
Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1



Abstract
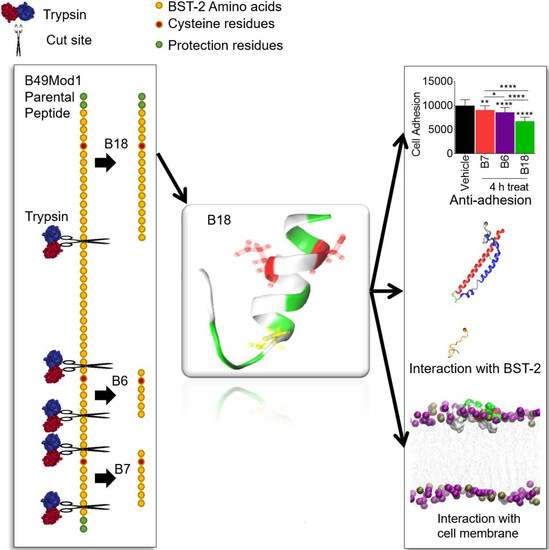
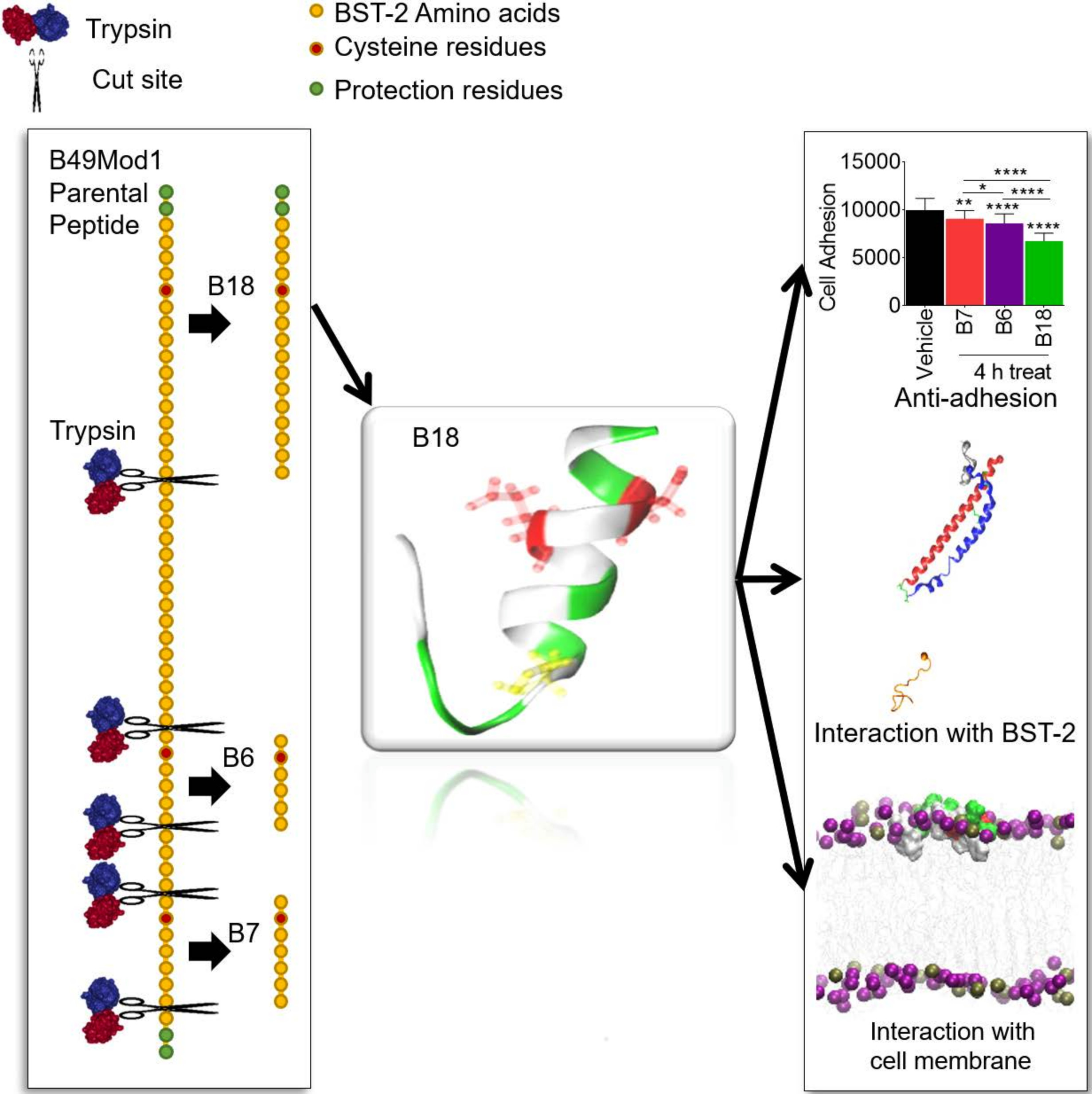
1. Introduction
2. Results
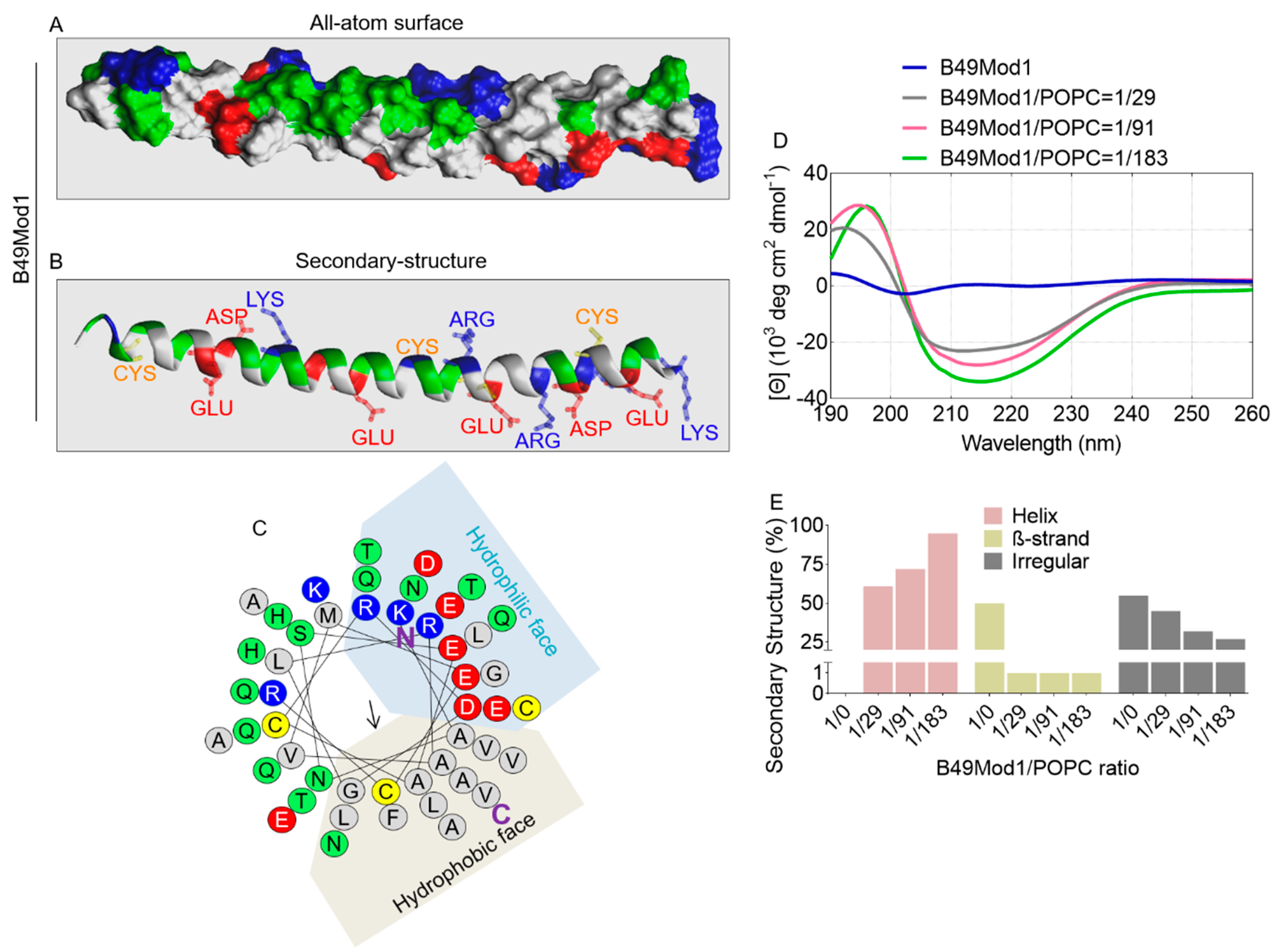
2.1. Structural Information of B49Mod1
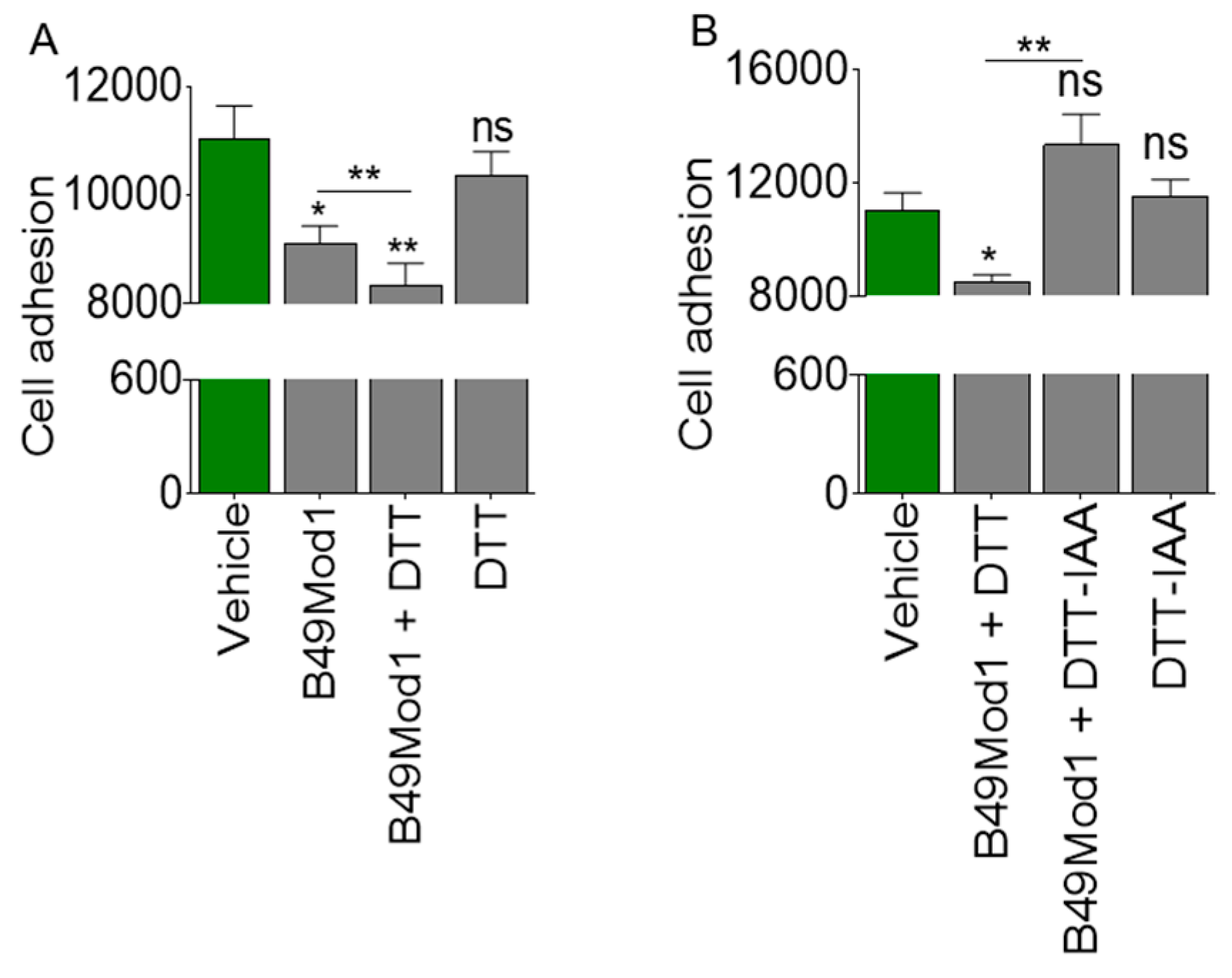
2.2. Cysteine-Linked Disulfide Bond is Important for Anti-Adhesion Activity of B49Mod1
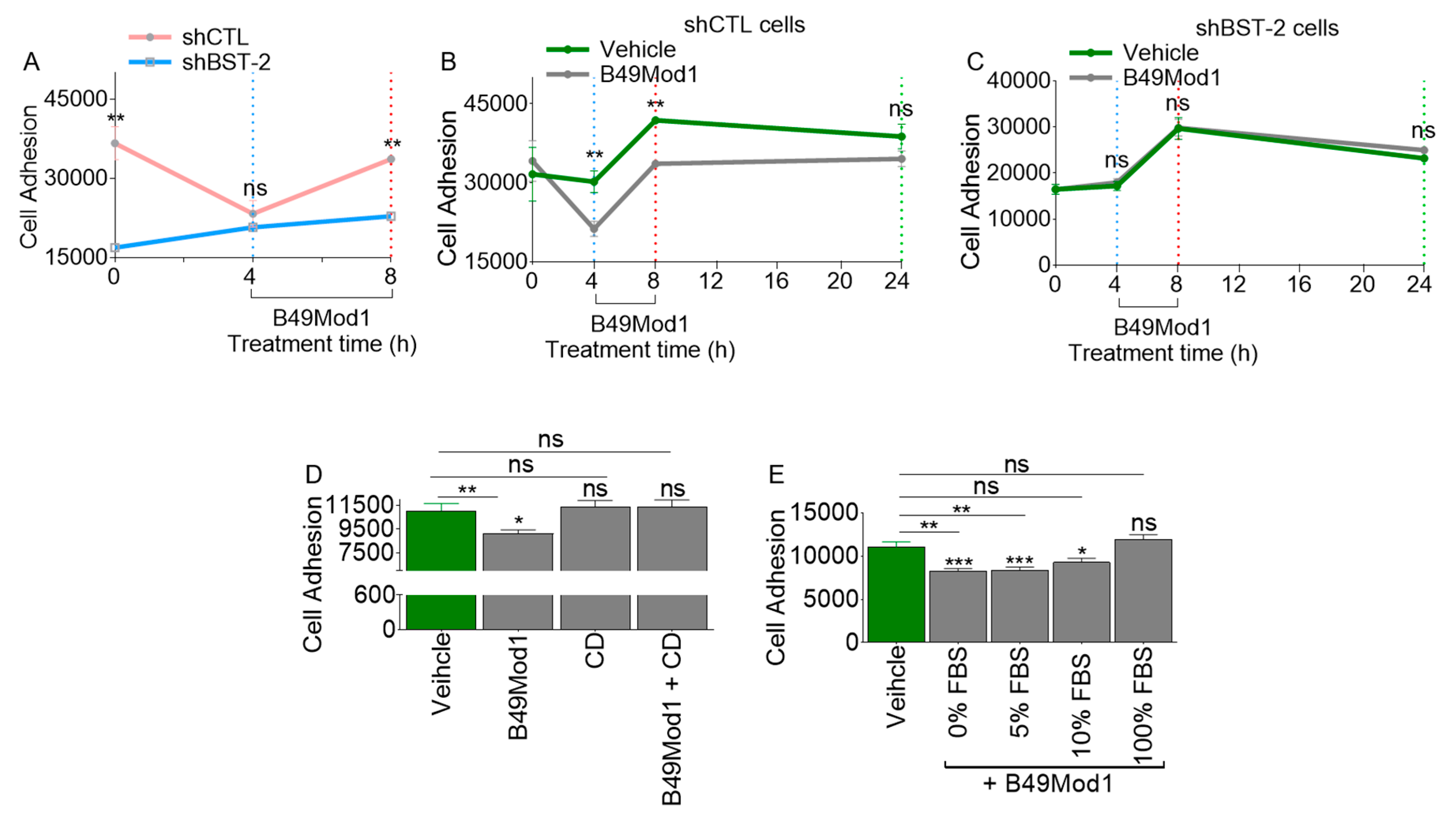
2.3. Extended Incubation Time Results in Loss of B49Mod1 Activity
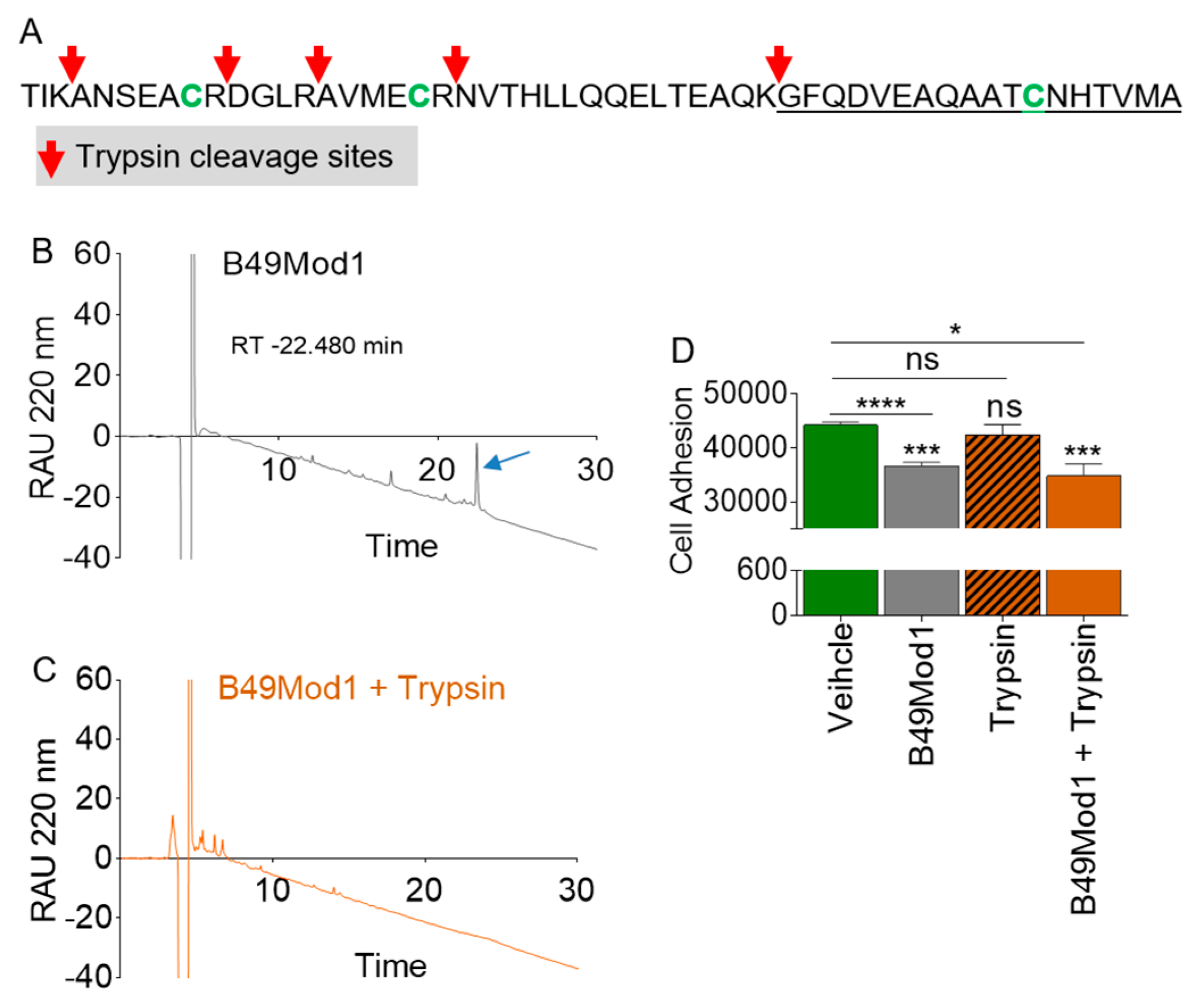
2.4. B49Mod1 is Susceptible to Proteolysis
2.5. Trypsin Proteolysis of B49Mod1
2.6. Identification of the Smallest and Most Potent B49Mod1 Fragment
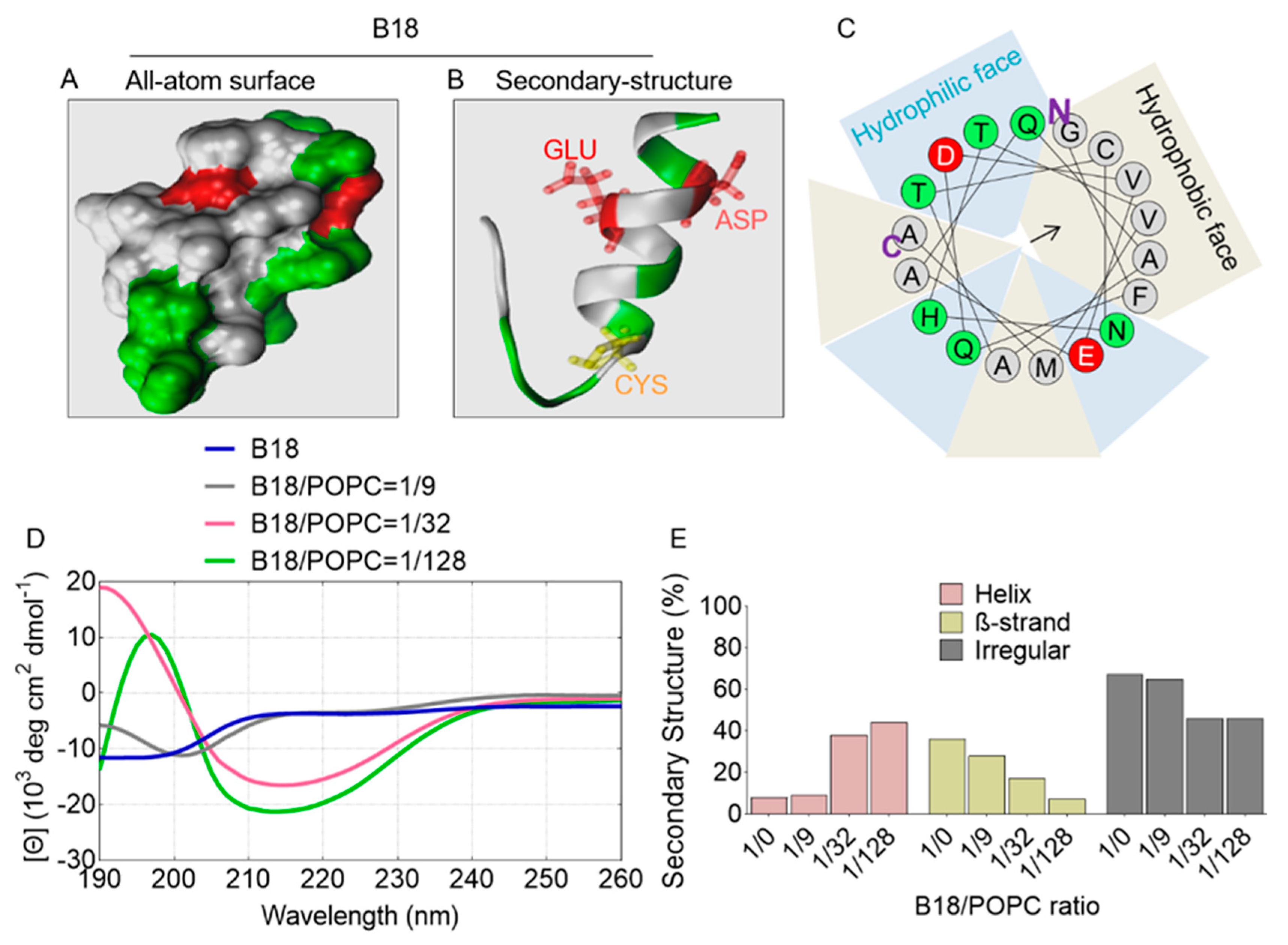
2.7. All-Atom and Secondary Structure of B18
2.8. B18 Binds BST-2
2.9. Identification of B18 Binding Sites
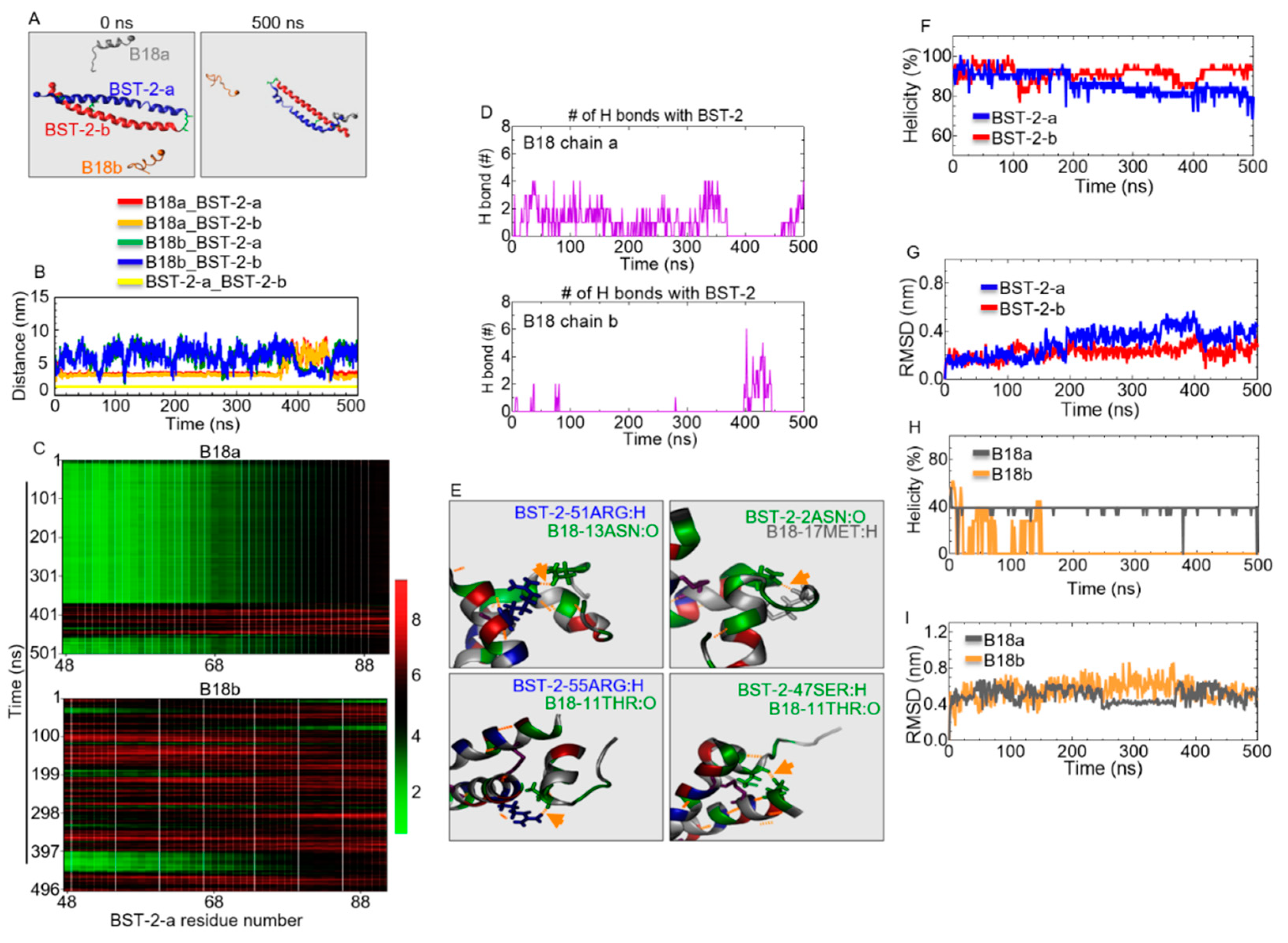
2.10. Structural Properties of B18 and BST-2 upon Binding
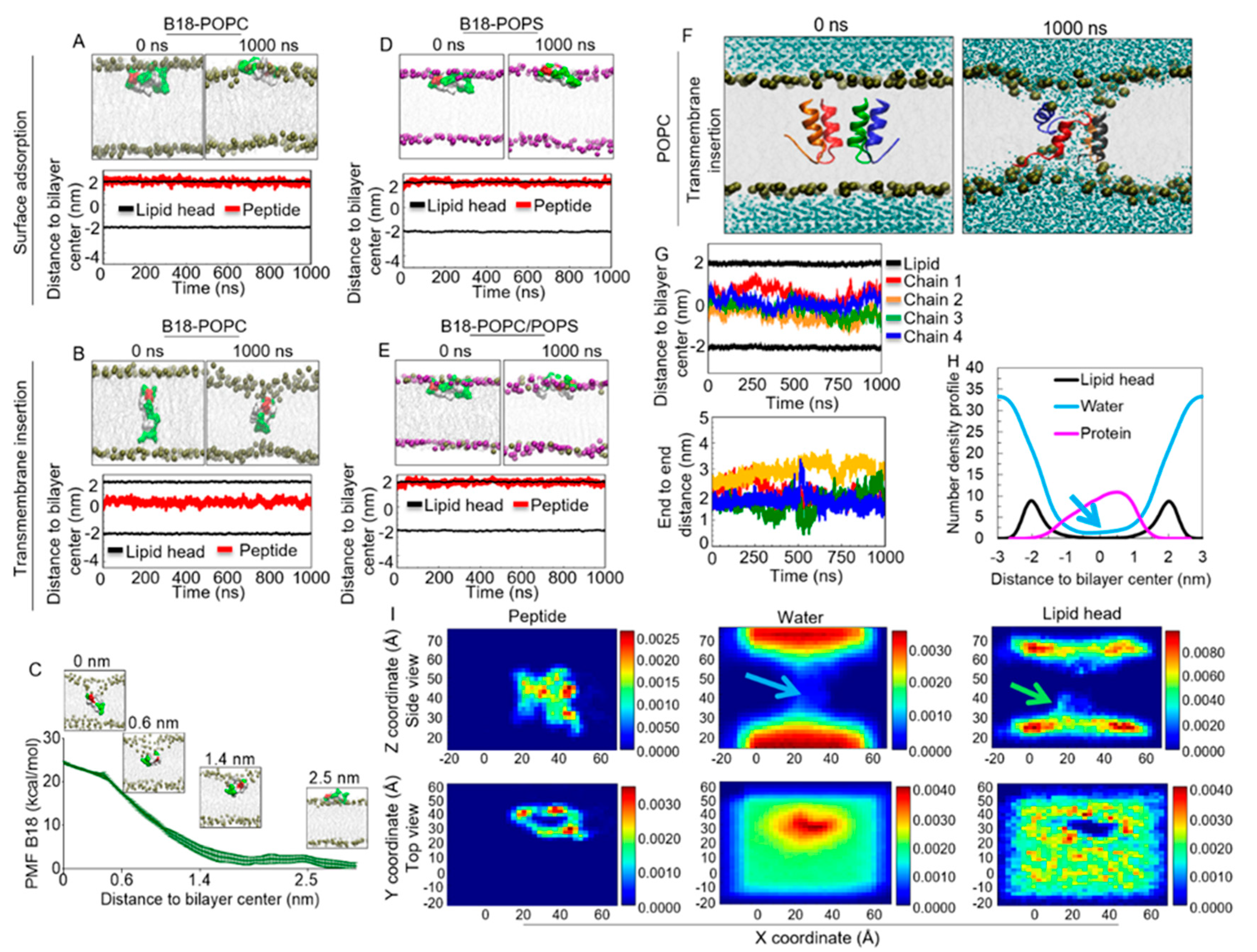
2.11. B18 Binds Zwitterionic Membrane
2.12. Energetics of B18 Insertion into the POPC Lipid Bilayers
2.13. B18 Binds Anionic POPS Lipid Bilayer
2.14. B18 forms Transmembrane Pores
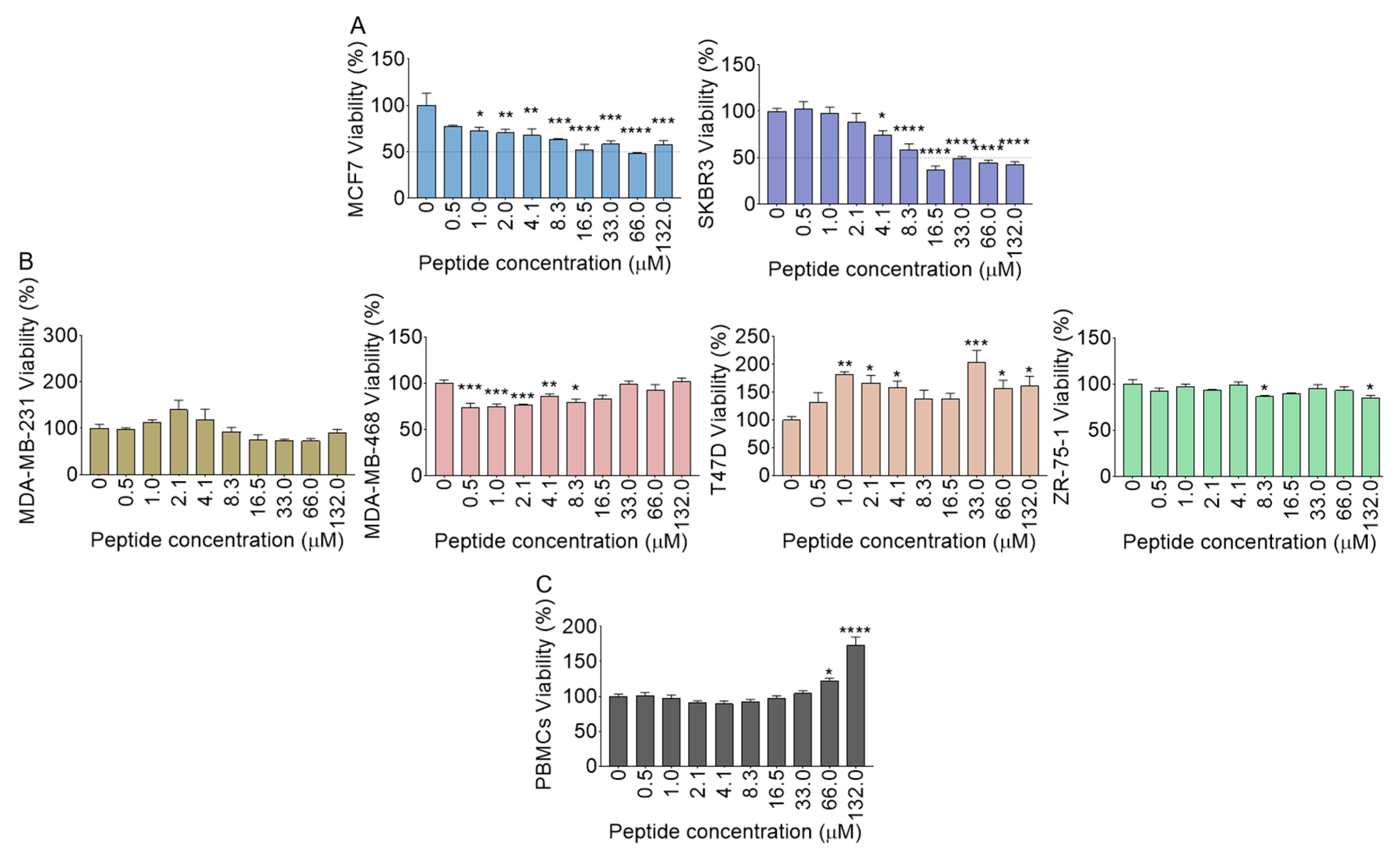
2.15. Polyfunctional Effects of B18 on Cell Viability
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef]
- Kim, E.S.; Kim, S.Y.; Koh, M.; Lee, H.M.; Kim, K.; Jung, J.; Kim, H.S.; Moon, W.K.; Hwang, S.; Moon, A. Creactive protein binds to integrin alpha2 and Fcgamma receptor I, leading to breast cell adhesion and breast cancer progression. Oncogene 2018, 37, 28–38. [Google Scholar] [CrossRef]
- Ishikawa, J.; Kaisho, T.; Tomizawa, H.; Lee, B.O.; Kobune, Y.; Inazawa, J.; Oritani, K.; Itoh, M.; Ochi, T.; Ishihara, K.; et al. Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 1995, 26, 527–534. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. The role of BST-2/Tetherin in host protection and disease manifestation. Immun. Inflamm. Dis. 2016, 4, 4–23. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. BST-2: at the crossroads of viral pathogenesis and oncogenesis. Future Virol. 2016, 11, 127–140. [Google Scholar] [CrossRef]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.J.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef]
