Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study

 , , , and
, , , and
Abstract
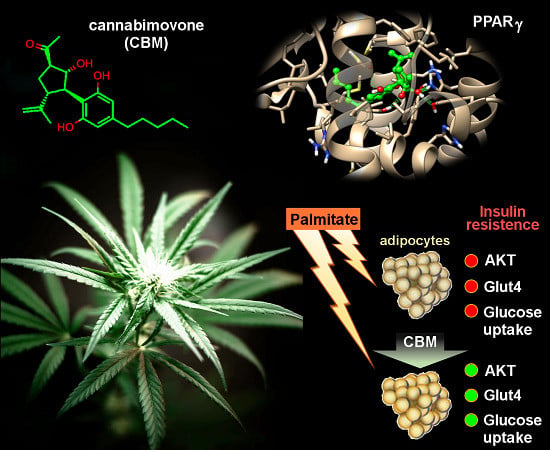
1. Introduction
2. Results
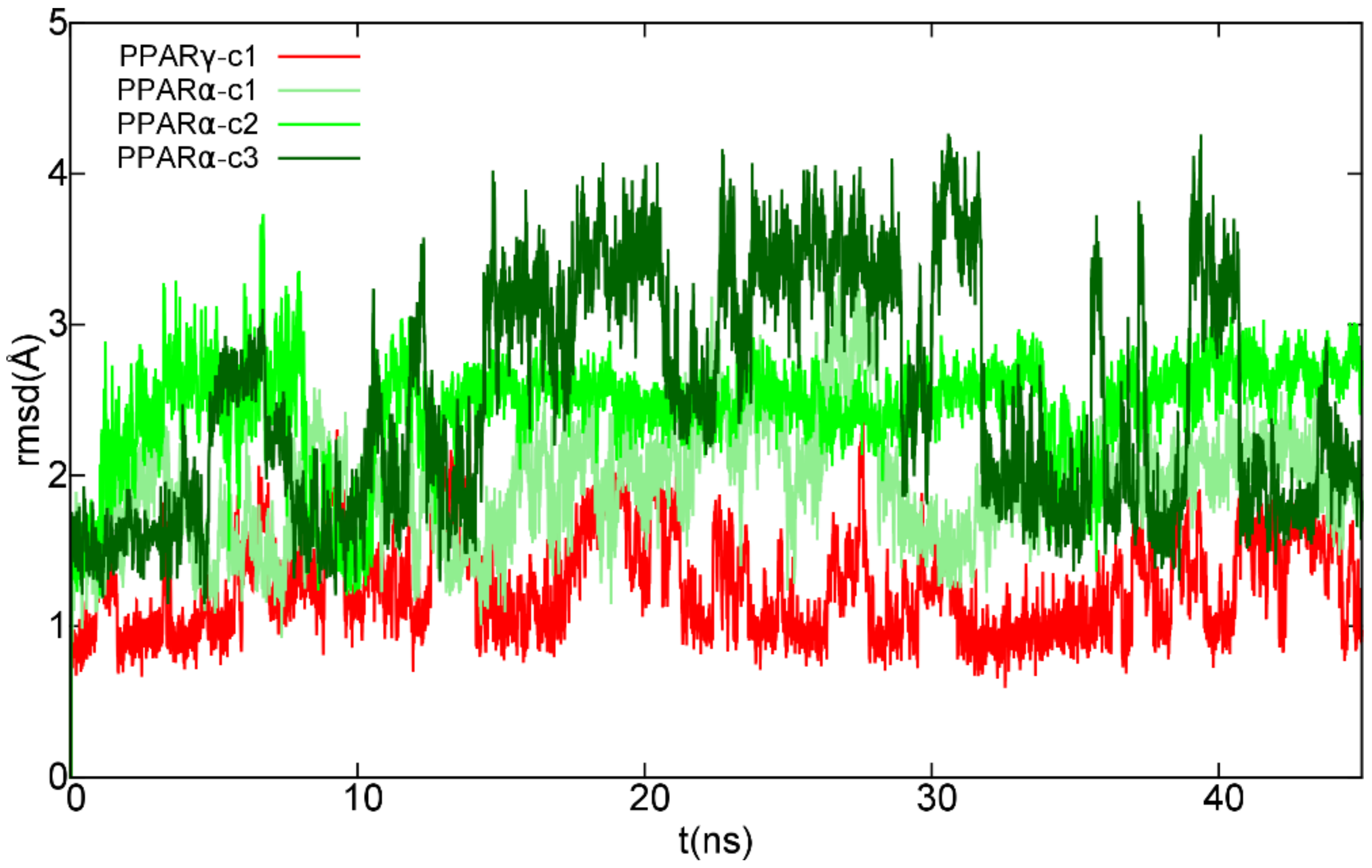
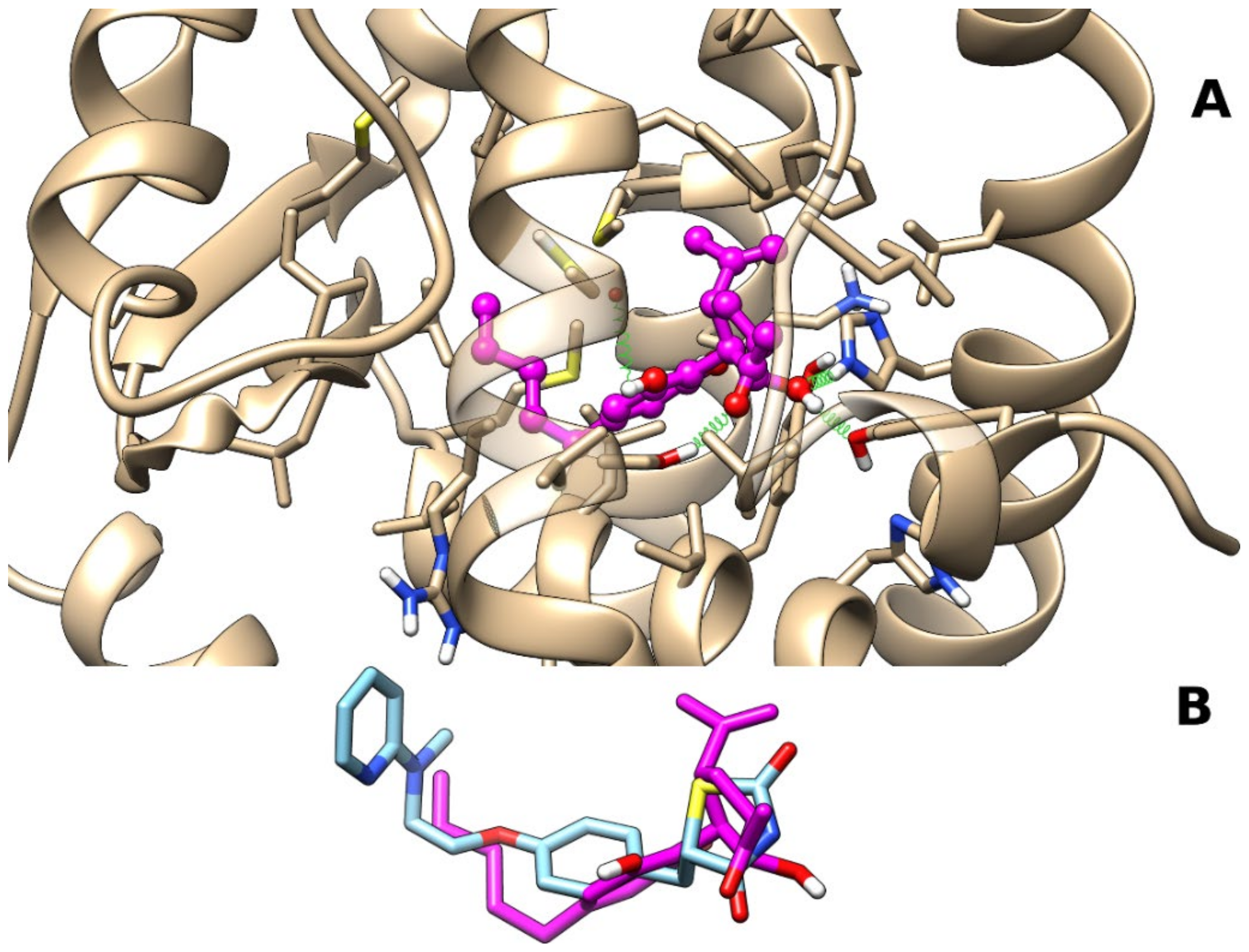
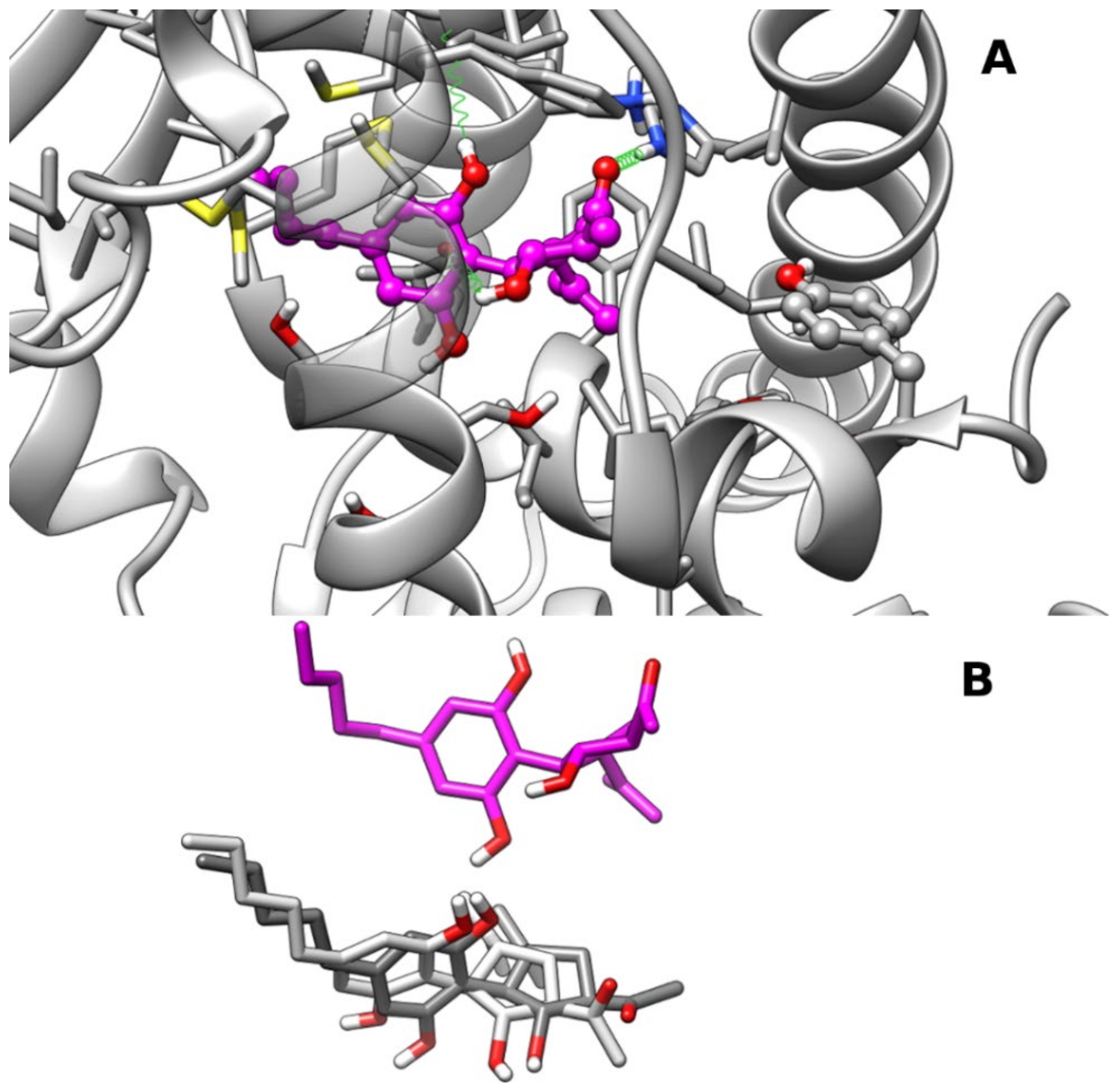
2.1. Molecular Docking and Molecular Dynamics
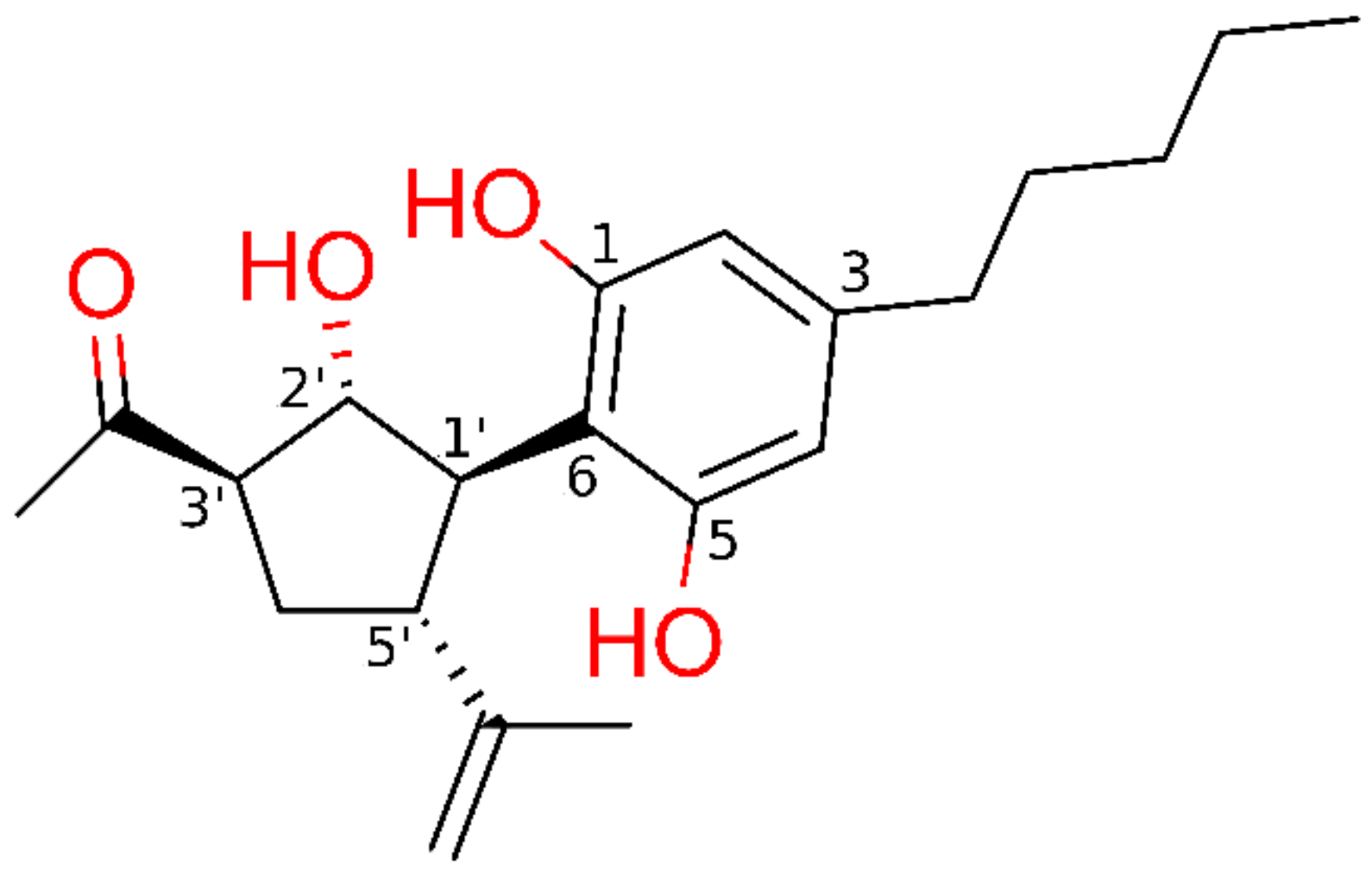
2.1.1. Theoretical Complexes of PPARγ with Cannabimovone (CBM)
2.1.2. Theoretical Complexes of PPARα with Cannabimovone (CBM)
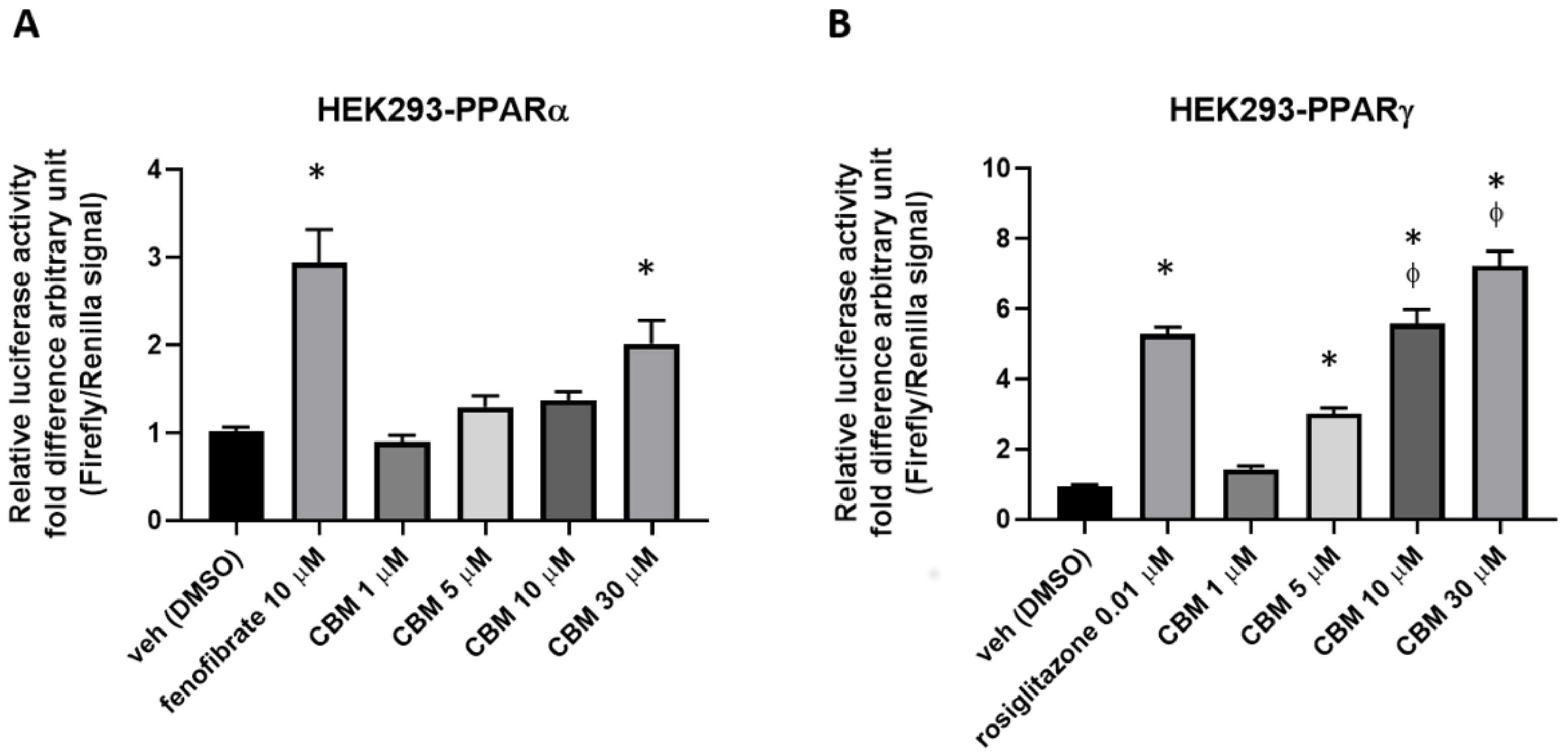
2.2. Luciferase Assays
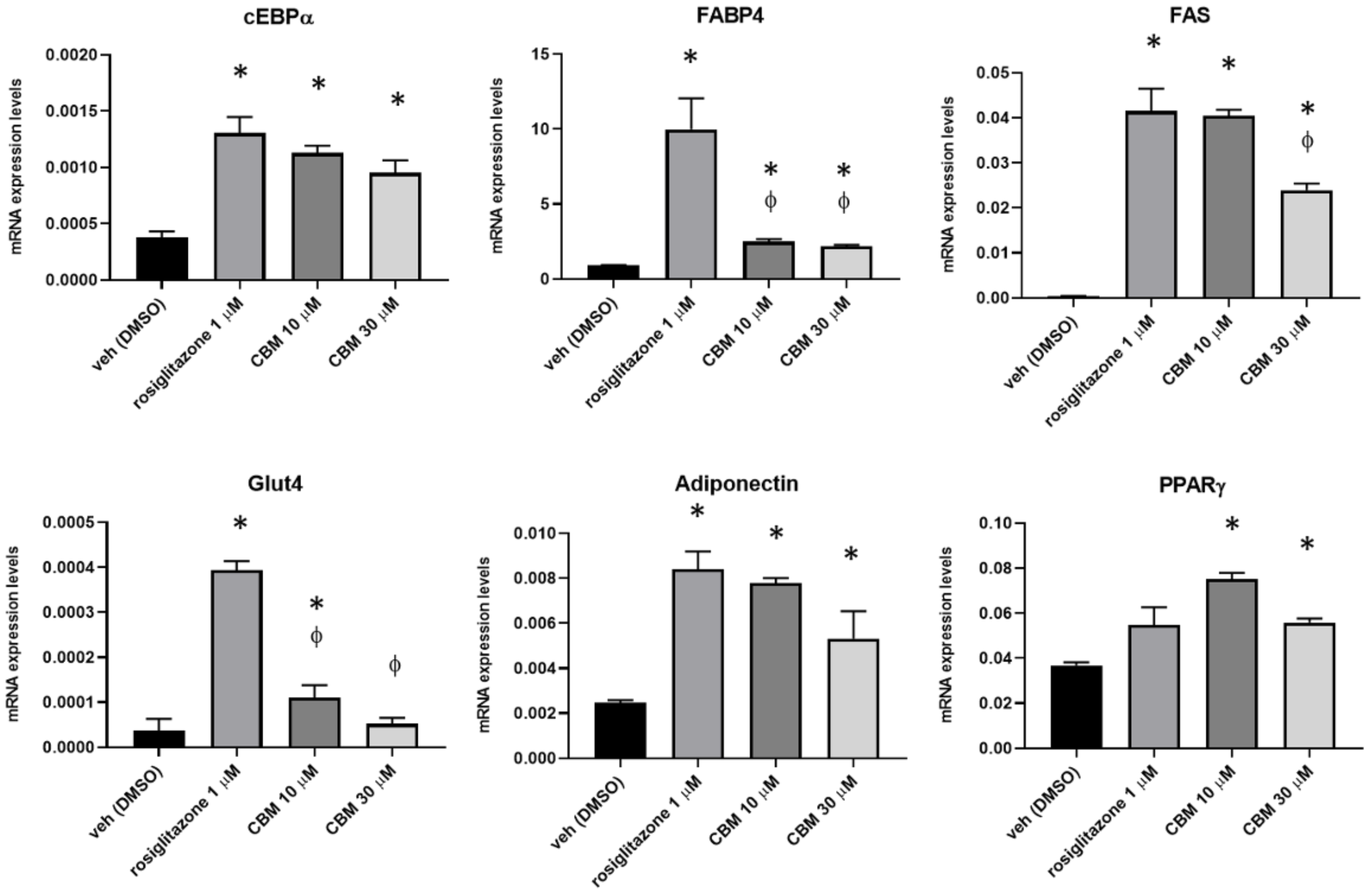
2.3. CBM Upregulates Genes Involved in Adipocyte Differentiation and Energy Metabolism
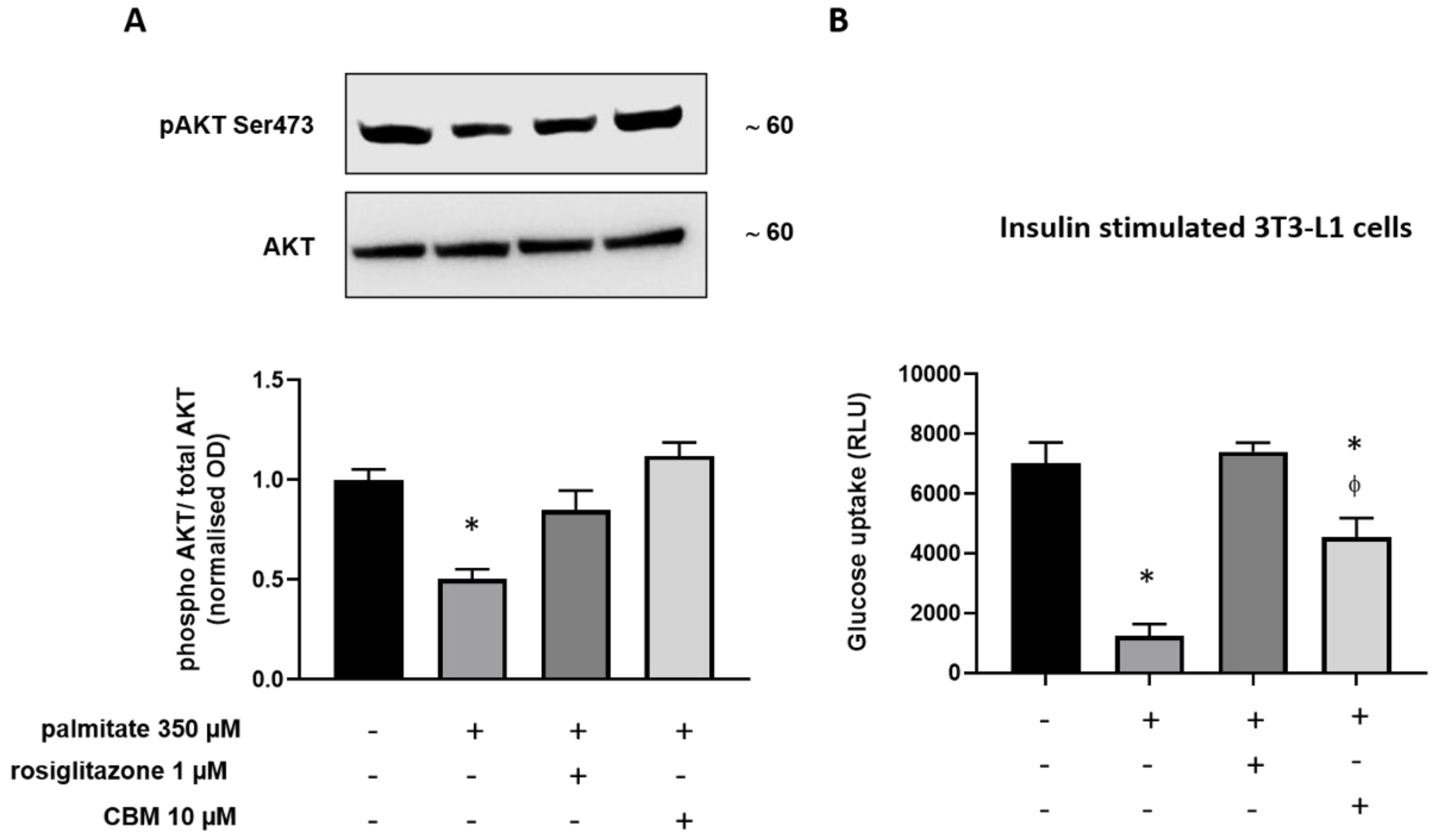
2.4. CBM Improves Insulin Sensitivity in Differentiating 3T3-L1 cells
3. Discussion
4. Methods
4.1. Purification of Cannabimovone
4.2. Molecular Docking and Molecular Dynamics
4.3. Cell Culture, Transfection, and Luciferase Assay
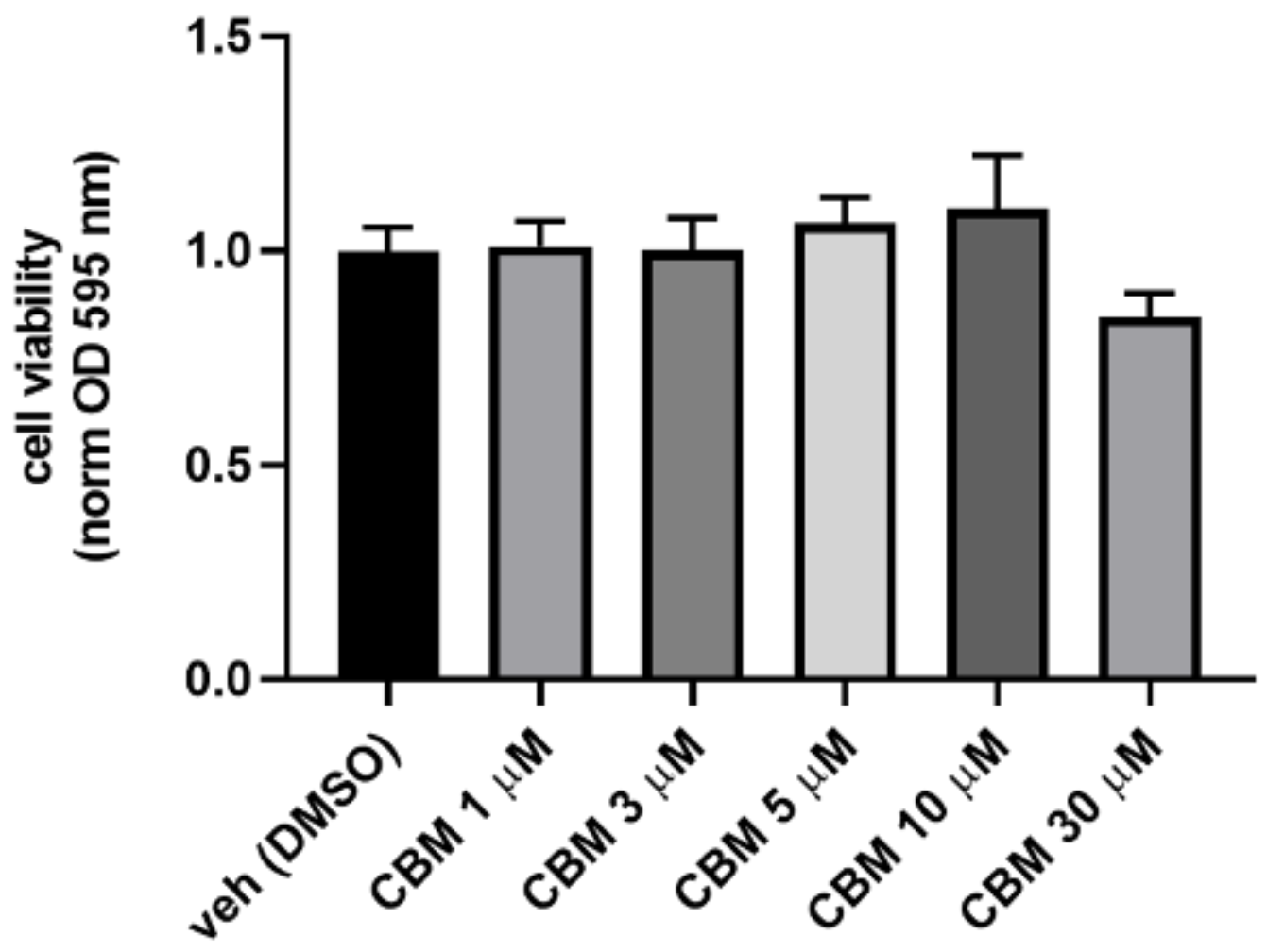
4.4. MTT Assay
4.5. RNA Purification and Quantitative Real-Time PCR (qPCR)
4.6. Western Blotting Analysis
4.7. Glucose Uptake Assay
4.8. Chemical Reagents
