
Multidimensional Liquid Chromatography Employing a Graphene Oxide Capillary Column as the First Dimension: Determination of Antidepressant and Antiepileptic Drugs in Urine


Abstract
1. Introduction
2. Results and Discussion
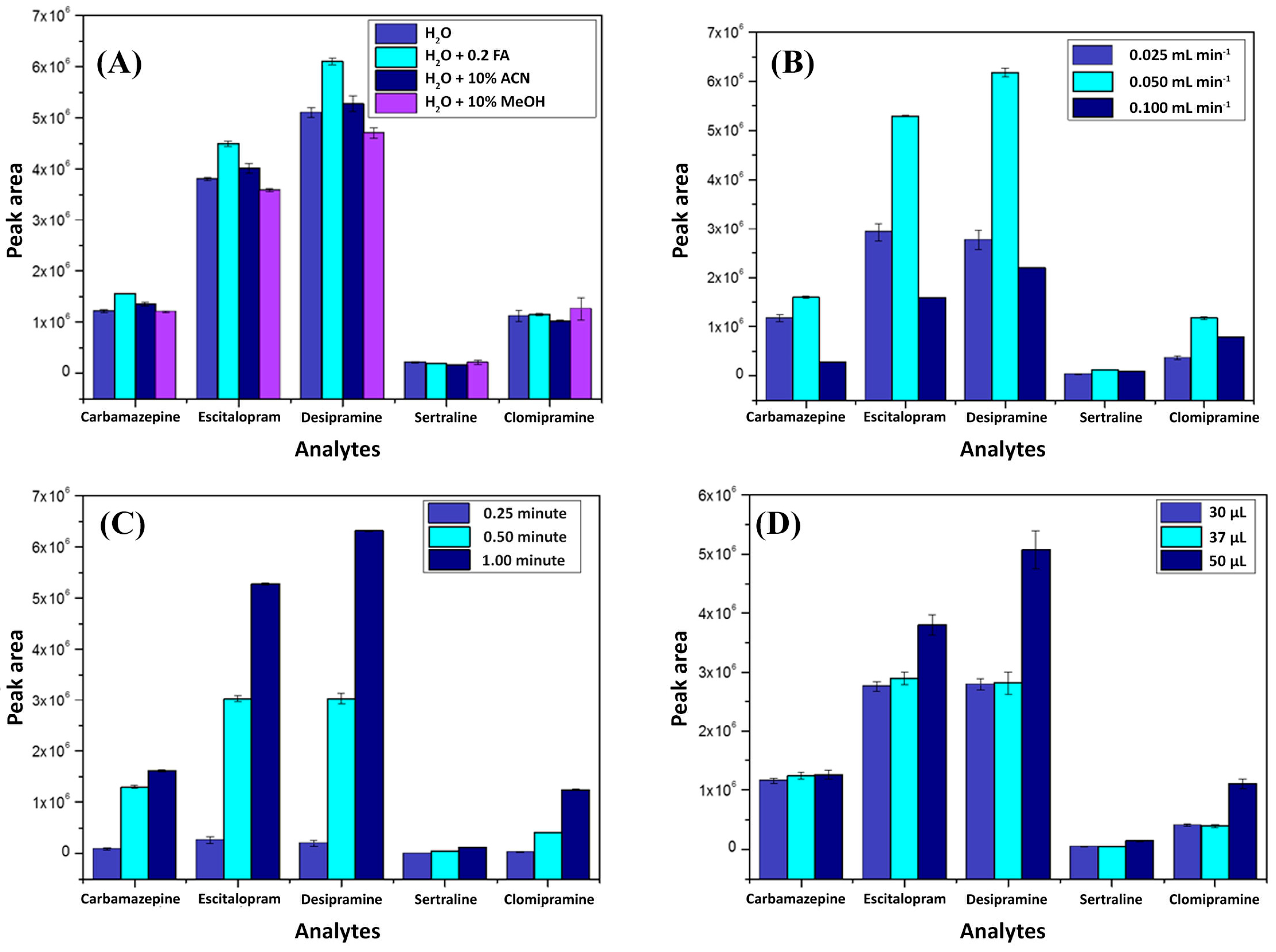
2.1. Method Enhancement
2.1.1. Chromatographic Separation
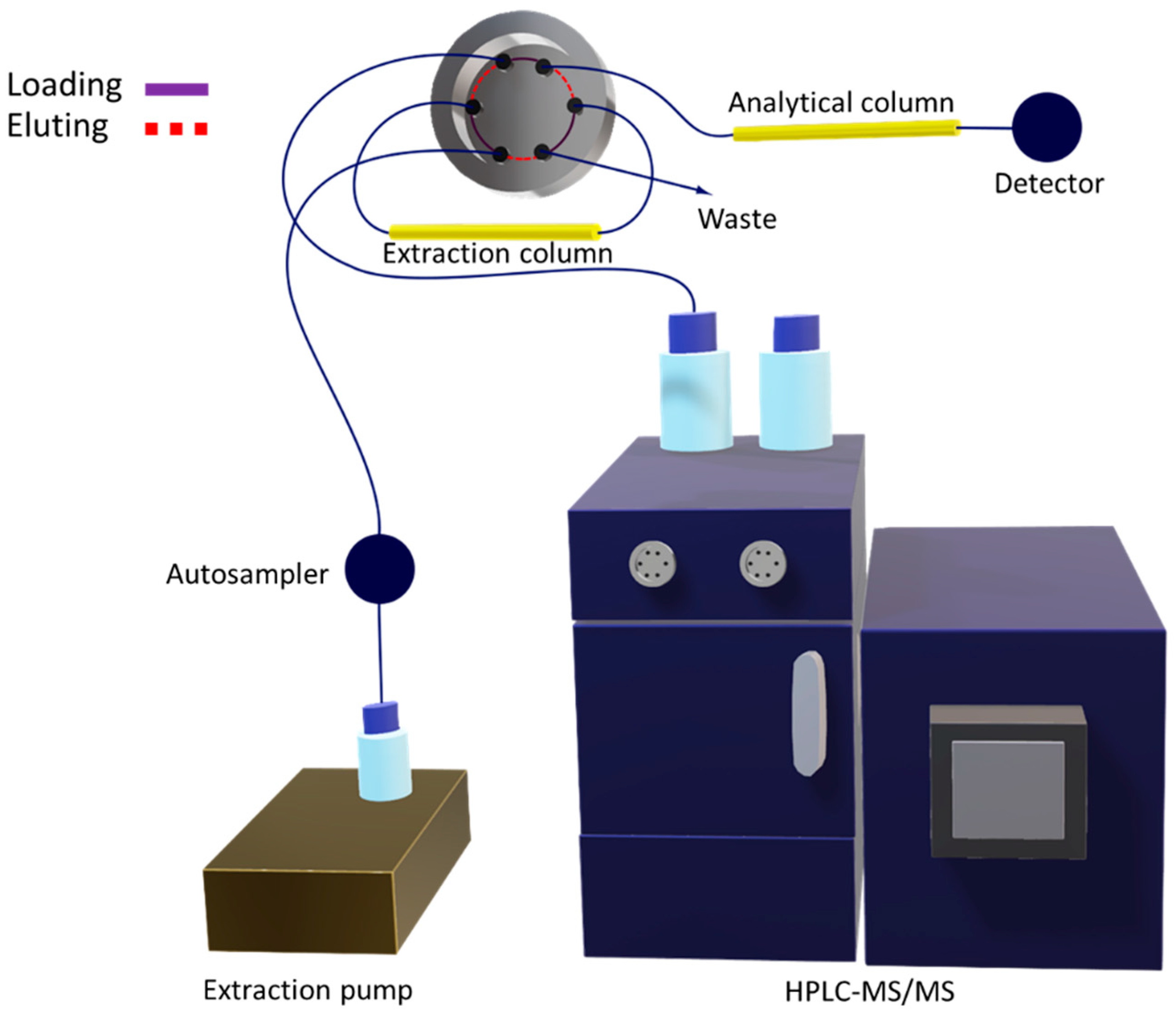
2.1.2. Multidimensional Automated Procedure
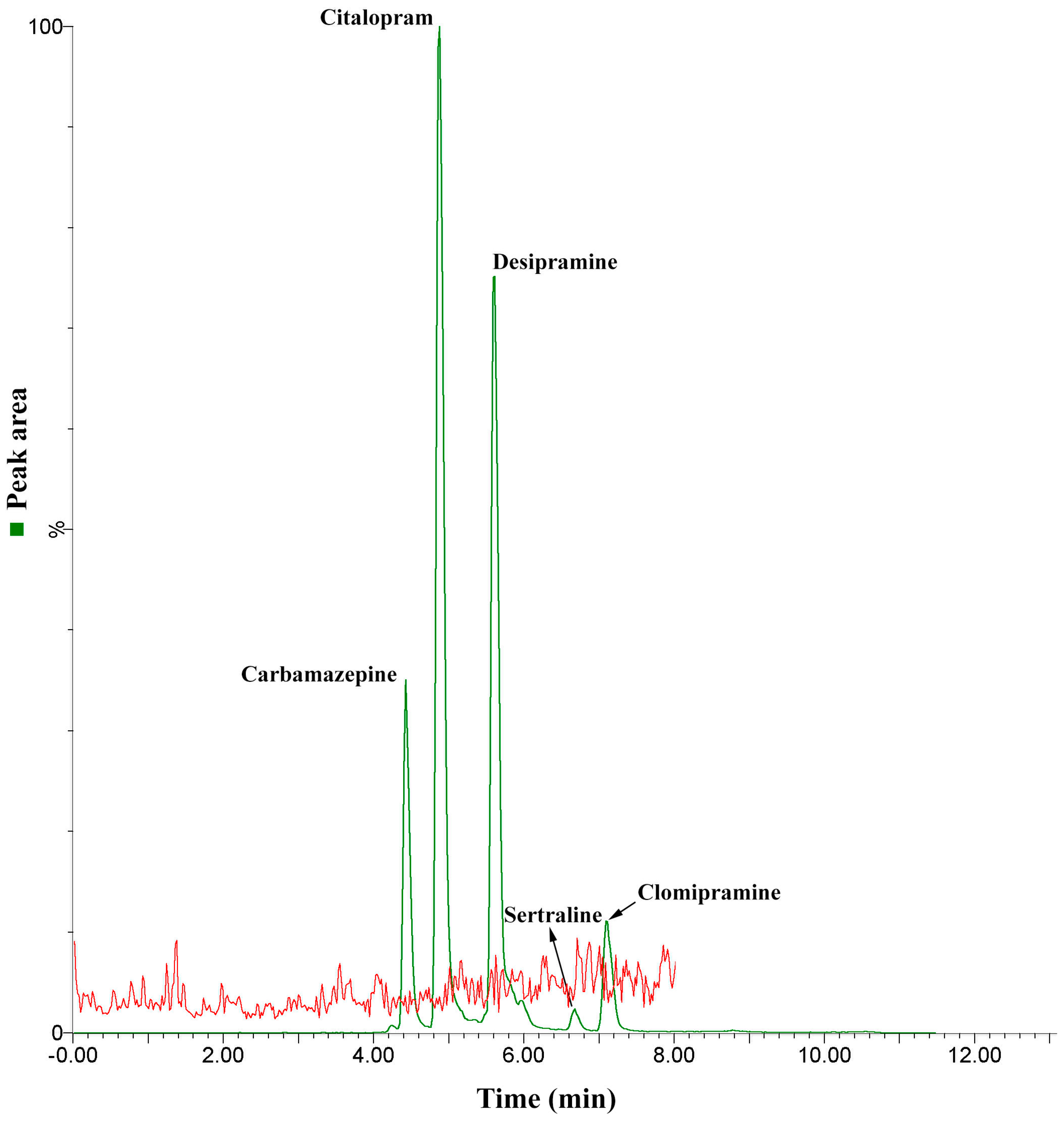
2.2. Figures of Merit
2.3. Overall Method Performance
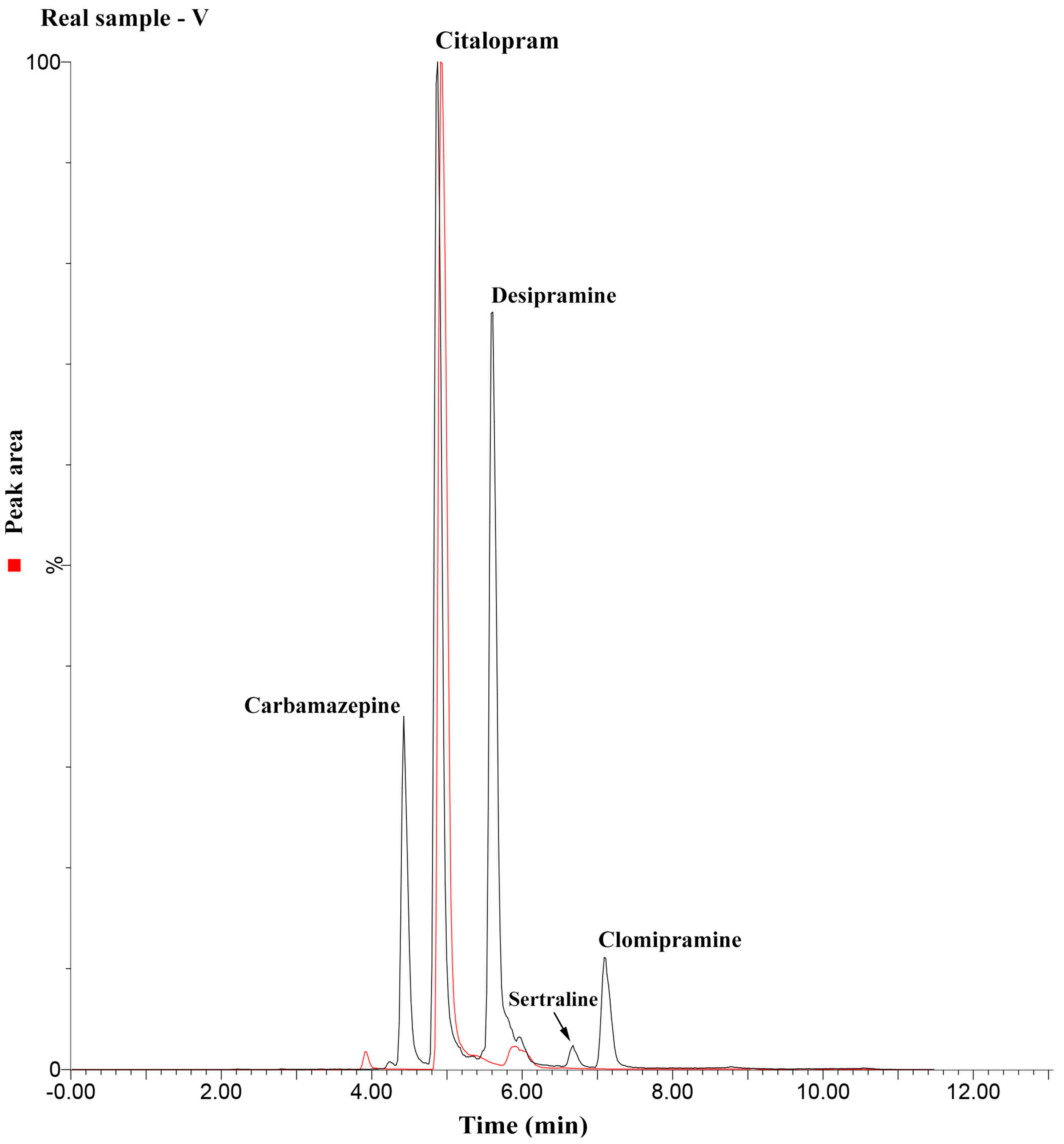
2.4. Method Application
3. Experimental
3.1. Reagents and Standard Solutions
3.2. Extraction Column Preparation
3.3. Instrumentation
3.4. Multidimensional Analytical Method
3.5. Method Enhancement
3.6. Figures of Merit
3.7. Method Application
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Murtada, K.; de Andrés, F.; Zougagh, M.; Ríos, Á. Strategies for antidepressants extraction from biological specimens using nanomaterials for analytical purposes: A review. Microchem. J. 2019, 150, 104193. [Google Scholar] [CrossRef]