- Jones, P.H.; Mehta, H.V.; Maric, M.; Roller, R.J.; Okeoma, C.M. Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology 2012, 9, 10. [Google Scholar] [CrossRef]
- Casartelli, N.; Sourisseau, M.; Feldmann, J.; Guivel-Benhassine, F.; Mallet, A.; Marcelin, A.G.; Guatelli, J.; Schwartz, O. Tetherin restricts productive HIV-1 cell-to-cell transmission. PLoS Pathog. 2010, 6, e1000955. [Google Scholar] [CrossRef]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; Mccarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef]
- Schubert, H.L.; Zhai, Q.; Sandrin, V.; Eckert, D.M.; Garcia-Maya, M.; Saul, L.; Sundquist, W.I.; Steiner, R.A.; Hill, C.P. Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc. Natl. Acad. Sci. USA 2010, 107, 17951–17956. [Google Scholar] [CrossRef]
- Hotter, D.; Sauter, D.; Kirchhoff, F. Emerging role of the host restriction factor tetherin in viral immune sensing. J. Mol. Biol. 2013, 425, 4956–4964. [Google Scholar] [CrossRef]
- Andrew, A.J.; Berndsen, C.E.; Kao, S.; Strebel, K. The size and conservation of a coiled-coil structure in the ectodomain of human BST-2/tetherin is dispensable for inhibition of HIV-1 virion release. J. Biol. Chem. 2012, 287, 44278–44288. [Google Scholar] [CrossRef]
- Andrew, A.J.; Miyagi, E.; Kao, S.; Strebel, K. The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology 2009, 6, 80. [Google Scholar] [CrossRef]
- Fitzpatrick, K.; Skasko, M.; Deerinck, T.J.; Crum, J.; Ellisman, M.H.; Guatelli, J. Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. Plos Pathog. 2010, 6, e1000701. [Google Scholar] [CrossRef]
- Chu, H.; Wang, J.J.; Qi, M.; Yoon, J.J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 is essential for the formation of the intracellular virus-containing compartment in HIV-infected macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar] [CrossRef]
- Wang, W.; Nishioka, Y.; Ozaki, S.; Jalili, A.; Abe, S.; Kakiuchi, S.; Kishuku, M.; Minakuchi, K.; Matsumoto, T.; Sone, S. HM1.24 (CD317) is a novel target against lung cancer for immunotherapy using anti-HM1.24 antibody. Cancer Immunol. Immunother. 2009, 58, 967–976. [Google Scholar] [CrossRef]
