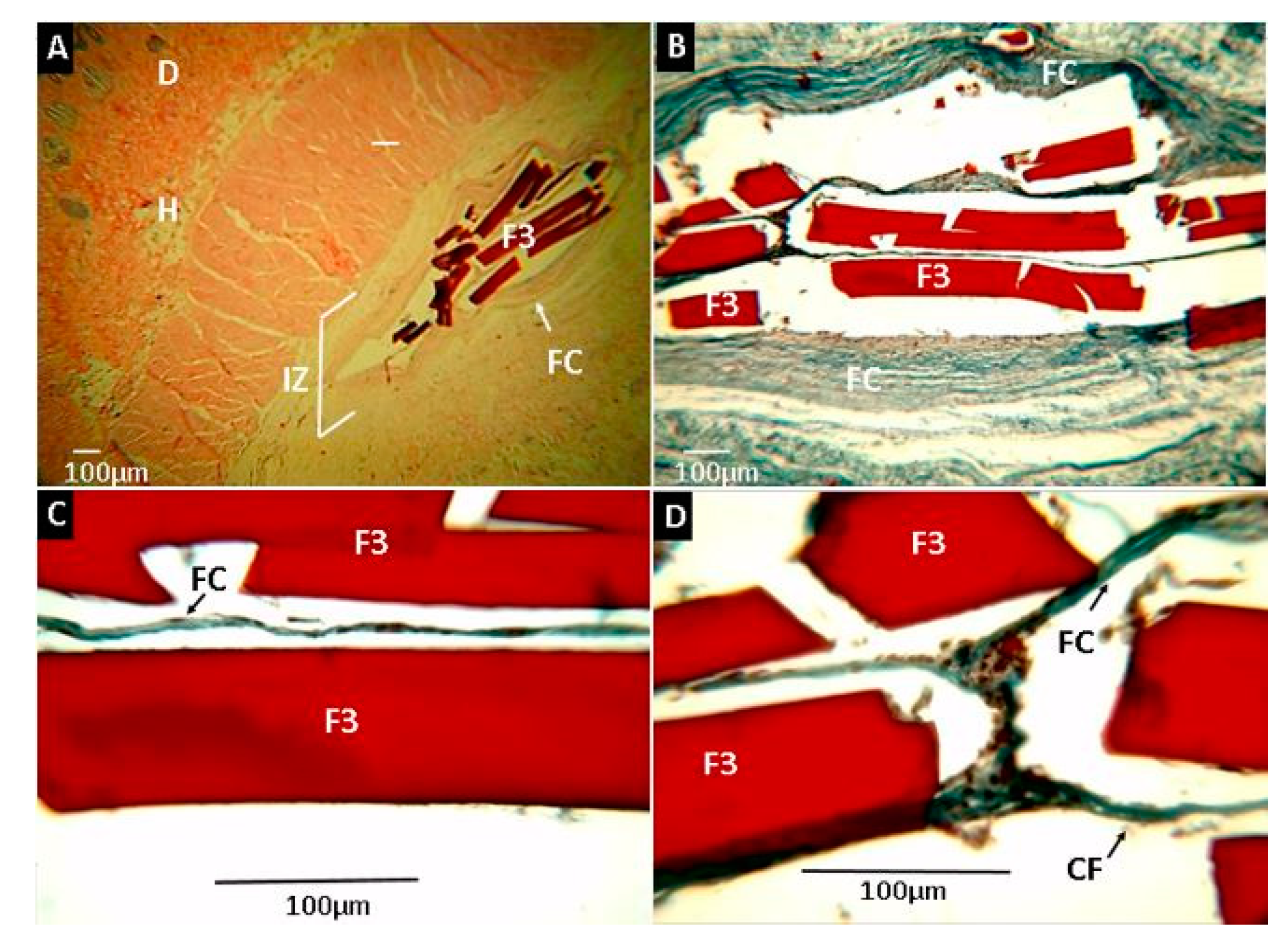
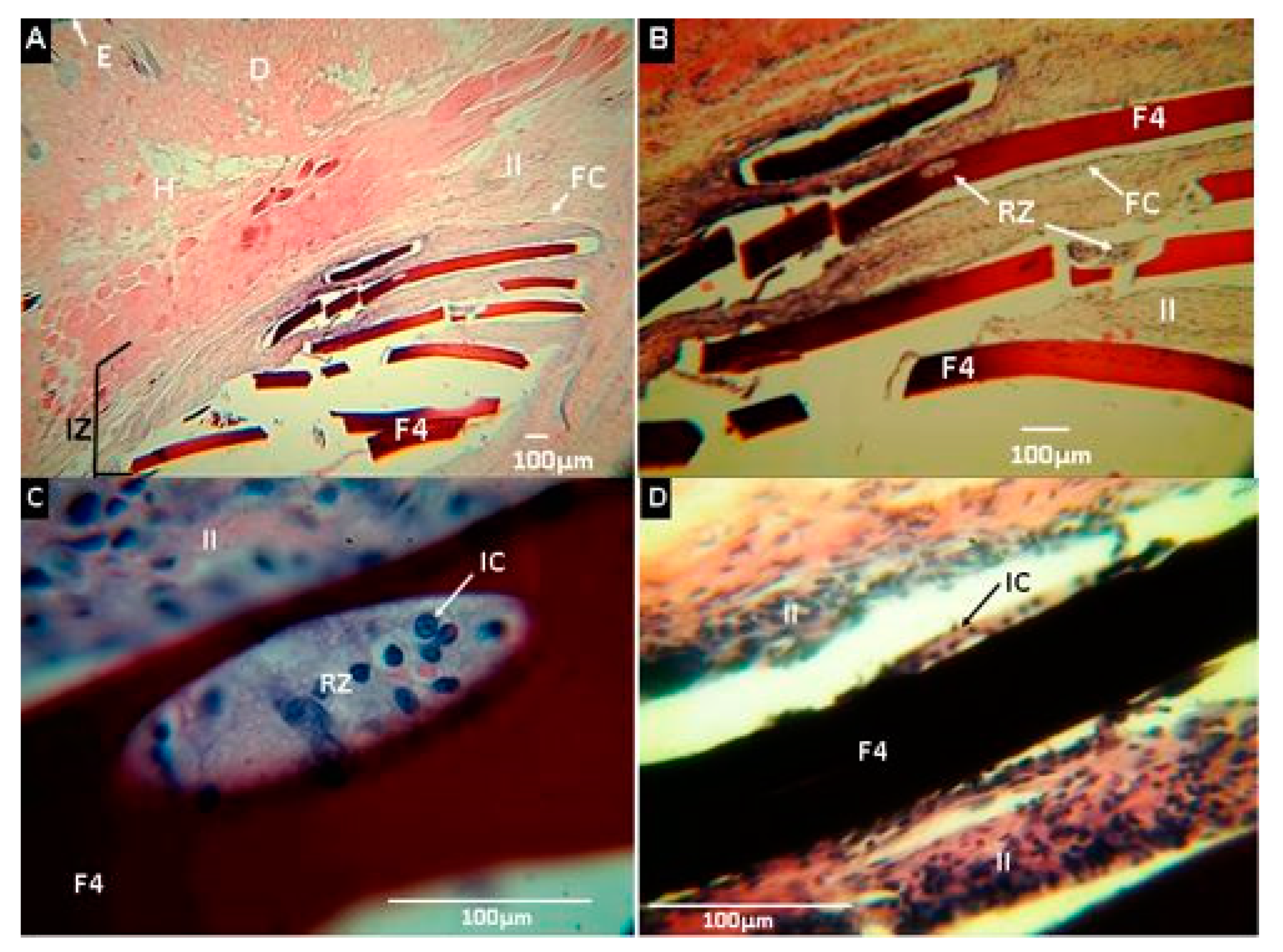
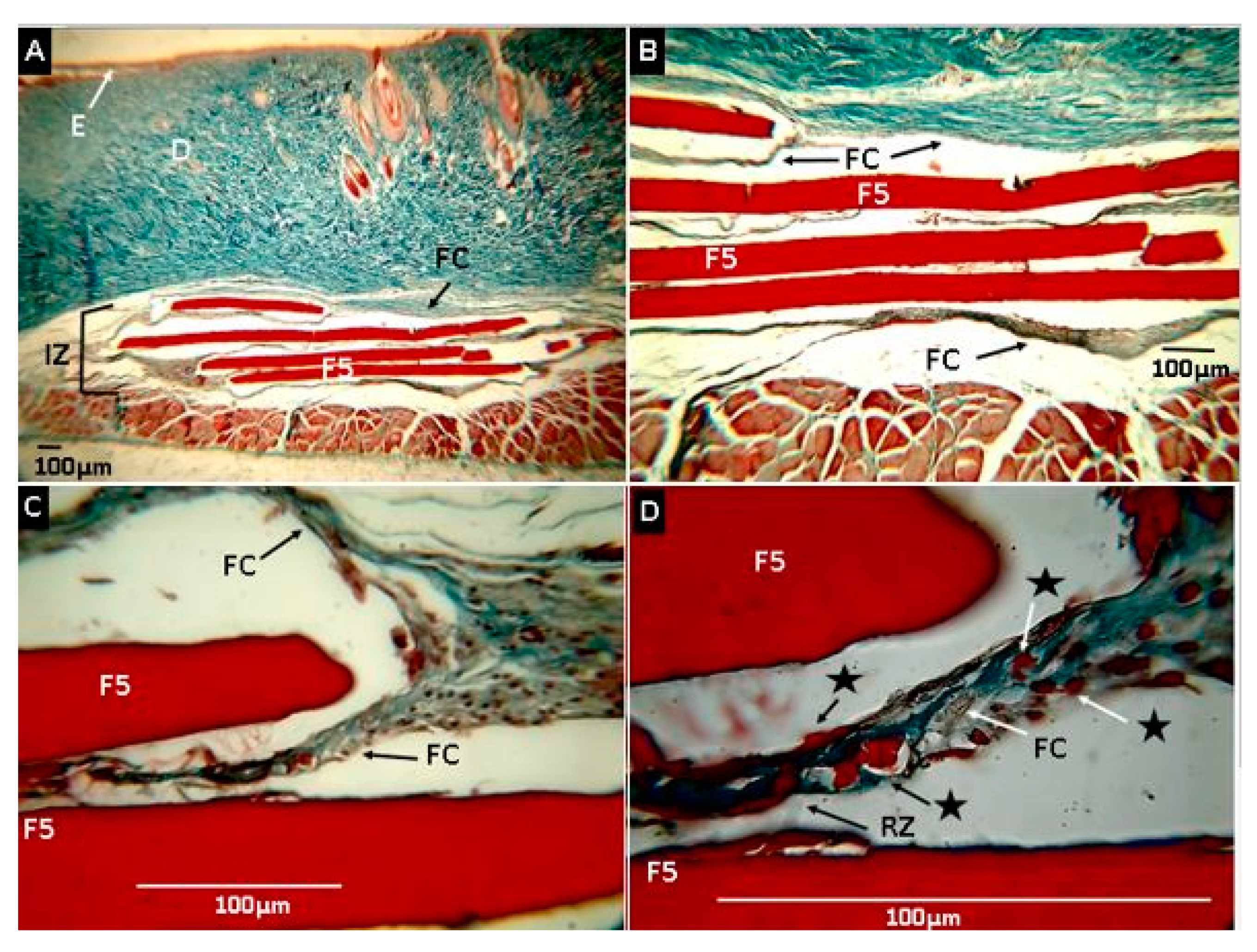
2.2.7. Biomodel In Vivo Tests
3. Materials and Methods
3.1. Materials
3.2. Methods
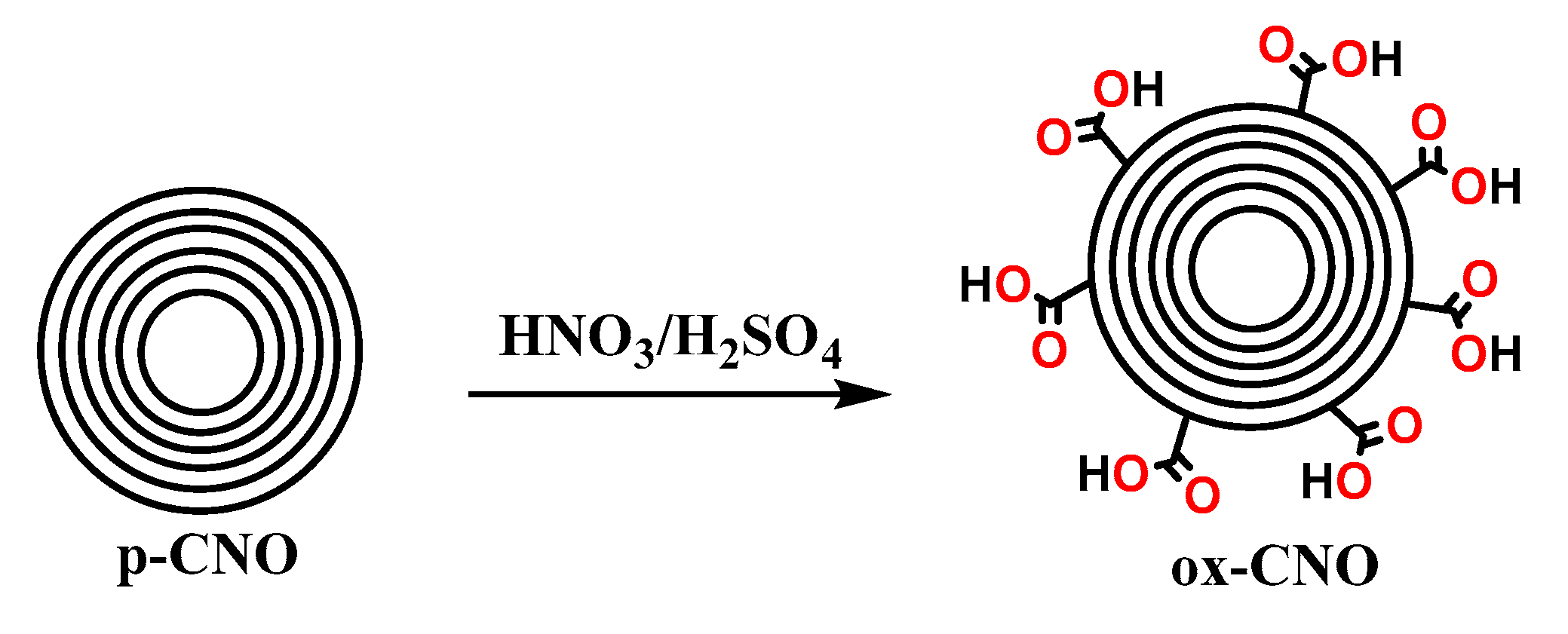
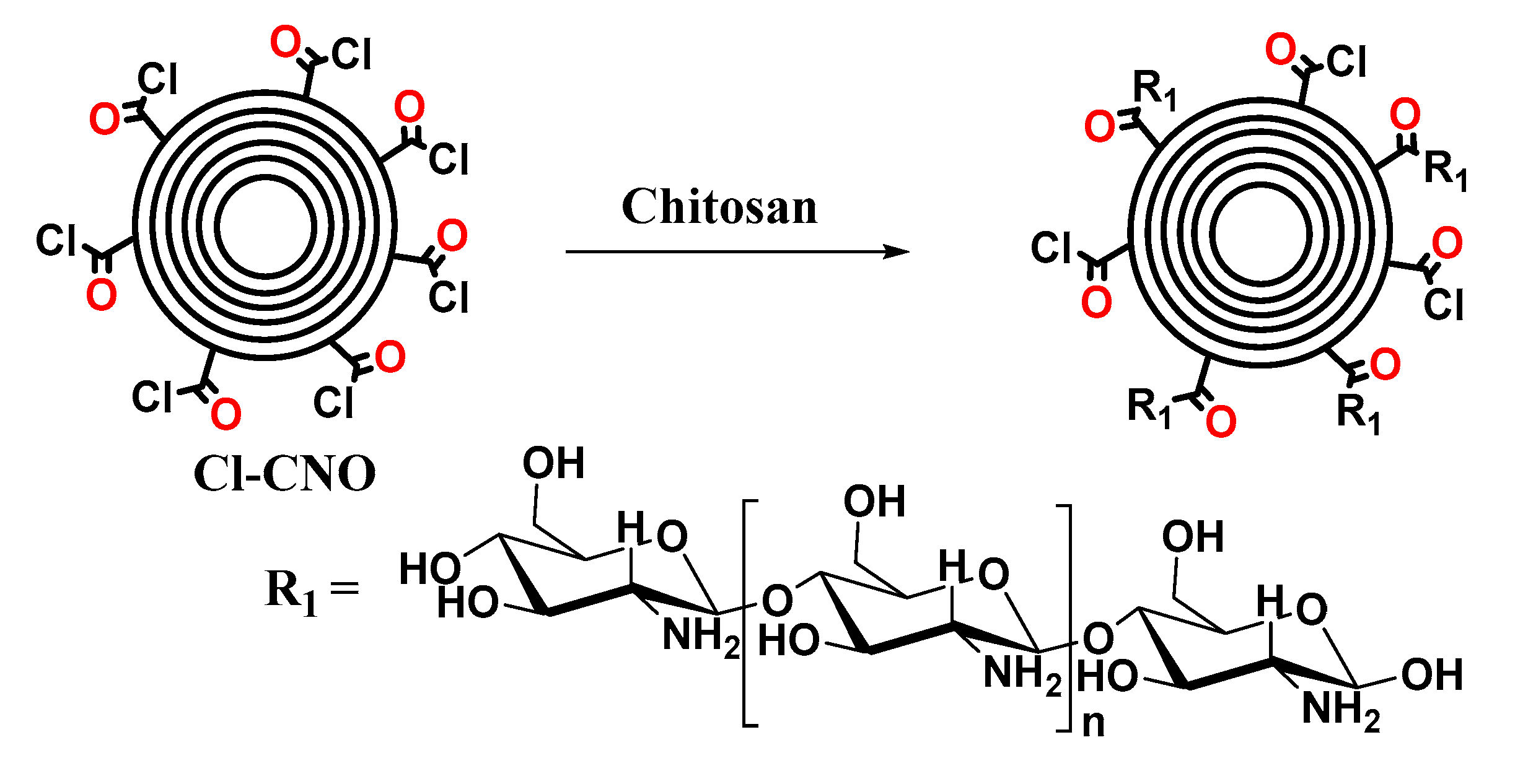
3.2.1. Synthesis of ox-CNO, Cl-CNO, and CS-g-CNO
3.2.2. Characterization of ox-CNO and CS-g-CNO
3.2.3. Nanocomposite Film Preparation
3.2.4. Film Characterization
Fourier-Transform Infrared Spectroscopy (FTIR)
X-ray Diffraction (XRD)
Scanning Electron Microscope (SEM)
Tensile Strength of Films
Thermal Studies
Degradation in Simulated Biological Fluid (SBF)
Biomodel In Vivo tests
Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hollister, S.J. Porous scaffold design for tissue engineering. Nat. Mater. 2005, 4, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ushida, T.; Tateishi, T. Scaffold design for tissue engineering. Macromol. Biosci. 2002, 2, 67–77. [Google Scholar] [CrossRef]
- Eivazzadeh-Keihan, R.; Maleki, A.; de la Guardia, M.; Bani, M.S.; Chenab, K.K.; Pashazadeh-Panahi, P.; Baradaran, B.; Mokhtarzadeh, A.; Hamblin, M.R. Carbon based nanomaterials for tissue engineering of bone: Building new bone on small black scaffolds: A review. J. Adv. Res. 2019, 18, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.S.; Atala, A. Carbon nanotube applications for tissue engineering. Biomaterials 2007, 28, 344–353. [Google Scholar] [CrossRef]
- Gerasimenko, A.Y.; Ichkitidze, L.P.; Podgaetsky, V.M.; Selishchev, S.V. Biomedical applications of promising nanomaterials with carbon nanotubes. Biomed. Eng. 2015, 48, 310–314. [Google Scholar] [CrossRef]
- Shin, S.R.; Li, Y.-C.; Jang, H.L.; Khoshakhlagh, P.; Akbari, M.; Nasajpour, A.; Zhang, Y.S.; Tamayol, A.; Khademhosseini, A. Graphene-based materials for tissue engineering. Adv. Drug Deliv. Rev. 2016, 105, 255–274. [Google Scholar] [CrossRef]
- Yu, X.; Tang, X.; Gohil, S.V.; Laurencin, C.T. Biomaterials for bone regenerative engineering. Adv. Healthc. Mater. 2015, 4, 1268–1285. [Google Scholar] [CrossRef]