- Gotlib, I.H.; Joormann, J. Cognition and Depression: Current Status and Future Directions. Annu. Rev. Clin. Psychol. 2010, 6, 285–312. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.T.; Haigh, E.A.P. Advances in Cognitive Theory and Therapy: The Generic Cognitive Model. Annu. Rev. Clin. Psychol. 2014, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Alves, V.; Gonçalves, J.; Conceição, C.; Câmara, H.M.T.; Câmara, J.S. An improved analytical strategy combining microextraction by packed sorbent combined with ultra high pressure liquidchromatography for the determination of fluoxetine, clomipramineand their active metabolites in human urine. J. Chromatogr. A 2015, 1408, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Dome, P.; Tombor, L.; Lazary, J.; Gonda, X.; Rihmer, Z. Natural health products, dietary minerals and over-the-counter medications as add-on therapies to antidepressants in the treatment of major depressive disorder: A review. Brain Res. Bull. 2019, 146, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Ghoraba, Z.; Aibaghi, B.; Soleymanpour, A. Application of cation-modified sulfur nanoparticles as an efficient sorbent for separation and preconcentration of carbamazepine in biological and pharmaceutical samples prior to its determination by high-performance liquid chromatography. J. Chromatogr. B 2017, 1063, 245–252. [Google Scholar] [CrossRef]
- Dear, J.W.; Bateman, D.N. Antidepressants. Medicine 2016, 44, 135–137. [Google Scholar] [CrossRef]
- Liveri, K.; Constantinou, M.A.; Afxentiou, M.; Kanari, P. A fatal intoxication related to MDPV and pentedrone combined with antipsychotic and antidepressant substances in Cyprus. Forensic Sci. Int. 2016, 265, 160–165. [Google Scholar] [CrossRef]
- Zheng, M.M.; Wang, S.T.; Hu, W.K.; Feng, Y.Q. In-tube solid-phase microextraction based on hybrid silica monolith coupled to liquid chromatography-mass spectrometry for automated analysis of ten antidepressants in human urine and plasma. J. Chromatogr. A 2010, 1217, 7493–7501. [Google Scholar] [CrossRef]
- Rodríguez-Flores, J.; Salcedo, A.M.C.; Fernández, L.M. Rapid quantitative analysis of letrozole, fluoxetine and their metabolites in biological and environmental samples by MEKC. Electrophoresis 2009, 30, 624–632. [Google Scholar] [CrossRef]