- Silveira, N.J.; Varuzza, L.; Machado-Lima, A.; Lauretto, M.S.; Pinheiro, D.G.; Rodrigues, R.V.; Severino, P.; Nobrega, F.G.; Silva, W.A., Jr.; De, B.P.C.A.; et al. Searching for molecular markers in head and neck squamous cell carcinomas (HNSCC) by statistical and bioinformatic analysis of larynx-derived SAGE libraries. Bmc Med. Genom. 2008, 1, 56. [Google Scholar] [CrossRef]
- Fang, K.H.; Kao, H.K.; Chi, L.M.; Liang, Y.; Liu, S.C.; Hseuh, C.; Liao, C.T.; Yen, T.C.; Yu, J.S.; Chang, K.P. Overexpression of BST2 is associated with nodal metastasis and poorer prognosis in oral cavity cancer. Laryngoscope 2014, 124, E354–E360. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Balyasnikova, I.V.; Han, Y.; Lesniak, M.S. The expression of BST2 in human and experimental mouse brain tumors. Exp. Mol. Pathol. 2011, 91, 440–446. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Lo, K.W.; Yim, S.F.; Siu, N.S.; Chan, S.C.; Ho, T.W.; Wong, K.W.; Yu, M.Y.; Wang, V.W.; et al. Identification of molecular markers and signaling pathway in endometrial cancer in Hong Kong Chinese women by genome-wide gene expression profiling. Oncogene 2007, 26, 1971–1982. [Google Scholar] [CrossRef]
- Schliemann, C.; Roesli, C.; Kamada, H.; Borgia, B.; Fugmann, T.; Klapper, W.; Neri, D. In vivo biotinylation of the vasculature in B-cell lymphoma identifies BST-2 as a target for antibody-based therapy. Blood 2010, 115, 736–744. [Google Scholar] [CrossRef]
- Cai, D.; Cao, J.; Li, Z.; Zheng, X.; Yao, Y.; Li, W.; Yuan, Z. Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. Bmc Cancer 2009, 9, 102. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Demali, K.A.; Olivier, A.K.; Okeoma, C.M. Bone marrow stromal antigen 2 expressed in cancer cells promotes mammary tumor growth and metastasis. Breast Cancer Res. Bcr 2014, 16, 493. [Google Scholar] [CrossRef]
- Gu, G.; Zhao, D.; Yin, Z.; Liu, P. BST-2 binding with cellular MT1-MMP blocks cell growth and migration via decreasing MMP2 activity. J. Cell Biochem. 2012, 113, 1013–1021. [Google Scholar] [CrossRef]