- Stratton, S.; Shelke, N.B.; Hoshino, K.; Rudraiah, S.; Kumbar, S.G. Bioactive polymeric scaffolds for tissue engineering. Bioact. Mater. 2016, 1, 93–108. [Google Scholar] [CrossRef]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric scaffolds in tissue engineering application: A review. Int. J. Polym. Sci. 2011, 2011, 290602. [Google Scholar] [CrossRef]
- Venkatesan, J.; Bhatnagar, I.; Manivasagan, P.; Kang, K.-H.; Kim, S.-K. Alginate composites for bone tissue engineering: A review. Int. J. Biol. Macromol. 2015, 72, 269–281. [Google Scholar] [CrossRef]
- Saravanan, S.; Leena, R.S.; Selvamurugan, N. Chitosan based biocomposite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R.; Koushik, C.; Saravanan, S.; Moorthi, A.; Vairamani, M.; Selvamurugan, N. A novel injectable temperature-sensitive zinc doped chitosan/β-glycerophosphate hydrogel for bone tissue engineering. Int. J. Biol. Macromol. 2013, 54, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Shui, W.; Zhang, W.; Yin, L.; Nan, G.; Liao, Z.; Zhang, H.; Wang, N.; Wu, N.; Chen, X.; Wen, S. Characterization of scaffold carriers for BMP9-transduced osteoblastic progenitor cells in bone regeneration. J. Biomed. Mater. Res. Part. A 2014, 102, 3429–3438. [Google Scholar] [CrossRef]
- McFadden, T.M.; Duffy, G.P.; Allen, A.B.; Stevens, H.Y.; Schwarzmaier, S.M.; Plesnila, N.; Murphy, J.M.; Barry, F.P.; Guldberg, R.E.; O’brien, F.J. The delayed addition of human mesenchymal stem cells to pre-formed endothelial cell networks results in functional vascularization of a collagen–glycosaminoglycan scaffold in vivo. Acta Biomater. 2013, 9, 9303–9316. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Chang, Y.-H.; Li, K.-C.; Lu, C.-H.; Sung, L.-Y.; Yeh, C.-L.; Lin, K.-J.; Huang, S.-F.; Yen, T.-C.; Hu, Y.-C. The use of ASCs engineered to express BMP2 or TGF-β3 within scaffold constructs to promote calvarial bone repair. Biomaterials 2013, 34, 9401–9412. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, Y.; Liu, Q.; Gao, T.; Feng, J.Q.; Dechow, P.; D’Souza, R.N.; Qin, C.; Liu, X. Biomimetic engineering of nanofibrous gelatin scaffolds with noncollagenous proteins for enhanced bone regeneration. Tissue Eng. Part. A. 2013, 19, 1754–1763. [Google Scholar] [CrossRef]