- Truta, L.; Castro, A.L.; Tarelho, S.; Costa, P.; Sales, M.G.F.; Teixeira, H.M. Antidepressants detection and quantification in whole blood samples by GC–MS/MS, for forensic purposes. J. Pharm. Biomed. Anal. 2016, 128, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Mohebbi, A.; Farajzadeh, M.A.; Yaripour, S.; Mogaddam, M.R.A. Determination of tricyclic antidepressants in human urine samples by the three–step sample pretreatment followed by hplc–uv analysis: An efficient analytical method for further pharmacokinetic and forensic studies. EXCLI J. 2018, 17, 952–963. [Google Scholar] [PubMed]
- Niu, Z.; Zhang, W.; Yu, C.; Zhang, J.; Wen, Y. Recent advances in biological sample preparation methods coupled with chromatography, spectrometry and electrochemistry analysis techniques. TrAC Trends Anal. Chem. 2018, 102, 123–146. [Google Scholar] [CrossRef]
- Maciel, E.V.S.; de Toffoli, A.L.; Neto, E.S.; Nazario, C.E.D.; Lanças, F.M. New materials in sample preparation: Recent advances and future trends. TrAC Trends Anal. Chem. 2019, 119, 115633. [Google Scholar] [CrossRef]
- Maciel, E.V.S.; de Toffoli, A.L.; Lanças, F.M. Current status and future trends on automated multidimensional separation techniques employing sorbent-based extraction columns. J. Sep. Sci. 2019, 42, 258–272. [Google Scholar] [CrossRef] [PubMed]
- De Toffoli, A.L.; Maciel, E.V.S.; Lanças, F.M. Evaluation of the tubing material and physical dimensions on the performance of extraction columns for on-line sample preparation-LC–MS/MS. J. Chromatogr. A 2019, 1597, 18–27. [Google Scholar] [CrossRef]
- De Toffoli, A.L.; Maciel, E.V.S.; Fumes, B.H.; Lanças, F.M. The role of graphene-based sorbents in modern sample preparation techniques. J. Sep. Sci. 2018, 41, 288–302. [Google Scholar] [CrossRef]
- Vasconcelos Soares Maciel, E.; Henrique Fumes, B.; Lúcia de Toffoli, A.; Mauro Lanças, F. Graphene particles supported on silica as sorbent for residue analysis of tetracyclines in milk employing microextraction by packed sorbent. Electrophoresis 2018, 39, 2047–2055. [Google Scholar] [CrossRef]
- Ich Validation of Analytical Procedures: Text and Methodology Q2(R1) 2005, 1–17. Available online: https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf (accessed on 17 February 2020).