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; Collins, J.L.; Pearce, K.H. The Nuclear Receptor Superfamily and Drug Discovery. ChemMedChem 2006, 1, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Gearing, K.L.; Göttlicher, M.; Teboul, M.; Widmark, E.; Gustafsson, J.A. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Willson, T.M.; Lambert, M.H.; Kliewer, S.A. Peroxisome Proliferator–Activated Receptor γ and Metabolic Disease. Annu. Rev. Biochem. 2001, 70, 341–367. [Google Scholar] [CrossRef]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene—A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Staels, B.; Fruchart, J.-C. Therapeutic Roles of Peroxisome Proliferator-Activated Receptor Agonists. Diabetes 2005, 54, 2460–2470. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Horita, S.; Nakamura, M.; Satoh, N.; Suzuki, M.; Seki, G. Thiazolidinediones and Edema: Recent Advances in the Pathogenesis of Thiazolidinediones-Induced Renal Sodium Retention. PPAR Res. 2015, 2015, 1–7. [Google Scholar] [CrossRef]
- Devinsky, O.; Cilio, M.R.; Cross, H.; Fernandez-Ruiz, J.; French, J.; Hill, C.; Katz, R.; Di Marzo, V.; Jutras-Aswad, D.; Notcutt, W.G.; et al. Cannabidiol: Pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Devinsky, O.; Whalley, B.J.; Di Marzo, V. Cannabinoids in the Treatment of Neurological Disorders. Neurotherapeutics 2015, 12, 689–691. [Google Scholar] [CrossRef] [PubMed]
- Boggs, D.L.; Nguyen, J.D.; Morgenson, D.; Taffe, M.A.; Ranganathan, M. Clinical and Preclinical Evidence for Functional Interactions of Cannabidiol and Δ9-Tetrahydrocannabinol. Neuropsychopharmacology 2018, 43, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Ibeas Bih, C.; Chen, T.; Nunn, A.V.W.; Bazelot, M.; Dallas, M.; Whalley, B.J. Molecular Targets of Cannabidiol in Neurological Disorders. Neurotherapeutics 2015, 12, 699–730. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef]
- D’Aniello, E.; Fellous, T.; Iannotti, F.A.; Gentile, A.; Allarà, M.; Balestrieri, F.; Gray, R.; Amodeo, P.; Vitale, R.M.; Di Marzo, V. Identification and characterization of phytocannabinoids as novel dual PPARα/γ agonists by a computational and in vitro experimental approach. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 586–597. [Google Scholar] [CrossRef]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef]
- Taglialatela-Scafati, O.; Pagani, A.; Scala, F.; De Petrocellis, L.; Di Marzo, V.; Grassi, G.; Appendino, G. Cannabimovone, a Cannabinoid with a Rearranged Terpenoid Skeleton from Hemp. Eur. J. Org. Chem. 2010, 2010, 2067–2072. [Google Scholar] [CrossRef]
- Azhar, S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol. 2010, 6, 657–691. [Google Scholar] [CrossRef]
- Ciudin, A.; Hernandez, C.; Simo, R. Update on Cardiovascular Safety of PPARgamma Agonists and Relevance to Medicinal Chemistry and Clinical Pharmacology. Curr. Top. Med. Chem. 2012, 12, 585–604. [Google Scholar] [CrossRef]
- Moseti, D.; Regassa, A.; Kim, W.-K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef]
- Rosen, E.D. C/EBPalpha induces adipogenesis through PPARgamma: A unified pathway. Genes Dev. 2002, 16, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, X.; Lan, F.; Zhou, T.; Cai, H.; Sun, H.; Kong, W.; Kong, W. Hydrogen Sulphide Treatment Increases Insulin Sensitivity and Improves Oxidant Metabolism through the CaMKKbeta-AMPK Pathway in PA-Induced IR C2C12 Cells. Sci. Rep. 2017, 7, 13248. [Google Scholar] [CrossRef] [PubMed]