- Saravanan, S.; Nethala, S.; Pattnaik, S.; Tripathi, A.; Moorthi, A.; Selvamurugan, N. Preparation, characterization and antimicrobial activity of a bio-composite scaffold containing chitosan/nano-hydroxyapatite/nano-silver for bone tissue engineering. Int. J. Biol. Macromol. 2011, 49, 188–193. [Google Scholar] [CrossRef]
- Khor, E.; Lim, L.Y. Implantable applications of chitin and chitosan. Biomaterials 2003, 24, 2339–2349. [Google Scholar] [CrossRef]
- Soundarya, S.P.; Menon, A.H.; Chandran, S.V.; Selvamurugan, N. Bone tissue engineering: Scaffold preparation using chitosan and other biomaterials with different design and fabrication techniques. Int. J. Biol. Macromol. 2018, 119, 1228–1239. [Google Scholar] [CrossRef]
- Dhivya, S.; Keshav Narayan, A.; Logith Kumar, R.; Viji Chandran, S.; Vairamani, M.; Selvamurugan, N. Proliferation and differentiation of mesenchymal stem cells on scaffolds containing chitosan, calcium polyphosphate and pigeonite for bone tissue engineering. Cell Prolif. 2018, 51, e12408. [Google Scholar] [CrossRef]
- Shamekhi, M.A.; Mirzadeh, H.; Mahdavi, H.; Rabiee, A.; Mohebbi-Kalhori, D.; Eslaminejad, M.B. Graphene oxide containing chitosan scaffolds for cartilage tissue engineering. Int. J. Biol. Macromol. 2019, 127, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Kashi, M.; Baghbani, F.; Moztarzadeh, F.; Mobasheri, H.; Kowsari, E. Green synthesis of degradable conductive thermosensitive oligopyrrole/chitosan hydrogel intended for cartilage tissue engineering. Int. J. Biol. Macromol. 2018, 107, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Manzoor, K.; Ahmad, S.; Akram, N.; Ikram, S. Chitosan-based nanocomposites for cardiac, liver, and wound healing applications. In Applications of Nanocomposite Materials in Orthopedics; Elsevier: Amsterdam, The Netherlands, 2019; pp. 253–262. [Google Scholar]