- Naushad, W.; Mahauad-Fernandez, W.D.; Okeoma, C.M. Structural determinant of BST-2-mediated regulation of breast cancer cell motility: a role for cytoplasmic tail tyrosine residues. Oncotarget 2017, 8, 110221–110233. [Google Scholar] [CrossRef]
- Yi, E.H.; Yoo, H.; Noh, K.H.; Han, S.; Lee, H.; Lee, J.K.; Won, C.; Kim, B.H.; Kim, M.H.; Cho, C.H.; et al. BST-2 is a potential activator of invasion and migration in tamoxifen-resistant breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 435, 685–690. [Google Scholar] [CrossRef]
- Sayeed, A.; Luciani-Torres, G.; Meng, Z.; Bennington, J.L.; Moore, D.H.; Dairkee, S.H. Aberrant regulation of the BST2 (Tetherin) promoter enhances cell proliferation and apoptosis evasion in high grade breast cancer cells. PLoS ONE 2013, 8, e67191. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. Cysteine-linked dimerization of BST-2 confers anoikis resistance to breast cancer cells by negating proapoptotic activities to promote tumor cell survival and growth. Cell Death Dis. 2017, 8, e2687. [Google Scholar] [CrossRef]
- Yoo, H.; Park, S.H.; Ye, S.K.; Kim, M. IFN-gamma-induced BST2 mediates monocyte adhesion to human endothelial cells. Cell Immunol. 2011, 267, 23–29. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Naushad, W.; Panzner, T.D.; Bashir, A.; Lal, G.; Okeoma, C.M. BST-2 promotes survival in circulation and pulmonary metastatic seeding of breast cancer cells. Sci. Rep. 2018, 8, 17608. [Google Scholar] [CrossRef]
- Mahauad-Fernandez, W.D.; Okeoma, C.M. B49, a BST-2-based peptide, inhibits adhesion and growth of breast cancer cells. Sci. Rep. 2018, 8, 4305. [Google Scholar] [CrossRef] [PubMed]
- Okeoma, C.M.; Mahauad-Fernandez, W.D. BST-2 as a therapeutic target and a predictive/prognostic bio-signature for breast cancer growth and metastasis. WO 2017011375, 19 January 2017. [Google Scholar]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, 449–454. [Google Scholar] [CrossRef] [PubMed]