- Fellous, T.; De Maio, F.; Kalkan, H.; Carannante, B.; Boccella, S.; Petrosino, S.; Maione, S.; Di Marzo, V.; Iannotti, F.A. Phytocannabinoids promote viability and functional adipogenesis of bone marrow-derived mesenchymal stem cells through different molecular targets. Biochem. Pharmacol. 2020, 175, 113859. [Google Scholar] [CrossRef] [PubMed]
- D’Aniello, E.; Iannotti, F.; Falkenberg, L.; Martella, A.; Gentile, A.; De Maio, F.; Ciavatta, M.; Gavagnin, M.; Waxman, J.; Di Marzo, V.; et al. In Silico Identification and Experimental Validation of (−)-Muqubilin A, a Marine Norterpene Peroxide, as PPARα/γ-RXRα Agonist and RARα Positive Allosteric Modulator. Mar. Drugs 2019, 17, 110. [Google Scholar] [CrossRef]
- Vitale, R.; D’Aniello, E.; Gorbi, S.; Martella, A.; Silvestri, C.; Giuliani, M.; Fellous, T.; Gentile, A.; Carbone, M.; Cutignano, A.; et al. Fishing for Targets of Alien Metabolites: A Novel Peroxisome Proliferator-Activated Receptor (PPAR) Agonist from a Marine Pest. Mar. Drugs 2018, 16, 431. [Google Scholar] [CrossRef]
- Carreras, J.; Kirillova, M.S.; Echavarren, A.M. Synthesis of (−)-Cannabimovone and Structural Reassignment of Anhydrocannabimovone through Gold(I)-Catalyzed Cycloisomerization. Angew. Chemie Int. Ed. 2016, 55, 7121–7125. [Google Scholar] [CrossRef]