- Wu, G.; Deng, X.; Song, J.; Chen, F. Enhanced biological properties of biomimetic apatite fabricated polycaprolactone/chitosan nanofibrous bio-composite for tendon and ligament regeneration. J. Photochem. Photobiol. B Biol. 2018, 178, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Yang, L.; Ye, C.; Zhang, W.; Ran, J.; Xue, D.; Wang, Z.; Pan, Z.; Hu, Q. An asymmetric chitosan scaffold for tendon tissue engineering: In vitro and in vivo evaluation with rat tendon stem/progenitor cells. Acta Biomater. 2018, 73, 377–387. [Google Scholar] [CrossRef]
- Qasim, S.; Zafar, M.; Najeeb, S.; Khurshid, Z.; Shah, A.; Husain, S.; Rehman, I. Electrospinning of chitosan-based solutions for tissue engineering and regenerative medicine. Int. J. Mol. Sci. 2018, 19, 407. [Google Scholar] [CrossRef]
- González-Quevedo, D.; Martínez-Medina, I.; Campos, A.; Campos, F.; Carriel, V. Tissue engineering strategies for the treatment of tendon injuries: A systematic review and meta-analysis of animal models. Bone Jt. Res. 2018, 7, 318–324. [Google Scholar] [CrossRef]
- Ueno, H.; Mori, T.; Fujinaga, T. Topical formulations and wound healing applications of chitosan. Adv. Drug Deliv. Rev. 2001, 52, 105–115. [Google Scholar] [CrossRef]
- Ratner, B.D.; Hoffman, A.S.; Schoen, F.J.; Lemons, J.E. Biomaterials Science: An Introduction to Materials in Medicine; Elsevier: Amsterdam, The Netherlands, 2004; ISBN 008047036X. [Google Scholar]
- Thakur, V.K.; Voicu, S.I. Recent advances in cellulose and chitosan based membranes for water purification: A concise review. Carbohydr. Polym. 2016, 146, 148–165. [Google Scholar] [CrossRef]
- He, Y.; Miao, J.; Chen, S.; Zhang, R.; Zhang, L.; Tang, H.; Yang, H. Preparation and characterization of a novel positively charged composite hollow fiber nanofiltration membrane based on chitosan lactate. Rsc Adv. 2019, 9, 4361–4369. [Google Scholar] [CrossRef]
- Medina, V.F.; Griggs, C.S.; Mattei-Sosa, J.; Petery, B.; Gurtowski, L. Advanced Filtration Membranes Using Chitosan and Graphene Oxide. U.S. Patent Application 20190039026, 7 February 2019. [Google Scholar]