- J Dufourc, E.; Buchoux, S.; Toupé, J.; Sani, M.-A.; Jean-François, F.; Khemtémourian, L.; Grélard, A.; Loudet-Courrèges, C.; Laguerre, M.; Elezgaray, J. Membrane interacting peptides: from killers to helpers. Curr. Protein Pept. Sci. 2012, 13, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. Febs Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide–membrane interactions of three related antimicrobial peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef]
- Syed, H.; Tauseef, M.; Ahmad, Z. A connection between antimicrobial properties of venom peptides and microbial ATP synthase. Int. J. Biol. Macromol. 2018, 119, 23–31. [Google Scholar] [CrossRef]
- Yang, C.-H.; Chen, Y.-C.; Peng, S.-Y.; Tsai, A.P.-Y.; Lee, T.J.-F.; Yen, J.-H.; Liou, J.-W. An engineered arginine-rich α-helical antimicrobial peptide exhibits broad-spectrum bactericidal activity against pathogenic bacteria and reduces bacterial infections in mice. Sci. Rep. 2018, 8, 14602. [Google Scholar] [CrossRef]
- Andreev, O.A.; Karabadzhak, A.G.; Weerakkody, D.; Andreev, G.O.; Engelman, D.M.; Reshetnyak, Y.K. pH (low) insertion peptide (pHLIP) inserts across a lipid bilayer as a helix and exits by a different path. Proc. Natl. Acad. Sci. USA 2010, 107, 4081–4086. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Zhang, M.; Lyu, Y.; Zhu, X.; Wang, J.; Jin, Z.; Narsimhan, G. Enhanced solubility and antimicrobial activity of alamethicin in aqueous solution by complexation with γ-cyclodextrin. J. Funct. Foods 2018, 40, 700–706. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; Mackerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; Mackerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, R.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Durell, S.R.; Brooks, B.R.; Ben-Naim, A. Solvent-induced forces between two hydrophilic groups. J. Phys. Chem. 1994, 98, 2198–2202. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Le, W.; Chen, B.; Cui, Z.; Liu, Z.; Shi, D. Detection of cancer cells based on glycolytic-regulated surface electrical charges. Biophys. Rep. 2019, 5, 10–18. [Google Scholar] [CrossRef]