- Mueckler, M. Insulin resistance and the disruption of Glut4 trafficking in skeletal muscle. J. Clin. Invest. 2001, 107, 1211–1213. [Google Scholar] [CrossRef]
- Martinez, L.; Berenguer, M.; Bruce, M.C.; Le Marchand-Brustel, Y.; Govers, R. Rosiglitazone increases cell surface GLUT4 levels in 3T3-L1 adipocytes through an enhancement of endosomal recycling. Biochem. Pharmacol. 2010, 79, 1300–1309. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate Induces Insulin Resistance in H4IIEC3 Hepatocytes through Reactive Oxygen Species Produced by Mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef]
- Feng, X.-T.; Wang, T.-Z.; Leng, J.; Chen, Y.; Liu, J.-B.; Liu, Y.; Wang, W.-J. Palmitate Contributes to Insulin Resistance through Downregulation of the Src-Mediated Phosphorylation of Akt in C2C12 Myotubes. Biosci. Biotechnol. Biochem. 2012, 76, 1356–1361. [Google Scholar] [CrossRef]
- Mo, J.; Zhou, Y.; Yang, R.; Zhang, P.; He, B.; Yang, J.; Li, S.; Shen, Z.; Chen, P. Ginsenoside Rg1 ameliorates palmitic acid-induced insulin resistance in HepG2 cells in association with modulating Akt and JNK activity. Pharmacol. Rep. 2019, 71, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Acton, A.; Banck, M.; Bréfort, J.; Cruz, M.; Curtis, D.; Hassinen, T.; Heikkilä, V.; Hutchison, G.; Huuskonen, J.; Jensen, J.; et