- Sun, T.; Guo, X.; Zhong, R.; Ma, L.; Li, H.; Gu, Z.; Guan, J.; Tan, H.; You, C.; Tian, M. Interactions of oligochitosan with blood components. Int. J. Biol. Macromol. 2019, 124, 304–313. [Google Scholar] [CrossRef]
- Heise, K.; Hobisch, M.; Sacarescu, L.; Maver, U.; Hobisch, J.; Reichelt, T.; Sega, M.; Fischer, S.; Spirk, S. Low-molecular-weight sulfonated chitosan as template for anticoagulant nanoparticles. Int. J. Nanomed. 2018, 13, 4881–4894. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, T.; Zhong, R.; Ma, L.; You, C.; Tian, M.; Li, H.; Wang, C. Effects of chitosan oligosaccharides on human blood components. Front. Pharmacol. 2018, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Dimassi, S.; Tabary, N.; Chai, F.; Blanchemain, N.; Martel, B. Sulfonated and sulfated chitosan derivatives for biomedical applications: A review. Carbohydr. Polym. 2018, 202, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Al-Kinani, A.A.; Arshad, M.S.; Singh, N.; van der Merwe, S.M.; Chang, M.-W.; Alany, R.G.; Ahmad, Z. Engineering and development of chitosan-based Nanocoatings for Ocular Contact Lenses. J. Pharm. Sci. 2019, 108, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ahmed, S. A review on chitosan and its nanocomposites in drug delivery. Int. J. Biol. Macromol. 2018, 109, 273–286. [Google Scholar] [CrossRef]
- Ahsan, S.M.; Thomas, M.; Reddy, K.K.; Sooraparaju, S.G.; Asthana, A.; Bhatnagar, I. Chitosan as biomaterial in drug delivery and tissue engineering. Int. J. Biol. Macromol. 2018, 110, 97–109. [Google Scholar] [CrossRef]
- Gomillion, C.T. Assessing the potential of chitosan/polylactide nanoparticles for delivery of therapeutics for triple-negative breast cancer treatment. Regen. Eng. Transl. Med. 2019, 5, 61–73. [Google Scholar]
- Raval, R.; Rangnekar, R.H.; Raval, K. Optimization of chitosan nanoparticles synthesis and its applications in fatty acid absorption. In Materials, Energy and Environment Engineering; Springer: Berlin/Heidelberg, Germany; pp. 253–256.