- Cirac, A.D.; Moiset, G.; Mika, J.T.; Koçer, A.; Salvador, P.; Poolman, B.; Marrink, S.J.; Sengupta, D. The molecular basis for antimicrobial activity of pore-forming cyclic peptides. Biophys. J. 2011, 100, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Pino-Angeles, A.; Leveritt Iii, J.M.; Lazaridis, T. Pore structure and synergy in antimicrobial peptides of the magainin family. Plos Comput. Biol. 2016, 12, e1004570. [Google Scholar] [CrossRef]
- Song, C.; Weichbrodt, C.; Salnikov, E.S.; Dynowski, M.; Forsberg, B.O.; Bechinger, B.; Steinem, C.; De Groot, B.L.; Zachariae, U.; Zeth, K. Crystal structure and functional mechanism of a human antimicrobial membrane channel. Proc. Natl. Acad. Sci. 2013, 110, 4586–4591. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Lyu, Y.; Fitriyanti, M.; Hou, H.; Jin, Z.; Zhu, X.; Narsimhan, G. Understanding the antimicrobial activity of water soluble γ-cyclodextrin/alamethicin complex. Colloids Surf. B Biointerfaces 2018, 172, 451–458. [Google Scholar] [CrossRef]
- Lyu, Y.; Zhu, X.; Xiang, N.; Narsimhan, G. Molecular dynamics study of pore formation by melittin in a 1, 2-Dioleoyl-sn-glycero-3-phosphocholine and 1, 2-di (9 z-octadecenoyl)-sn-glycero-3-phospho-(1′-rac-glycerol) mixed lipid bilayer. Ind. Eng. Chem. Res. 2015, 54, 10275–10283. [Google Scholar] [CrossRef]
- Lyu, Y.; Xiang, N.; Mondal, J.; Zhu, X.; Narsimhan, G. Characterization of Interactions between Curcumin and Different Types of Lipid Bilayers by Molecular Dynamics Simulation. J. Phys. Chem. B 2018, 122, 2341–2354. [Google Scholar] [CrossRef]
- Bird, G.H.; Madani, N.; Perry, A.F.; Princiotto, A.M.; Supko, J.G.; He, X.; Gavathiotis, E.; Sodroski, J.G.; Walensky, L.D. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. USA 2010, 107, 14093–14098. [Google Scholar] [CrossRef]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Xiang, N.; Lyu, Y.; Zhu, X.; Bhunia, A.K.; Narsimhan, G. Methodology for identification of pore forming antimicrobial peptides from soy protein subunits β-conglycinin and glycinin. Peptides 2016, 85, 27–40. [Google Scholar] [CrossRef]
- Irudayam, S.J.; Pobandt, T.; Berkowitz, M.L. Free energy barrier for melittin reorientation from a membrane-bound state to a transmembrane state. J. Phys. Chem. B 2013, 117, 13457–13463. [Google Scholar] [CrossRef][Green Version]