- Murtada, K.; de Andrés, F.; Ríos, A.; Zougagh, M. Determination of antidepressants in human urine extracted by magnetic multiwalled carbon nanotube poly(styrene-co-divinylbenzene) composites and separation by capillary electrophoresis. Electrophoresis 2018, 39, 1808–1815. [Google Scholar] [CrossRef]
- Fuentes, A.M.A.; Fernández, P.; Fernández, A.M.; Carro, A.M.; Lorenzo, R.A. Microextraction by packed sorbent followed by ultra high performance liquid chromatography for the fast extraction and determination of six antidepressants in urine. J. Sep. Sci. 2019, 42, 2053–2061. [Google Scholar] [CrossRef]
- Farsimadan, S.; Goudarzi, N.; Chamjangali, M.A.; Bagherian, G. Optimization of ultrasound-assisted dispersive liquid-liquid microextraction based on solidification of floating organic droplets by experimental design methodologies for determination of three anti-anxiety drugs in human serum and urine samples by high p. Microchem. J. 2016, 128, 47–54. [Google Scholar] [CrossRef]
- Resende, S.; Deschrijver, C.; van de Velde, E.; Verstraete, A. Development and validation of an analytical method for quantification of 15 non-tricyclic antidepressants in serum with UPLC-MS/MS. Toxicol. Anal. Clin. 2016, 28, 294–302. [Google Scholar] [CrossRef]
- Cai, J.; Zhu, G.T.; He, X.M.; Zhang, Z.; Wang, R.Q.; Feng, Y.Q. Polyoxometalate incorporated polymer monolith microextraction for highly selective extraction of antidepressants in undiluted urine. Talanta 2017, 170, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.C.; de Toffoli, A.L.; Maciel, E.V.S.; Lanças, F.M. Online fully automated SPE-HPLC-MS/MS determination of ceftiofur in bovine milk samples employing a silica-anchored ionic liquid as sorbent. Electrophoresis 2018, 39, 2210–2217. [Google Scholar] [CrossRef] [PubMed]
- El Sherbiny, D.; Wahba, M.E.K. Micellar liquid chromatographic method for the simultaneous determination of citalopram hydrobromide with its two demethylated metabolites. Utility as a diagnostic tool in forensic toxicology. J. Pharm. Biomed. Anal. 2019, 164, 173–180. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Linear Equation | R2 | LOD (µg L−1) | LOQ (µg L−1) |
|---|---|---|---|---|
| Carbamazepine | y = 1681.7 + 2835.6x | 0.999 | 0.01 | 0.5 |
| Citalopram | y = −2542.9 + 6904.3x | 0.997 | 0.04 | 0.5 |
| Clomipramine | y = −14200.5 + 1167.5x | 0.985 | 0.5 | 25 |
| Desipramine | y = −1593.3 + 7048.4x | 0.994 | 0.01 | 0.5 |
| Sertraline | y = −1614.1 + 128.4x | 0.985 | 2.0 | 20 |
| Analytes | Enrichment Factor | Accuracy (%) | Precision (% RSD) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | |||||||||||
| L | M | H | L | M | H | L | M | H | L | M | H | |
| Carbamazepine | 4.7 | 5.3 | 5.1 | 83.2 | 95.8 | 98.8 | 12.3 | 2.3 | 3.6 | 13.6 | 2.1 | 1.4 |
| Citalopram | 6.8 | 7.6 | 7.0 | 125.3 | 89.7 | 99.1 | 2.0 | 1.9 | 3.2 | 6.8 | 2.9 | 5.5 |
| Clomipramine | 17.3 | 18.1 | 17.4 | 98.7 | 117.6 | 102.4 | 5.2 | 6.1 | 4.0 | 9.2 | 3.2 | 4.8 |