- Berkland, C.; Qian, J.; Sullivan, B.P. Micelle Sequestering Polymers. U.S. Patent Application 20150216896, 6 August 2015. [Google Scholar]
- Hamedi, H.; Moradi, S.; Hudson, S.M.; Tonelli, A.E. Chitosan based hydrogels and their applications for drug delivery in wound dressings: A review. Carbohydr. Polym. 2018, 199, 445–460. [Google Scholar] [CrossRef]
- Mohebbi, S.; Nezhad, M.N.; Zarrintaj, P.; Jafari, S.H.; Gholizadeh, S.S.; Saeb, M.R.; Mozafari, M. Chitosan in biomedical engineering: A critical review. Curr. Stem Cell Res. Ther. 2019, 14, 93–116. [Google Scholar] [CrossRef]
- Cazón, P.; Vázquez, M. Applications of Chitosan as Food Packaging Materials. In Sustainable Agriculture Reviews 36; Springer: Berlin/Heidelberg, Germany, 2019; pp. 81–123. [Google Scholar]
- Wang, H.; Qian, J.; Ding, F. Emerging chitosan-based films for food packaging applications. J. Agric. Food Chem. 2018, 66, 395–413. [Google Scholar] [CrossRef]
- HPS, A.K.; Saurabh, C.K.; Adnan, A.S.; Fazita, M.R.N.; Syakir, M.I.; Davoudpour, Y.; Rafatullah, M.; Abdullah, C.K.; Haafiz, M.K.M.; Dungani, R. A review on chitosan-cellulose blends and nanocellulose reinforced chitosan biocomposites: Properties and their applications. Carbohydr. Polym. 2016, 150, 216–226. [Google Scholar]
- Koosha, M.; Mirzadeh, H.; Shokrgozar, M.A.; Farokhi, M. Nanoclay-reinforced electrospun chitosan/PVA nanocomposite nanofibers for biomedical applications. Rsc Adv. 2015, 5, 10479–10487. [Google Scholar] [CrossRef]
- Umeyama, T.; Imahori, H. Photofunctional hybrid nanocarbon materials. J. Phys. Chem. C 2012, 117, 3195–3209. [Google Scholar] [CrossRef]
- Rettenbacher, A.S.; Elliott, B.; Hudson, J.S.; Amirkhanian, A.; Echegoyen, L. Preparation and functionalization of multilayer fullerenes (carbon nano-onions). Chem. Eur. J. 2006, 12, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Igarashi, M.; Kaito, T. Study on solid lubricant properties of carbon onions produced by heat treatment of diamond clusters or particles. Tribol. Int. 2004, 37, 899–905. [Google Scholar] [CrossRef]
- Ibáñez-Redín, G.; Furuta, R.H.M.; Wilson, D.; Shimizu, F.M.; Materon, E.M.; Arantes, L.M.R.B.; Melendez, M.E.; Carvalho, A.L.; Reis, R.M.; Chaur, M.N. Screen-printed interdigitated electrodes modified with nanostructured carbon nano-onion films for detecting the cancer biomarker CA19-9. Mater. Sci. Eng. C 2019, 99, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Stilwell, J.; Zhang, T.; Elboudwarej, O.; Jiang, H.; Selegue, J.P.; Cooke, P.A.; Gray, J.W.; Chen, F.F. Molecular characterization of the cytotoxic mechanism of multiwall carbon nanotubes and nano-onions on human skin fibroblast. Nano Lett. 2005, 5, 2448–2464. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Grande, C.D.; Rodrigues, D.F. Biodegradation of graphene oxide-polymer nanocomposite films in wastewater. Environ. Sci. Nano 2017, 4, 1808–1816. [Google Scholar] [CrossRef]
- Grande, C.D.; Mangadlao, J.; Fan, J.; De Leon, A.; Delgado-Ospina, J.; Rojas, J.G.; Rodrigues, D.F.; Advincula, R. Chitosan cross-linked graphene oxide nanocomposite films with antimicrobial activity for application in food industry. Macromol. Symp. 2017, 374, 1600114. [Google Scholar] [CrossRef]
- Ruiz, S.; Tamayo, A.J.; Delgado Ospina, J.; Navia Porras, P.D.; Valencia Zapata, E.M.; Mina Hernandez, H.J.; Valencia, H.C.; Zuluaga, F.; Grande Tovar, D.C. Antimicrobial films based on nanocomposites of chitosan/poly(vinyl alcohol)/graphene oxide for biomedical applications. Biomolecules 2019, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- López Tenorio, D.; Valencia, H.C.; Valencia, C.; Zuluaga, F.; Valencia, E.M.; Mina, H.J.; Grande Tovar, D.C. Evaluation of the biocompatibility of CS-Graphene oxide compounds in vivo. Int. J. Mol. Sci. 2019, 20, 1572. [Google Scholar] [CrossRef]
- Valencia, C.; Valencia, C.; Zuluaga, F.; Valencia, M.; Mina, J.; Grande-Tovar, C. Synthesis and application of scaffolds of chitosan-graphene oxide by the freeze-drying method for tissue regeneration. Molecules 2018, 23, 2651. [Google Scholar] [CrossRef]
- Tamayo Marín, A.J.; Londoño, R.S.; Delgado, J.; Navia Porras, P.D.; Valencia Zapata, E.M.; Mina Hernandez, H.J.; Valencia, H.C.; Grande Tovar, D.C. Biocompatible and antimicrobial electrospun membranes based on nanocomposites of chitosan/poly (vinyl alcohol)/graphene oxide. Int. J. Mol. Sci. 2019, 20, 2987. [Google Scholar] [CrossRef] [PubMed]