- Lyu, Y.; Xiang, N.; Zhu, X.; Narsimhan, G. Potential of mean force for insertion of antimicrobial peptide melittin into a pore in mixed DOPC/DOPG lipid bilayer by molecular dynamics simulation. J. Chem. Phys. 2017, 146, 155101. [Google Scholar] [CrossRef]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein–protein interactions: mimicking peptide binding epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Matsuura, K. Peptide Nanomaterials Designed from Natural Supramolecular Systems. Chem. Rec. 2019, 19, 843–858. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The use of therapeutic peptides to target and to kill cancer cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef] [PubMed]
- Mcgregor, D.P. Discovering and improving novel peptide therapeutics. Curr. Opin. Pharmacol. 2008, 8, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ni, J.; Chen, L.; Yu, L.; Xu, J.; Ding, J. Encapsulation of cell-adhesive RGD peptides into a polymeric physical hydrogel to prevent postoperative tissue adhesion. J. Biom. Mate. Res. B. Appl. Biom. 2012, 100, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Ukawala, M.; Rajyaguru, T.; Chaudhari, K.; Manjappa, A.S.; Murthy, R.S.; Gude, R. EILDV-conjugated, etoposide-loaded biodegradable polymeric micelles directing to tumor metastatic cells overexpressing alpha4beta1 integrin. Cancer Nanotechnol. 2011, 2, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Fitriyanti, M.; Narsimhan, G. Nucleation and growth of pores in 1, 2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC)/cholesterol bilayer by antimicrobial peptides melittin, its mutants and cecropin P1. Colloids Surf. B Biointerfaces 2019, 173, 121–127. [Google Scholar] [CrossRef]
- Zhou, L.; Narsimhan, G.; Wu, X.; Du, F. Pore formation in 1, 2-dimyristoyl-sn-glycero-3-phosphocholine/cholesterol mixed bilayers by low concentrations of antimicrobial peptide melittin. Colloids Surf. B Biointerfaces 2014, 123, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, C.; Bellstedt, P.; Görlach, M. CAPITO—a web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 