al. Chemical 2.00; Department of Chemistry, University of Kuopio: Kuopio, Finland, 2006. [Google Scholar]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Application of the RESP Methodology in the Parametrization of Organic Solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Iannotti, F.A.; Panza, E.; Barrese, V.; Viggiano, D.; Soldovieri, M.V.; Taglialatela, M. Expression, Localization, and Pharmacological Role of Kv7 Potassium Channels in Skeletal Muscle Proliferation, Differentiation, and Survival after Myotoxic Insults. J. Pharmacol. Exp. Ther. 2010, 332, 811–820. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) |
|---|---|---|
| PPARγ | GTCGGTTTCAGAAGTGCCTTG | GCTTTGGTCAGCGGGAAG |
| FABP4 | TGTGATGCCTTTGTGGCAACCTG | TATGATGCTCTTCACCTTCCTGTCG |
| C/EBPα | CAAGAACAGCAACGAGTACCG | GTCACTGGTCAACTCCAGCAC |
| S16 | CTGGAGCCTGTTTTGCTTCTG | TGAGATGGACTGTCGGATGG |
| GLUT4 | GCTCTGACGATGGGGAAC | CCAACACGGCCAAGACATTG |
| FAS | GGAGGTGGTGATAGCCGGTAT | TGGGTAATCCATAGAGCCCAG |