- Valencia Zapata, E.M.; Mina Hernandez, H.J.; Grande Tovar, D.C.; Valencia Llano, H.C.; Diaz Escobar, A.J.; Vázquez-Lasa, B.; San Román, J.; Rojo, L. Novel bioactive and antibacterial acrylic bone cement nanocomposites modified with graphene oxide and chitosan. Int. J. Mol. Sci. 2019, 20, 2938. [Google Scholar] [CrossRef] [PubMed]
- Grande Tovar, C.D.; Castro, J.I.; Valencia, C.H.; Navia Porras, D.P.; Hernandez, M.; Herminsul, J.; Valencia, M.E.; Velásquez, J.D.; Chaur, M.N. Preparation of chitosan/poly (vinyl alcohol) nanocomposite films incorporated with oxidized carbon nano-onions (multi-layer fullerenes) for tissue-engineering applications. Biomolecules 2019, 9, 684. [Google Scholar] [CrossRef]
- Wu, Z.; Feng, W.; Feng, Y.; Liu, Q.; Xu, X.; Sekino, T.; Fujii, A.; Ozaki, M. Preparation and characterization of chitosan-grafted multiwalled carbon nanotubes and their electrochemical properties. Carbon 2007, 45, 1212–1218. [Google Scholar] [CrossRef]
- Osswald, S.; Havel, M.; Gogotsi, Y. Monitoring oxidation of multiwalled carbon nanotubes by Raman spectroscopy. J. Raman Spectrosc. 2007, 38, 728–736. [Google Scholar] [CrossRef]
- Shriner, R.L.; Fuson, R.C.; Curtin, D.Y. The systematic identification of organic compounds; John Wiley Sons: New York, NY, USA, 1948; pp. 202–207. [Google Scholar]
- Ke, G.; Guan, W.; Tang, C.; Guan, W.; Zeng, D.; Deng, F. Covalent functionalization of multiwalled carbon nanotubes with a low molecular weight chitosan. Biomacromolecules 2007, 8, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Mallakpour, S.; Zadehnazari, A. A facile, efficient, and rapid covalent functionalization of multi-walled carbon nanotubes with natural amino acids under microwave irradiation. Prog. Org. Coat. 2014, 77, 679–684. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Mukherjee, A.; Chakraborty, S.; Kang, J.; Loos, P.J.; Kelly, K.F.; Schmidt, H.K.; Billups, W.E. Exfoliated soluble graphite. Carbon 2009, 47, 2945–2949. [Google Scholar] [CrossRef]
- Bustos-Ramírez, K.; Martínez-Hernández, A.L.; Martínez-Barrera, G.; Icaza, M.D.; Castaño, V.M.; Velasco-Santos, C. Covalently bonded chitosan on graphene oxide via redox reaction. Materials 2013, 6, 911–926. [Google Scholar] [CrossRef]
- Cioffi, C.T.; Palkar, A.; Melin, F.; Kumbhar, A.; Echegoyen, L.; Melle-Franco, M.; Zerbetto, F.; Rahman, G.M.A.; Ehli, C.; Sgobba, V. A carbon nano-onion–ferrocene donor–acceptor system: Synthesis, characterization and properties. Chem. Eur. J. 2009, 15, 4419–4427. [Google Scholar] [CrossRef] [PubMed]
- Carson, L.; Kelly-Brown, C.; Stewart, M.; Oki, A.; Regisford, G.; Luo, Z.; Bakhmutov, V.I. Synthesis and characterization of chitosan–carbon nanotube composites. Mater. Lett. 2009, 63, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Srinivasa, P.C.; Ramesh, M.N.; Kumar, K.R.; Tharanathan, R.N. Properties and sorption studies of chitosan–polyvinyl alcohol blend films. Carbohydr. Polym. 2003, 53, 431–438. [Google Scholar] [CrossRef]
- Pandele, A.M.; Ionita, M.; Crica, L.; Dinescu, S.; Costache, M.; Iovu, H. Synthesis, characterization, and in vitro studies of graphene oxide/chitosan-polyvinyl alcohol films. Carbohydr. Polym. 2014, 102, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.-T.; Gong, J.; Gu, X.-H.; Kim, H.-Y.; Dong, J.; Shen, X.-Y. Fabrication and characterization of poly (vinyl alcohol)/chitosan blend nanofibers produced by electrospinning method. Carbohydr. Polym. 2007, 67, 403–409. [Google Scholar] [CrossRef]
- Lu, L.; Peng, F.; Jiang, Z.; Wang, J. Poly(vinyl alcohol)/chitosan blend membranes for pervaporation of benzene/cyclohexane mixtures. J. Appl. Polym. Sci. 2006, 101, 167–173. [Google Scholar] [CrossRef]
- Yadav, I.; Nayak, S.K.; Rathnam, V.S.S.; Banerjee, I.; Ray, S.S.; Anis, A.; Pal, K. Reinforcing effect of graphene oxide reinforcement on the properties of poly (vinyl alcohol) and carboxymethyl tamarind gum based phase-separated film. J. Mech. Behav. Biomed. Mater. 2018, 81, 61–71. [Google Scholar] [CrossRef]
- Yang, X.; Tu, Y.; Li, L.; Shang, S.; Tao, X. Well-dispersed chitosan/graphene oxide nanocomposites. Acs Appl. Mater. Interfaces 2010, 2, 1707–1713. [Google Scholar] [CrossRef]