2013, 29, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Xiang, N.; Lyu, Y.; Zhu, X.; Narsimhan, G. Investigation of the interaction of amyloid beta peptide (11–42) oligomers with a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) membrane using molecular dynamics simulation. Phys. Chem. Chem. Phys. 2018, 20, 6817–6829. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Mackerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Silvius, J.R. Thermotropic phase transitions of pure lipids in model membranes and their modifications by membrane proteins. Lipid-Protein Interact. 1982, 2, 239–281. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
Sample Availability: The Research Foundation of State University of New York, Stony Brook University holds the IP for the peptides. The peptides will be available through license use. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | Number of Cysteine | Charge | H (kcal/mol) | µH (kcal/mol) | MW (kDa) |
|---|---|---|---|---|---|---|
| B3 | TIK | 0 | 1 | 0.357 | 0.762 | 0.360 |
| B7 | ANSEACR | 1 | 0 | −0.167 | 0.248 | 0.750 |
| B4 | DGLR | 0 | 0 | −0.020 | 0.722 | 0.460 |
| B6 | AVMECR | 1 | 0 | 0.442 | 0.295 | 0.708 |
| B15 | NVTHLLQQELTEAQK | 0 | −1 | 0.250 | 0.360 | 1.752 |
| B18 | GFQDVEAQAATCNHTVMA | 1 | −2 | 0.358 | 0.222 | 1.893 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, Y.; Mahauad-Fernandez, W.D.; Okeoma, C.M. Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1. Molecules 2020, 25, 1188. https://doi.org/10.3390/molecules25051188
Lyu Y, Mahauad-Fernandez WD, Okeoma CM. Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1. Molecules. 2020; 25(5):1188. https://doi.org/10.3390/molecules25051188
Chicago/Turabian StyleLyu, Yuan, Wadie D. Mahauad-Fernandez, and Chioma M. Okeoma. 2020. "Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1" Molecules 25, no. 5: 1188. https://doi.org/10.3390/molecules25051188
APA StyleLyu, Y., Mahauad-Fernandez, W. D., & Okeoma, C. M. (2020). Development and Characterization of the Shortest Anti-Adhesion Peptide Analogue of B49Mod1. Molecules, 25(5), 1188. https://doi.org/10.3390/molecules25051188