| Desipramine | 18.2 | 16.4 | 15.0 | 105.8 | 114.8 | 102.3 | 6.9 | 3.5 | 11.8 | 11.2 | 4.5 | 2.4 |
| Sertraline | 21.2 | 59.4 | 13.1 | 98.7 | 117.6 | 102.4 | 12.8 | 4.5 | 6.5 | 8.1 | 4.1 | 1.4 |
| Analyte | Precursor Ion (m/z) | Product Ion (m/z) | Cone Voltage (V) | Collision Energy (V) | Dwell Time (ms) |
|---|---|---|---|---|---|
| Carbamazepine | 253 | 152 | 24 | 42 | 0.075 |
| 167 | 24 | 44 | 0.075 | ||
| 180 | 24 | 32 | 0.075 | ||
| Desipramine | 267 | 72 | 22 | 14 | 0.075 |
| 193 | 22 | 42 | 0.075 | ||
| 208 | 22 | 24 | 0.075 | ||
| Sertraline | 306 | 123 | 16 | 48 | 0.075 |
| 159 | 16 | 30 | 0.075 | ||
| 275 | 16 | 14 | 0.075 | ||
| Clomipramine | 315 | 58 | 24 | 30 | 0.075 |
| 86 | 24 | 18 | 0.075 | ||
| 227 | 24 | 42 | 0.075 | ||
| Citalopram | 325 | 109 | 32 | 30 | 0.075 |
| 234 | 32 | 26 | 0.075 | ||
| 262 | 32 | 20 | 0.075 |
| Event | Time (min) | Solvent Composition (Extraction Column) | Solvent Composition (Analytical Column) |
|---|---|---|---|
| Loading | 0.00–1.00 | H2O + 0.2% FA | H2O (A)/ACN (B) * (30%:70%) |
| Eluting | 1.00–3.00 | H2O (A)/ACN (B) * (30%:70% → 35%:65%) | H2O (A)/ACN (B) * (30%:70% → 35%:65%) |
| 3.00–6.00 | H2O (A)/ACN (B) * (35%:65% → 40:60%) | H2O (A)/ACN (B) * (35%:65% → 40:60%) | |
| 5.00–6.00 | H2O (A)/ACN (B) * (40%:60%) | H2O (A)/ACN (B) * (40%:60%) | |
| Cleaning | 6.00–7.00 | H2O (A)/ACN (B) * (40%:60% → 50%:50%) | H2O (A)/ACN (B) * (40%:60% → 50%:50%) |
| 7.00–7.66 | H2O (A)/ACN (B) * (50%:50% → 10%:90%) | H2O (A)/ACN (B) * (50%:50% → 10%:90%) | |
| 7.66–8.60 | H2O + 0.2% FA | H2O (A)/ACN (B) * (10%:90%) | |
| Conditioning | 8.60–11.50 | H2O + 0.2% FA | H2O (A)/ACN (B) * (30%:70%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maciel, E.V.S.; Toffoli, A.L.d.; Alves, J.d.S.; Lanças, F.M. Multidimensional Liquid Chromatography Employing a Graphene Oxide Capillary Column as the First Dimension: Determination of Antidepressant and Antiepileptic Drugs in Urine. Molecules 2020, 25, 1092. https://doi.org/10.3390/molecules25051092
Maciel EVS, Toffoli ALd, Alves JdS, Lanças FM. Multidimensional Liquid Chromatography Employing a Graphene Oxide Capillary Column as the First Dimension: Determination of Antidepressant and Antiepileptic Drugs in Urine. Molecules. 2020; 25(5):1092. https://doi.org/10.3390/molecules25051092
Chicago/Turabian StyleMaciel, Edvaldo Vasconcelos Soares, Ana Lúcia de Toffoli, Jussara da Silva Alves, and Fernando Mauro Lanças. 2020. "Multidimensional Liquid Chromatography Employing a Graphene Oxide Capillary Column as the First Dimension: Determination of Antidepressant and Antiepileptic Drugs in Urine" Molecules 25, no. 5: 1092. https://doi.org/10.3390/molecules25051092
APA StyleMaciel, E. V. S., Toffoli, A. L. d., Alves, J. d. S., & Lanças, F. M. (2020). Multidimensional Liquid Chromatography Employing a Graphene Oxide Capillary Column as the First Dimension: Determination of Antidepressant and Antiepileptic Drugs in Urine. Molecules, 25(5), 1092. https://doi.org/10.3390/molecules25051092