- Espigares, I.; Elvira, C.; Mano, J.F.; Vázquez, B.; San Román, J.; Reis, R.L. New partially degradable and bioactive acrylic bone cements based on starch blends and ceramic fillers. Biomaterials 2002, 23, 1883–1895. [Google Scholar] [CrossRef]
- Herath, H.M.T.U.; Di Silvio, L.; Evans, J.R.G. Biological evaluation of solid freeformed, hard tissue scaffolds for orthopedic applications. J. Appl. Biomater. Biomech. 2010, 8, 89–96. [Google Scholar] [PubMed]
- Figueira Maldonado, E. Degradación hidrolítica a diferentes pH de un material compuesto Poli(ácido láctico)/Quitosano, Proyecto de grado; Universidad Simón Bolívar, Sartenejas: Caracas, Venezuela, 2008. [Google Scholar]
- Depan, D.; Shah, J.S.; Misra, R.D.K. Degradation mechanism and increased stability of chitosan-based hybrid scaffolds cross-linked with nanostructured carbon: Process-structure-functional property relationship. Polym. Degrad. Stab. 2013, 98, 2331–2339. [Google Scholar] [CrossRef]
- Maruyama, M.; Ito, M. In vitro properties of a chitosan-bonded self-hardening paste with hydroxyapatite granules. J. Biomed. Mater. Res. 1996, 32, 527–532. [Google Scholar] [CrossRef]
- Tomihata, K.; Ikada, Y. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 1997, 18, 567–575. [Google Scholar] [CrossRef]
- Pella, M.C.G.; Lima-Tenório, M.K.; Tenorio-Neto, E.T.; Guilherme, M.R.; Muniz, E.C.; Rubira, A.F. Chitosan-based hydrogels: From preparation to biomedical applications. Carbohydr. Polym. 2018, 196, 233–245. [Google Scholar] [CrossRef]
- Fujita, M.; Ishihara, M.; Simizu, M.; Obara, K.; Ishizuka, T.; Saito, Y.; Yura, H.; Morimoto, Y.; Takase, B.; Matsui, T. Vascularization in vivo caused by the controlled release of fibroblast growth factor-2 from an injectable chitosan/non-anticoagulant heparin hydrogel. Biomaterials 2004, 25, 699–706. [Google Scholar] [CrossRef]
- Sok, V.; Fragoso, A. Preparation and characterization of alkaline phosphatase, horseradish peroxidase, and glucose oxidase conjugates with carboxylated carbon nano-onions. Prep. Biochem. Biotechnol. 2018, 48, 136–143. [Google Scholar] [CrossRef]
- Vatanpour, V.; Safarpour, M.; Khataee, A.; Zarrabi, H.; Yekavalangi, M.E.; Kavian, M. A thin film nanocomposite reverse osmosis membrane containing amine-functionalized carbon nanotubes. Sep. Purif. Technol. 2017, 184, 135–143. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * | ox-CNO (wt %) | CS-g-CNO (wt %) | Td3% (°C) | Tg (°C) | Young’s Modulus (GPa) ** | Tensile Strength (MPa) ** |
|---|---|---|---|---|---|---|
| F1 | 0 | 0 | 67.3 | 19.9 | 3.69 ± 0.45 a | 38.2 ± 2.88 a |
| F2 | 0.50 | 0 | 50.8 | 80.2 | 3.60 ± 0.74 a | 38.3 ± 3.43 a |
| F3 | 1.00 | 0 | 85.0 | 79.3 | 3.67 ± 0.77 a | 43.1 ± 3.23 a |
| F4 | 0 | 0.50 | 78.3 | 51.6 | 1.36 ± 0.26 b | 23.7 ± 3.42 b |
| F5 | 0 | 1.00 | 79.9 | 38.3 | 2.26 ± 0.15 c | 28.0 ± 2.27 b |
| Component | F1 | F2 | F3 | F4 | F5 |
|---|---|---|---|---|---|
| CS (%) | 30.00 | 29.50 | 29.00 | 29.50 | 29.00 |
| PVA (%) | 70.00 | 70.00 | 70.00 | 70.00 | 70.00 |
| ox-CNO (%) | 0 | 0.50 | 1.00 | 0 | 0 |
| CS-g-CNO (%) | 0 | 0 | 0 | 0.50 | 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grande Tovar, C.D.; Castro, J.I.; Valencia, C.H.; Navia Porras, D.P.; Herminsul Mina Hernandez, J.; Valencia Zapata, M.E.; Chaur, M.N. Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications. Molecules 2020, 25, 1203. https://doi.org/10.3390/molecules25051203
Grande Tovar CD, Castro JI, Valencia CH, Navia Porras DP, Herminsul Mina Hernandez J, Valencia Zapata ME, Chaur MN. Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications. Molecules. 2020; 25(5):1203. https://doi.org/10.3390/molecules25051203
Chicago/Turabian StyleGrande Tovar, Carlos David, Jorge Iván Castro, Carlos Humberto Valencia, Diana Paola Navia Porras, José Herminsul Mina Hernandez, Mayra Eliana Valencia Zapata, and Manuel N. Chaur. 2020. "Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications" Molecules 25, no. 5: 1203. https://doi.org/10.3390/molecules25051203
APA StyleGrande Tovar, C. D., Castro, J. I., Valencia, C. H., Navia Porras, D. P., Herminsul Mina Hernandez, J., Valencia Zapata, M. E., & Chaur, M. N. (2020). Nanocomposite Films of Chitosan-Grafted Carbon Nano-Onions for Biomedical Applications. Molecules, 25(5), 1203. https://doi.org/10.3390/molecules25051203