| Adiponectin | TGACGACACCAAAAGGGCTC | GAGTGCCATCTCTGCCATCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannotti, F.A.; De Maio, F.; Panza, E.; Appendino, G.; Taglialatela-Scafati, O.; De Petrocellis, L.; Amodeo, P.; Vitale, R.M. Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study. Molecules 2020, 25, 1119. https://doi.org/10.3390/molecules25051119
Iannotti FA, De Maio F, Panza E, Appendino G, Taglialatela-Scafati O, De Petrocellis L, Amodeo P, Vitale RM. Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study. Molecules. 2020; 25(5):1119. https://doi.org/10.3390/molecules25051119
Chicago/Turabian StyleIannotti, Fabio Arturo, Fabrizia De Maio, Elisabetta Panza, Giovanni Appendino, Orazio Taglialatela-Scafati, Luciano De Petrocellis, Pietro Amodeo, and Rosa Maria Vitale. 2020. "Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study" Molecules 25, no. 5: 1119. https://doi.org/10.3390/molecules25051119
APA StyleIannotti, F. A., De Maio, F., Panza, E., Appendino, G., Taglialatela-Scafati, O., De Petrocellis, L., Amodeo, P., & Vitale, R. M. (2020). Identification and Characterization of Cannabimovone, a Cannabinoid from Cannabis sativa, as a Novel PPARγ Agonist via a Combined Computational and Functional Study. Molecules, 25(5), 1119. https://doi.org/10.3390/molecules25051119